
Abstract
Stephania tetrandra S. Moore (S. tetrandra) is distributed widely in tropical and subtropical regions of Asia and Africa. The root of this plant is known in Chinese as “Fen Fang Ji”. It is commonly used in traditional Chinese medicine to treat arthralgia caused by rheumatism, wet beriberi, dysuria, eczema and inflamed sores. Although promising reports have been published on the various chemical constituents and activities of S. tetrandra, no review comprehensively summarizes its traditional uses, phytochemistry, pharmacology and toxicology. Therefore, the review aims to provide a critical and comprehensive evaluation of the traditional use, phytochemistry, pharmacological properties, pharmacokinetics and toxicology of S. tetrandra in China, and meaningful guidelines for future investigations.
Graphic abstract

Similar content being viewed by others

Introduction
Stephania tetrandra S. Moore (S. tetrandra) is a traditional medicine used in China, Japan and South Korea. S. tetrandra is a perennial liana of the genus Stephania, belonging to the Menispermaceae family. The official name of S. tetrandra is “Fen Fang Ji” in Chinese. “Fang Ji” or “Han Fang Ji” or “Guang Fang Ji” or “Mu Fang Ji” in Chinese is also usually used as the folk name of S. tetrandra. However, Aristolochia fangchi Y.C. Wu ex L.D. Chow & S.M. Hwang (A. fangchi) (Aristolochiaceae) and Aristolochia heterophyla Hemsl (A. heterophyla) (Aristolochiaceae) are two separate species also used in traditional Chinese medicine (TCM) that are also called “Fang ji”, which can cause confusion. A. fangchi and A. heterophyla contain high concentrations of aristolochic acids that can result in nephrotoxicity. When used medicinally, the morphological similarities between the two roots pose a potential identification problem that may result in accidental substitution (Tankeu et al. 2016). Thus, a comprehensive reference of available information on S. tetrandra would prevent confusion in its usage.
S. tetrandra is principally used in TCM as a diuretic, anti-inflammatory and an antirheumatic treatment (Joshi et al. 2008; Chinese Pharmacopoeia Commission 2015; Jiangsu New Medical College 2006; Chinese Botany Editorial Committee 1996). Over the previous three decades, extensive chemical analysis and modern activity evaluation have been conducted because its traditional uses are of interest to researchers. To date, phytochemical studies have identified a total of 67 alkaloids, two bisflavones, two phytosterols and n-pentatriacontane from the roots and aerial parts of S. tetrandra.
The use of herbal medicines has gained popularity worldwide due to their long history of traditional use, their cost-effectiveness, accessibility and reportedly lower incidence of adverse effects (Mullaicharam 2011). Yet now the phrase “Chinese-herb nephropathy” has recently attracted attention because the aristolochic acid present in a number of TCMs can cause nephropathy (Nortier et al. 2015; Meyer et al. 2000; Okada 1999). “Yao Xing Lun” is an ancient TCM book from the Tang Dynasty of China that reported that “Fang Ji” had “little toxicity”. Although other ancient TCM books have labeled “Fang Ji” as non-toxic, it is not suitable for long-term use in large doses. In fact, there are published reports that S. tetrandra has toxic effects in the kidneys and liver (Liang et al. 2010). It has not yet been established whether the “little toxicity” is a result of aristolochic acids or the other constituents in S. tetrandra. To fully evaluate this traditional medicine, it is important to systematically review its traditional uses, phytochemistry, pharmacological properties, and toxicity. This will assist in establishing the profile of therapeutic and adverse clinical effects.
Ethnobotany of S. tetrandra
Botany
S. tetrandra is a perennial herbaceous liana of the subgenus Botryodiscia of the genus Stephania and a member of the Menispermaceae family. According to “The Plant List” (www.theplantlist.org), Stephania tetrandra S. Moore (S. tetrandra) is the only accepted name for the plant, and no synonyms have been reported for it.
S. tetrandra grows to a height of 1–3 m with a root that is fleshy and cylindrical. It has a long cylindrical stem that is straight and voluminous. The leaves are triangular in nature with a convex tip and a petiole that is 3–7 cm in length (Fig. 1). Its inflorescence consists of yellow-white flowers that form a capitulum between May and June, with staminate flowers that are radially symmetrical. The fruit is red and nearly spherical, ripening between July and September (Jiangsu New Medical College 2006; Chinese Botany Editorial Committee 1996; Feng et al. 1983; Luo 1982; Xie 2014).

Stephania tetrandra: a and b stem, flower and leaf, and c a longitudinal section of the roots
S. tetrandra grows on hillsides, foothills and the edges of grasslands and shrublands. In China, it is principally found in the provinces of Zhejiang, Anhui, Fujian, Taiwan, Hunan, Jiangxi, Guangxi, Guangdong and Hainan. Although the plant is robust, it has a slow growth cycle. Three years of growth are required before the roots of S. tetrandra can be used for medicinal purposes (Jiangsu New Medical College 2006; Chinese Botany Editorial Committee 1996; Zhu et al. 1983; Xie 2014). Currently, the majority of S. tetrandra used for medicinal purposes are wild-growing rather than cultivated.
Traditional use
“Fang Ji" was firstly recorded as medicine in an ancient Chinese medicinal book of "Shen Nong Ben Cao Jing" during the Han Dynasty (Huang et al. 2015). However, “Fang Ji” could refer to Aristolochia heterophyla Hemsl (“Hang Fang Ji”) or Cocculus trilobus (L.) DC (“Mu Fang Ji”) in ancient China (Huang et al. 2015; Hu 2009). Although “Fang Ji” used as traditional Chinese medicine over a thousand year, the medicinal history of S. tetrandra is few hundreds years (Huang et al. 2015; Hu 2009). Huang et al. speculated that S. tetrandra used as “Fang Ji” medicine in Ming Dynast for the first time (Huang et al. 2015), but the study of Chen et al. investigated that S. tetrandra was the main medicinal plant source of “Fang Ji” in the Republic China period (Chen 2006). Anyhow, S. tetrandra is officiallized as “Fangji” by the authoritative Chinese medicine book, “Pharmacopoeia of People’s Republic of China, 2015 version”.
The medicinal component of “Fang Ji” is its root, which was firstly recorded in the ancient Chinese book “Sheng Nong Ben Cao Jing” written during the Han Dynasty. A number of ancient Chinese medicinal books from Han Dynasty to Qing Dynasty, such as “Min Yi Bie Lu”, “Ben Cao Gang Mu”, “Ben Cao Cong Xin”, recorded that “Fang Ji” was suitable as a treatment for “wind-cold”, “wind-swelling”, treats warm malaria, edema, typhoid fever and stroke, acts as a diuretic, and so on.
To summarize, “Fang Ji” has been used in China as a folk remedy for arthralgia associated with rheumatoid arthritis, wet beriberi, dysuria, eczema and inflamed sores (Jiangsu New Medical College 2006). Nevertheless, different species resulted in different traditional medicinal effects. “Fang Ji” could refer to “Han Fang Ji” or “Mu Fang Ji” before Ming and Qing Dynasty. Afterwards, “Fang Ji” could also refer to “Han Fang Ji” or “Mu Fang Ji” or “Feng Fang Ji” or “Guang Fang Ji”. As recored in two ancient Chinese medicinal books “Ben Cao Meng Quan” and “Ben Cao Shi Yi”, “Mu” was used for the treatment of “Wind”, such as “wind-cold”, “wind-swelling”, “Han” was used for the treatment of “Water”, such as wet beriberi, dysuria. Addtionally, the study of Hu indicated that C. trilobus was used for the treatment of “Wind”, A. heterophyla, A. fangchi and S. tetrandra were used for the treatment of “Water” (Hu 2009). Meanwhlie, crude preparations or extracts of S. tetrandra exhibit numerous therapeutic effects. Such crude preparations have been in recent use to safely treat arthritis (Tankeu et al. 2016; Jiangsu New Medical College 2006; Sekiya et al. 2004), hypertension (Jiangsu New Medical College 2006), apoplexy (Jiangsu New Medical College 2006), edema (Jiangsu New Medical College 2006), neuralgia (Kang et al. 1996) and hepatic fibrosis (Sun et al. 1999).
An additional important traditional use of S. tetrandra is in Chinese herbal formulations which consisting of two or more Chinese herbs. Han-Dan-Gan-Le is a Chinese medicinal preparation which comprising Salvia miltorrhiza, Radix paeoniae, Astragalus membranaceus, S. tetrandra and the dried leaves of Ginkgo biloba. Han-Dan-Gan-Le (0.5 and 1.0 g/kg, administered orally) has been used for many years to successfully treat fibrosis and cirrhosis of the liver (Li et al. 1998). The decoction of Fang-Ji-Di-Huang (administered orally) which comprising Rehmannia glutinosa (Gaetn.) Libosch. (30 g), S. tetrandra (10 g), Cinnamomum cassia Presl (10 g), Saposhnikovia divaricata (Turcz.) schischk (10 g) and Glycyrrhizae radix et rhizome (10 g), utilized in the treatment of rheumatic arthritis (Zhao 2018). Fang-Ji-Huang-Qi-Tang contains six medicinal herbs, including S. tetrandra, Radix astragali, Rhizoma atractylodis macrocephalae, Radix glycyrrhizae, Rhizoma zingiberis and Fructus ziziphi jujubae. Its extracts (25, 50 or 100 mg/kg, administered orally) is a common Chinese prescription for the treatment of painful and inflammatory disorders such as rheumatoid arthritis (Lin et al. 2015) and renal edema (Gao 2017). Fang-Ji-Fu-Ling-Tang comprises five herbs, including S. tetrandra (20 g), Poria cocos (Schw.) wolf (10 g), Radix astragali (10 g), Cinnamomum cassia Presl (10 g) and Glycyrrhizae radix et rhizome (6 g). Its extracts (administered orally) has been used in the management of post-thrombotic syndrome (Li et al. 2018a) and chronic heart failure (Wang and Lu 2015).
Phytochemistry of S. tetrandra
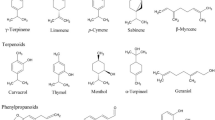
The majority of studies of the phytochemistry of S. tetrandra have been published within the last 30 years. The vast majority have reported that the major components of S. tetrandra are alkaloids, almost all identified within the roots. There appear to be only five non-alkaloid constituents isolated from the aerial part of S. tetrandra, two bisflavones (Si and Zhao 1993; Si et al. 2001), β-sitosterol (Si and Zhao 1993), stigmasterol (Liu et al. 2017a) and n-pentatriaeontane (Si and Zhao 1993). There have been no phytochemical studies that have reported on the flowers and fruits of S. tetrandra. To date, a total of 67 alkaloids and 5 non-alkaloids have been identified in S. tetrandra (Table 1). Alkaloids classified into six structural classes, including monobenzyltetrahydroisoquinolines, bisbenzyltetrahydroisoquinolines, aporphines, protoberberines and tetrahydroprotoberberines.
Monobenzyltetrahydroisoquinolines
Eight monobenzyltetrahydroisoquinolines have been reported to be present in S. tetrandra (Fig. 2): (1) N-methylcoclaurine (Xiao et al. 2018); (2) juziphine (Xiao et al. 2018); (3) coclaurine (Xiao et al. 2018; Sim et al. 2015); (4) protosinomenine (Xiao et al. 2018); (5) reticuline (Xiao et al. 2018); (6) coclaurine-7-O-β-d-glucopyranoside (Sim et al. 2015); (7) oblongine (Sim et al. 2015) and (8) oblongine chloride (Wino et al. 1988). Of these, N-methylcoclaurine, juziphine, coclaurine, protosinomenine, reticuline, coclaurine-7-O-β-d -glucopyranoside and oblongine have been tentatively identified using liquid chromatography-mass spectrometry (LC–MS) (Xiao et al. 2018; Sim et al. 2015).

Structures of 8 monobenzyltetrahydroisoquinolines
Bisbenzyltetrahydroisoquinolines
Bisbenzyltetrahydroisoquinolines are the major active constituents in S. tetrandra. To date, 30 bisbenzyltetrahydroisoquinolines have been identified as constituents (Xiao et al. 2018; Sim et al. 2013, 2015; Wino et al. 1988; Tsutsumi et al. 2003; Ogino et al. 1998; Li et al. 2009a; Si et al. 1992; Si and Zhao 1991; Deng et al. 1990; Tian 2014) (Fig. 3). Of these, homoaromaline (23), stephibaberine (24), obaberine (26), isotetrandrine (27), northalrugosdine (28) and 2-norcepharanthine (29) are the only compounds identified using LC–MS (Xiao et al. 2018). The 2 and/or 2′ N-position of bisbenzyltetrahydroisoquinolines in S. tetrandra is commonly oxidized or salified with a chloride ion (Wino et al. 1988; Tsutsumi et al. 2003; Ogino et al. 1998), as can be seen in compounds 11, 13, 15, 16, 17, 18, 30, 31, 32, 33, 34 and 35.

Structures of 30 bisbenzyltetrahydroisoquinolines
Aporphines
Fifteen aporphines have been reported to be present in S. tetrandra (Fig. 4). Only isoboldine (47), corytuberine (48), magnoflorine (49), dehydrocrebanine (50), dehydrodicentrine (51) and roemerine (52) have been tentatively identified using LC–MS (Xiao et al. 2018; Sim et al. 2013, 2015). The remaining 9 aporphines have been characterized using comprehensive spectroscopic techniques (Li et al. 2009a; Si et al. 1992; Si and Zhao 1991; Deng et al. 1990).

Structures of 15 aporphines
Protoberberines
Two protoberberines, stepharanine (54) and dehydrodiscretamine (55) (Fig. 5), have been identified in S. tetrandra using LC–MS (Xiao et al. 2018).

Structures of 2 protoberberines
Tetrahydroprotoberberines
In total, nine tetrahydroprotoberberines have been identified in S. tetrandra (Fig. 6). Only cyclanoline chloride (56) has been isolated from S. tetrandra following the application of comprehensive spectroscopic techniques (Wino et al. 1988; Tsutsumi et al. 2003). The others have been tentatively identified using LC–MS (Xiao et al. 2018).

Structures of 9 tetrahydroprotoberberines
Other alkaloids
Three aporphine alkaloids (Fig. 7), in which C6–C6a has been cleaved, have been isolated from S. tetrandra (Wino et al. 1988; Tsutsumi et al. 2003; Ogino et al. 1998).

Structures of 3 aporphine alkaloids with C6–C6a cleavage
Non-alkaloids
Only five non-alkaloid constituents in S. tetrandra were reported in literature (Fig. 8). The five non-alkaloid constituents all isolated from the aerial part of S. tetrandra, stephaflavone A (68) (Si and Zhao 1993; Si et al. 2001), Stephaflavone B (69) (Si and Zhao 1993; Si et al. 2001), β-sitosterol (70) (Si and Zhao 1993), stigmasterol (71) (Liu et al. 2017a) and n-pentatriaeontane (72) (Si and Zhao 1993).

Structures of 5 non-alkaloid constituents
Pharmacological properties
S. tetrandra has a wide range of pharmacological properties. To date, the published literature has focused principally on the alkaloids and extracts of S. tetrandra. The molecules that have received the greatest in-depth study of their pharmacological activity are tetrandrine (Yang et al. 2018a; Bhagya and Chandrashekar 2016; Wang et al. 2017a; Xing et al. 2014; Cai et al. 2011), fangchinoline (Desgrouas et al. 2014a; Wang et al. 2017a; Xing et al. 2014) and cepharanthine (Bailly 2019; Wang et al. 2017a). Not only are these compounds found in S. tetrandra, but also in other plants known to have medicinal properties (Table 2).
Antimicrobial effects
Tetrandrine represents an active bisbenyltetrahydroisoquinoline alkaloid found in S. tetrandra that displays direct and synergistic antimicrobial effects against bacteria, parasites, viruses and fungi.
Antiparasitic effects
To date, research of the antiparasitic effects of compounds in S. tetrandra has focused on their antiplasmodial activity. Furthermore, a number of in vivo studies and numerous in vitro reports have indicated that compounds in S. tetrandra, mostly compounds 5, 9, 10, 21, 25, 28, 29 and 52, display antiparasitic effects.
In vitro studies have demonstrated that cepharanthine (25) (50% inhibitory concentration (IC50) 1.2 μM), and roemerine (52) (IC50 17.9 μM) exhibit significant antiplasmodial activity with IC50 values that are relatively low and similar strong activity with control compound (Chloroquine, IC50 0.7 μM) (Baghdikian et al. 2013). But cepharanthine and roemerine also displayed the stronger cytotoxicity against K562S cells at IC50 of 3.3 μM and 16.1 μM, respectively, compared with control compound (Chloroquine, IC50 32 μM) (Baghdikian et al. 2013). Although reticuline (5) and cycleanine (21) showed no stronger activity than chloroquine (IC50 0.002 ± 0.09 μM), reticuline and cycleanine could significantly inhibit the growth of Plasmodium falciparum at IC50 1.18 ± 0.08 μM and 0.08 ± 0.06 μM, respectively (Fadaeinasab et al. 2015). Additionally, reticuline (IC50 148.0 ± 1.2 μM) and cycleanine (52.96 μg/ml) also showed weak activity against Leishmania infantum in vitro (García Díaz et al. 2019; González-Coloma et al. 2012). Nevertheless, these investigations were conducted without the use of positive control and also lack analysis of its mechanism. At the concentration of 1 × 10−5 μM, tetrandrine (9) has potent antimalarial effect on both chloroquine sensitive and resistant strains of Plasmodum falciparum in vitro (Ye et al. 1989; Ye and Dyke 1989). In vitro, fangchinoline (10) and cepharanthine (25) showed similar antiplasmodial activity (IC50 0.2 μM for cepharanthine, 0.3 μM for fangchinoline), but stronger antiplasmodial activity than the control chloroquine (IC50 0.6 μM) (Chea et al. 2010). Also in this study, fangchinoline and cepharanthine displayed stronger cytotoxicity than chloroquine to human THP1 cells (Chea et al. 2010). 2-Norcepharanthine (29) and cycleanine have strong antiplasmodial activities against P. falciparum D6 at similar IC50 about 80 nM compared with chloroquine (IC50 6.18 ± 0.71 nM) (Angerhofer et al. 1999). The result of cycleanine in this study was consistent with that of Fadaeinasab’s study (Fadaeinasab et al. 2015). Yet the cytotoxicity of 2-norcepharanthine and cycleanine need to be assessed. Northalrugosidine (28) showed the most potent in vitro activity against Leishmania donovani promastigotes (0.28 μM), and the highest selectivity (29.3-fold) versus its general cytotoxicity against HT-29 human colon adenocarcinoma cells (Naman et al. 2015). Meanwhile the reduction in parasite burden to the murine spleen and liver after the in vivo testing of northalrugosidine at 2.8, 5.6, and 11.1 mg/kg per animal when administered intravenously supported the idea that it could be further investigated as a potential new drug candidate or lead molecule for drug discovery (Naman et al. 2015). However, most of these studies lacked evaluation of compounds in vivo, and investigation of mechanism of action.
In addition to in vitro studies, tetrandrine (15 mg/kg, 30 mg/kg or 60 mg/kg) has been found to be three times more effective against chloroquine-resistant strains of P. falciparum than against the sensitive strain in Aotus monkeys, having even greater activity when combined with chloroquine or artemisinin (Ye et al. 2013). Moreover, tetrandrine (20 mg/kg) exhibited an effect greater than albendazole against Echinococcus multilocularis and combining the two compounds (20 mg/kg tetrandrine and 50 mg/kg albendazole) resulted in a synergistic effect (Chen et al. 2002, 2003). Whereas the mechanism by which this bioactivity occurs should be further investigated. The combination of cepharanthine (21 mg/kg or 63 mg /kg) with chloroquine (30 mg/kg) or amodiaquine (10 mg/kg) was demonstrated to significantly improve the survival of mice and delay parasitic recrudescence (Desgrouas et al. 2014b). The mechanism by which cepharanline acts against plasmodia is to block the development of P. falciparum in the ring stage via down-regulation of Maurer’s clefts (Desgrouas et al. 2014c). Notwithstanding, further research models are critically required to confirm this mechanism both in in vivo models and in humans.
Antibacterial effects
Only two compounds, 9 and 52, have been evaluated for their antibacterial effects. Tetrandrine (9) (200.9, 401.9 or 803.8 μM) exhibited a synergistic effect against methicillin-resistant Staphylococcus aureus (MRSA) when combined with the multi-drug resistant (MDR)-efflux pump substrate ethidium bromide, causing a two to fourfold reduction in the minimum inhibitory concentration (MIC) of ethidium bromide against MRSA using a combined test (Lee et al. 2011). Tetrandrine at concentrations of 200.9–803.8 μM inhibited both methicillin-susceptible and methicillin-resistant strains of Staphylococcus aureus (S. aureus), with a MIC of 401.9 μM (Lee et al. 2012). Meanwhile, it was demonstrated that tetrandrine-induced damage of bacterial cell walls, in combination with potential interference of cell wall biosynthesis via its direct binding to peptidoglycan, may underlie the synergy against S. aureus (Lee et al. 2012; Zhao et al. 2001). Though the compound exhibited potent inhibitory activity against S. aureus, the required dose of the two compounds was excessive in these studies, the experimental data not being relevant to clinical applications. Moreover, the two studies lacked appropriate positive controls. Tetrandrine (48.23 μM), an efflux pump inhibitor as verapamil, could reverse drug resistance in 82% of isoniazid and ethambutol-dual resistant clinical isolates of Mycobacterium tuberculosis, but did not show a direct antibacterial activity against Mycobacterium tuberculosis (Zhang et al. 2015). Quite on the contrary, another study revealed that tetrandrine at 12.86–51.44 μM exerted strong direct tuberculostatic activity against 16 Mycobacterium tuberculosis strains in vitro (Vichkanova et al. 1973). Yet these investigations were conducted without the use of positive control, and the direct antibacterial activity against Mycobacterium tuberculosis of tetrandrine need to be validated furtherly. In both in vitro and in vivo studies, roemerine (52) (MICs ranging from 114.7 μM to 229.4 μM in vitro; the concentration of 20 mg/kg was used in vivo) demonstrated antibacterial effects towards methicillin susceptible and methicillin-resistant strains, and increased bacterial cell membrane permeability in a concentration-dependent manner (Yin et al. 2015). More importantly, roemerine (229.4 μM) showed similar antibacterial effects with positive controls of vancomycin (11.0 μM) and oxacillin (638.4 μM) in this study (Yin et al. 2015). The antibacterial mechanisms of roemerine (from 268.8 to 358.4 μM) demonstrated that it accumulates intracellularly, but it is susceptible to being subject to efflux from cells through multidrug resistant transporter proteins (Avci et al. 2018). Roemerine was shown to be effective against Escherichia coli in vitro (Ayyildiz et al. 2017; Gokgoz and Akbulut 2015). By assessing these studies, the concentrations of roemerine were too high to be considered antibacterial effects towards methicillin susceptible and methicillin-resistant strains (Cos et al. 2006; Gertsch 2009). Moreover, roemerine was not be evaluated for cytoxicity at such high concentrations in these studies. Additionally, cycleanine showed broad antibactericidal effects on Bacillus subtilus, Cornynebacterium diphtheria, Klebsiella pneumonia, Pseudomonas aeruginosa, Salmonella typhi, Streptococcus pyogenes at the concenrtaion of 1607.7 μM (Lohombo-Ekomba et al. 2004). As the same above problem, it is still somewhat doubtful whether the broad antibactericidal effects of cycleanine found at such high concentration are meaningful. Moreover, this study had other shortages, including no evalution of solvent activity and a single dose of cycleanine, resulting into lower reliability and absence of dose-dependent effect.
Antifungal effects
To date, the antifungal effects of S. tetrandra have principally focused on Candida albicans. Tetrandrine (9) (48.2–64.3 μM) exhibits synergistic effects with fluconazole against both fluconazole-sensitive and fluconazole-resistant Candida albicans (C. albicans) (Li and Zhang 2006; Lv et al. 2014; Zhao et al. 2019; Benhamou et al. 2016; Zhang et al. 2009a, 2013) and also directly inhibits C. albicans in mice (Shi et al. 2016a). Nonetheless, these studies lacked appropriate positive controls and adequately broad dose range. Moreover, the synergistic effects of tetrandrine should be doubted at such high concentrations (Cos et al. 2006; Gertsch 2009). In a mouse candida vaginitis model, vaginal administration of 13 mg/kg/day of tetrandrine also had a synergistic antifungal effect when combined with ketoconazole against C. albicans (Zhang et al. 2010). But, only a single dose of tetrandrine was used throughout the study, limiting the reliability of the dose–response characterisitics. Moreover, this investigation was conducted without the use of a positive control. In addition, tetrandrine (20, 40 or 60 mg/kg/day) exhibits synergistic effects with posaconazole or itraconazole or voriconazole against Aspergillus fumigatus in vitro and in vivo, and the synergistic mechanism was related to inhibition of the drug efflux pump (Li et al. 2015a, 2017a). Magnoflorine (49) has demonstrated a substantial ability to inhibit the growth of Candida strains, as evidenced by a reduction in formation of a C. albicans biofilm at a MIC of 106.6 μM, based on microdilution antifungal susceptibility testing. Magnoflorine didn’t have any toxicity to HaCaT cells even in 600 μM of treatment (Kim et al. 2018). The main limitations of the study are that high doses of magnoflorine were used and the study lacked a positive control. Compared with positive control amphotericin B (0.04 µg/disc), α or β-magnoflorine (5.0 µg/disc or 10 µg/disc) showed weak antifungal activities against Penicillium avellaneum UC-4376 (Chen et al. 2009a). Furthermore, only a single dose of α or β-magnoflorine were used throughout the studies, and thus limited the information on the dose-dependent effect. Roemerine (52) at 3.58 µM was also shown to inhibit the formation of a C. albicans biofilm in vitro, possibly related to the cyclic adenosine monophosphate (cAMP) pathway (Ma et al. 2015). The toxicity of roemerine was investigated in this study that roemerine had no significant toxicity using Candida elegans worms, three cancer cells and one normal cell (Ma et al. 2015). However, roemerine in this study was not observed to have a fungicidal effect against planktonic C. albicans cells. Another experiment showed that roemerine had antifungal activities against C. albicans in vitro at IC50 16.1 µM and MIC 35.84 µM, and no cytotoxic effects towards mammalian kidney fibroblasts (the kidney cells of the African green monkey) up to a concentration of 85.3 μM (Agnihotri et al. 2008). Nonetheless, this investigation was conducted without the use of positive control. Cycleanine at the concentraion of 3215.4 μM showed broad antifungicidal effects on Trichophyton longiformis, Candida albicans, Aspergillus flavis, Microsperum canis, Fusanum solani var lycopersici-tomato, Emoniliform spp. when ketoconazole or amphotericin B flucytosine or benlate nabam being the control (Lohombo-Ekomba et al. 2004). At such high concentration, the result of broad antifungicidal effects of cycleanine is unreliable, and also cytotoxicity assay was not investigated in this study.
Antiviral effects
Limited research has been published regarding the antiviral activity of the alkaloids and extracts of S. tetrandra. Fangchinoline (10) exhibited antiviral activity against HIV-1 laboratory strains NL4-3, LAI and Bal in MT-4 and PM1 cells with a 50% effective concentration ranging from 0.8 to 1.7 μM by interfering with gp160 proteolytic processing (Wan et al. 2012). Nevertheless, this study lacked positive control in vitro, which further compromised the quality of the study. Compared with positive control drug (zidovudine, EC50 0.045 ± 0.056 μg/ml), coclaurine (3) demonstrated potent anti-HIV activity with an EC50 value of 0.8 μg/ml, and low cytotoxicity (IC50 > 100 μg/ml) (Kashiwada et al. 2005). However, an experimental animal model must also be established to further verify the anti-HIV activity of coclaurine. Cycleanine (21) has been observed to act against HIV-2 (EC50 = 2.94 μM) but it was at least tenfold less active against HIV-1 (Otshudi et al. 2005). Tetrandrine (9) (1–10 μM), dose-dependently suppressed dengue virus production in A549 cells by activating cycloxygenase-2 (COX-2) expression, and nuclear factor κB (NF-κB) and activator protein 1 (AP-1) activity (Liou et al. 2008). Although tetrandrine (intraperitoneal administration of 30 mg/kg/day) potently inhibited herpes simplex virus type-1-induced keratitis in BALB/c mice by modulating murine herpes simplex keratitis (HSK) development at least partly by modifying the host immune/inflammatory response to the virus in vivo (Hu et al. 1997), future studies should attempt to evaluate the toxicity of tetrandrine in vivo. In vitro, cepharanthine (0.21–26.4 μM) or its cepharanthine hydrochloride (0.01–100 μM) demonstrated potent inhibiting wild-type and lamivudine-resistant hepatitis B virus (HBV) clinical isolates, herpes simplex virus type-1 (HSV-1) and herpes simplex virus type-2 (HSV-2) (Zhou et al. 2012; Liu et al. 2004; Qian et al. 2010). The major limitation of these studies were only conducted in vitro, and lacked further study in mechasim of action. In vitro, cepharanthine had also antiviral activity against severe acute respiratory syndrome coronavirus, with IC50 values of the four treatments ranged between 9.9 and 15.67 μM (Zhang et al. 2005), but this study lack positive control and in vivo study. Tetrandrine, an L-type calcium channel inhibitor, has been shown to inhibit two-pore Ca2+ channels in the late endosome/lysosome and subsequently block Zaire ebola virus entry in vitro with an IC50 of 55 nM (Sakurai et al. 2015; Schafer et al. 2018). Tetrandrine was further evaluated in a mouse model of ebola infection and shown to significantly decrease morbidity, mortality, and viral titers (Sakurai et al. 2015; Rhein and Maury 2015). Still and all, the antiviral effects of S. tetrandra have not been studied in sufficient detail, additional research being required to assess the pharmacological and mechanistic characteristics of a greater number of its components.
Anti-inflammatory effects
Traditionally, S. tetrandra has been utilized as a TCM because of its anti-inflammatory properties. For this purpose, aqueous extracts of S. tetrandra are most commonly used. Such aqueous extracts and alkaloids of S. tetrandra (0.07813, 0.15625 or 0.31250 mg/ml) have been demonstrated to mediate against inflammation induced by lipopolysaccharide (LPS) in RAW264.7 cells, through inhibition of the release of nitric oxide, tumor necrosis factor (TNF)-α and interleukin (IL)-6 (Guo et al. 2015). Moreover, aqueous extracts and the alkaloids of S. tetrandra below the concentration of 0.31250 mg/ml did not exerted cytotoxicity to RAW264.7 cells (Guo et al. 2015). Interestingly, another study showed that total alkaloids from S. tetrandra decotion had the similar anti-inflammatory effects, mechanism, and cytotoxicity (Wang et al. 2016a). S. tetrandra extract, tetrandrine (9) (1.6–16.0 μM) and fangchinoline (10) (1.64–16.4 μM) also exert anti-inflammatory effects by interfering with reactive oxygen species (ROS) production and Ca2+ influx through G protein modulation to prevent macrophage adhesion molecule-1 (Mac-1) up-regulation in activated neutrophils (Shen et al. 2001). In the study, phytochemical analysis of S. tetrandra extracts was not conducted. Moreover, the study lack positive control, which further compromised the quality of the study. A lot of literature have reported anti-inflammatory effects of tetrandrine (concentrations of 0.1–100 μM for in vitro studies, concentrations of 6.25–80.0 mg/kg for in vivo studies) by various signalling pathways, such as nuclear factor κB (NF-κB) pathway, phosphoinositide 3-kinase (PI3K) pathway, extracellular regulated protein kinases (ERK) pathway, and signal transducers and activators of transcription 3 (STAT3) signaling pathway, etc. (Wu and Ng 2007; Xu et al. 2016a; Wu et al. 2015, 2011; Zhang et al. 2009b; Choi et al. 2000; Kang et al. 2014; Li et al. 2018b; Bao et al. 2016; Gao et al. 2016; Wang et al. 2014a; Xue et al. 2008). In these in vivo studies, tetrandrine displayed equivalent or stronger anti-inflammatory effects compared with their positive control drugs, such as indomethacin, methotrexate (Wu et al. 2015; Li et al. 2018b; Gao et al. 2016). However, most of these in vitro studies lacked positive controls, and the evaluation of anti-inflammatory effects in vivo (Wu and Ng 2007; Xu et al. 2016a; Wu et al. 2011; Zhang et al. 2009b; Choi et al. 2000; Kang et al. 2014; Bao et al. 2016; Wang et al. 2014a; Xue et al. 2008). Fangchinoline (10) and tetrandrine (9) are the major alkaloids in S. tetrandra traditionally used to treat inflammatory diseases in Asian countries, including China, Japan and Korea. Both fangchinoline (4.0 μM) and tetrandrine (6.0 μM) have demonstrated anti-inflammatory effects towards mouse ear edema induced by croton oil, although the two compounds have been shown to act through different mechanisms. The same concentration (12.5 μM) of fanchinoline and tetrandrine have demonstrated different effects on cyclooxygenase (COX), murine interleukin-5 (IL-5) and human IL-6 (Choi et al. 2000). Although fangchinoline exhibited stronger anti-inflammatory effect than tetrandrine, this study conducted without the use of a proper positive control. Tetrandrine (10–50 μM) exhibits anti-inflammatory effects through various mechanisms, including the suppression of extra-signal response kinase (ERK1/2), c-jun N-terminal kinase (JNK1/2), inhibition of the phosphorylation and degradation of inhibitor of kappa B (IκB) and activation of NF-κB (Kang et al. 2014). Although detailed research was performed into anti-inflammatory mechanisms in this study, a major limitation was lack of a positive control.
Tetrandrine at the concentration of 10 μM or 50 μM has also been shown to suppress the expression of COX-2 and inducible nitric oxide synthase (iNOS) (Wu and Ng 2007), and inhibit NF-κB and extracellular regulated protein kinase (ERK) signal transduction. It has also been shown to reduce activation of the STAT3 signaling pathway (Xu et al. 2016a). Furthermore, tetrandrine (administration of 40 mg/kg/day) has been shown to suppress a systemic inflammatory response and multiple organ dysfunction syndrome, thus protecting against damage (Wu et al. 2015). Nevertheless, dose-dependent effects were not observed in each case and some of the investigations lacked positive controls. Tetrandrine (3.21–16.07 μM) may play an important role in the prevention of glomerulonephritis by down-regulation of ERK/NF-κB signaling and inhibition of the activation of mesangial cells (Wu et al. 2011). Yet the authors of this study did not perform comparisons using a positive control. Moreover, the concentrations conducted in this study were lower than other anti-inflammatory effect studies of tetrandrine. Administration 40 mg/kg of tetrandrine not only decreased DNA binding by NF-κB but also reduced the accumulation of leukocytes, downregulated IL-1β and TNF-α production, limited the inflammatory response and thereby significantly ameliorated the severity of dextran sulfate sodium-induced colitis (Zhang et al. 2009b). However, the safety or side effects of tetrandrine were not addressed in this study, aspects which should be established via a toxicity study prior to use in a human study.
Anyhow we hold that tetrandrine could be a potent anti-inflammatory agent for further drug development after completing in vivo toxicity and in vivo metabolic assessment.
Reticuline (5) at the concentration of 0.5 mg/kg was shown to possess anti-inflammatory effects against xylene-induced ear edema and carrageenan-induced paw edema in mice and rats. The effects were mediated through inhibition of TNF-α and IL-6 mRNA expression, by reducing phosphorylation of Janus kinase 2 (JAK2) and STAT3 (Yang et al. 2018b). Nonetheless, this investigation was conducted without the use of positive control, and also lacks analysis of dose-effects relationship of reticuline. Cepharanthine (25), cycleanine (21) and tetrandrine (9) (each at a concentration of 5 μg/ml) significantly suppressed NO production, exerting anti-inflammatory activity (Kondo et al. 1993). However, only a single dose of these compounds was used throughout these studies, thus limiting data regarding the effect being dose-dependent. Cepharanthine at concentrations of 33.0 μM and 82.5 μM were also shown to inhibit chemotaxis of Jurkat T cells by reducing chemokine (C-X-C motif) ligand 10 (CXCL10) production, resulting in suppression of inflammation (Aota et al. 2018). It’s worth noting that this study also did not include a positive control. Meanwhile, this study conducted higher concentration of cepharanthine than that of Kondo’s study to evaluate the anti-inflammatory activity. Dicentrine (39) at the concentrations of 5.90, 7.52 or 10.0 μM was shown to suppress LPS-induced inflammation by blocking the activation of NF-κB and activator protein-1 (AP-1) through inhibition of mitogen-activated protein kinase (MAPK) and p-protein kinase B (Akt) signaling in RAW264.7 macrophages (Yodkeeree et al. 2018). Although this study assessed the cytotoxicity to RAW264.7 macrophages, anti-inflammatory effects and the mechanism of action, these authors did not compare the efficacy of dicentrine with a positive control. Magnoflorine (49) (3.125–50 μM) enhanced an LPS-activated pro-inflammatory response by upregulating TNF-α, IL-1β, prostaglandin E2 (PGE2) production and COX-2 protein expression in U937 macrophages. Compared to the positive control of levamisole, magnoflorine showed weaker the 50% effective dose (ED50) values of 7.89 and 10.28 µM for TNF-α and IL-1β, respectively. (Haque et al. 2018). In an in vivo trial, magnoflorine (5–20 mg/kg) ameliorated acute lung injury induced by LPS by suppressing the activation of NF-κB and MAPK (Guo et al. 2018). However, no clinical trials have been reported and so further evidence in humans is required to assess the anti-inflammatory effect of magnoflorine.
Multiple components in S. tetrandra demonstrate anti-inflammatory activity through various mechanisms. These results reinforce the use of this plant as an anti-inflammatory agent, complimenting the traditional use of this plant as a treatment for inflamed sores.
Anticancer/antiproliferative effects
A large body of literature has reported that S. tetrandra and its alkaloids have anticancer and antiproliferative effects in various malignant tumor cells. In this regard, fangchinoline (10) and tetrandrine (9) remain the most studied constituents of S. tetrandra. Tetrandrine has broad-spectrum antitumor effects (Bhagya and Chandrashekar 2018; Liu 2017; Liu et al. 2016) in many types of cancer cells, including human liver cancer (Zhang et al. 2018a; Yu and Ho 2013), osteosarcoma (Tian et al. 2017; Lu et al. 2017), oral cancer (Lien et al. 2017; Huang et al. 2013), colon carcinoma (Meng et al. 2004; Li et al. 2019), gastric cancer (Bai et al. 2018; Qin et al. 2013), brain glioblastoma multiforme GBM 8401 cancer (Jiang et al. 2019) and lung carcinoma (Chow et al. 2018; Cho et al. 2009). It also exhibits antineoplastic activity against gallbladder carcinoma (Zhu et al. 2014), thyroid carcinoma B-CPAP cells (Zhang et al. 2017a), prostatic carcinoma PC3 cells (Lan et al. 2018), esophageal cancer (Wang et al. 2012), breast cancer (Guo and Pei 2019), pancreatic cancer (Singh et al. 2018), nasopharyngeal carcinoma (Lin et al. 2016, 2017b), acute megakaryoblastic leukemia (Liu et al. 2017c), cervical cancer (Zhang et al. 2018b) and renal carcinoma (Chen et al. 2014, 2017a) cells. Although tetrandrine exhibited broad anti-tumor effects, a number of these studies lacked positive controls or anti-tumor evaluation of tetrandrine in vivo. It is a challenge to compare each other in quantitative data on these activities as a result of different tumor models and different cell lines of the same cancer.
As a result of the structural similarity with tetrandrine (9), fangchinoline (10) also has significant antitumor activity in various types of cancers, including carcinoma of the breast (Wang et al. 2014b, 2017b), prostate (Li et al. 2015b), bladder (Fan et al. 2017), lung (Luo et al. 2016), stomach (Chen et al. 2017b), pancreas (Lee et al. 2017) and liver (Wang et al. 2011). It also acts against osteosarcoma (Li et al. 2017b), glioblastoma (Guo et al. 2016), melanoma (Shi et al. 2017) and chronic myelogenous leukemia (Wang et al. 2018). Still and all, additional investigation using in vivo models with appropriate positive controls, dose, and standards is required.
In addition to fangchinoline (10) and tetrandrine (9), a number of other alkaloids in S. tetrandra have also reported anticancer/antiproliferative effects. Cepharanthine (25) was shown to possess potent cytotoxicity against a number of types of carcinoma cell, including those of the lung (Hua et al. 2015), breast (Gao et al. 2017), ovary (Payon et al. 2019), colon and rectum (Rattanawong et al. 2018). It also acts against choroidal melanoma cells (Zhu et al. 2017) and K562 human leukemia cells (Wu et al. 2001). These anticancer effects of cepharanthine are only in vitro investigation, whereas anti-tumor evaluation of cepharanthine in vivo need to be verified. N-methylcoclaurine (1) has demonstrated potential therapeutic effects against leukemia (IC50 < 10 μM) in studies evaluating its impact on the growth of murine leukemic L1210 cells, human leukemic CCRF-CEM cells and HL-60 cells (Tzeng et al. 1990). Cycleanine (21) with the IC50 values from 7 to 14 μM showed potential anti-cancer effects on various ovarian cancer cells by acting through the apoptosis pathway. Yet compared to the positive control, paclitaxel (IC50 values from 5 to 8 nM), anti-cancer effects on various ovarian cancer cells of cycleanine has no advantage. (Uche et al. 2016). Dicentrine (39) has been shown to IC50 values from 4 to 43 μM on different U87MG cell lines by inhibiting the over-expression of epidermal growth factor receptor cancer cells by intercalation of DNA and the introduction of DNA strand breaks. Unfortunately, this study did not use a positive control, which compromised the quality of the study (Konkimalla and Efferth 2010). In severe combined immunodeficiency mice, dicentrine (100 μg/mice) significantly inhibited the development of tumors in the leukemia cell line K562. Alternatively, dicentrine (14.7 μM), in vitro, could decrease the colony formation efficiency in both hepatoma cell lines, HuH-7 and MS-G2. (Huang et al. 1998). It is a pity that only one dose of dicentrine used in the in vivo and in vitro studies, and thus limited the information on the dose-dependent effect. And the dose of dicentrine should be used in μg/kg based on the weight of mice instead of μg/mice in the in vivo study. Dehydrocrebanine (50) has demonstrated strong activity against promyelocytic leukemia cells (HL-60), with an IC50 of 6.35 µM (Makarasen et al. 2011). Though, the mechanism of action and in vivo activity need further assessment. Roemerine (52) was shown to anti-prostate cancer effect in vivo in the mouse model of prostate cancer, while in vitro inhibit the proliferation and migration of the following prostate cancer cell lines: DU145, LNCaP, PC-3 and 22RV1. It also induced different degrees of apoptosis, particularly in LNCaP cells (Ma et al. 2017). The shortage of this study was that only a single dose of roemerine was used.
A number of mechanisms are involved in the manner in which the constituents of S. tetrandra act against various carcinoma cells, including induction of tumor cell apoptosis (Zhu et al. 2014; Zhang et al. 2017a; Bai et al. 2018; Liu et al. 2017b; Chen et al. 2014; Wang et al. 2017b; Fan et al. 2017), tumor cell autophagy (Zhang et al. 2018b; Lien et al. 2017; Huang et al. 2013; Bai et al. 2018; Guo and Pei 2019; Fan et al. 2017; Gao et al. 2017), tumor cell cycle blockade (Singh et al. 2018; Chen et al. 2014; Luo et al. 2016; Gao et al. 2017) and inhibition of tumor cell invasion and metastasis (Zhang et al. 2018a; Li et al. 2017b; Shi et al. 2017). Reversal of multidrug resistance is an additional important mechanism of the anticancer effects of tetrandine (9) and fangchinoline (10). These two compounds are effective in overcoming multidrug resistance in a variety of tumor cells in a synergistic manner via modulation of P-glycoprotein (Choi et al. 1998; Sun and Wink 2014; Lu et al. 2017), multidrug resistance-associated protein 1 (Wang et al. 2012), multidrug efflux transporters (Yao et al. 2017), Akt/NF-κB/matrix metalloprotein (MMP)-9 (Chen et al. 2017a) and β-catenin expression (Zhang et al. 2017b). Tetrandine has also been shown to enhance radiation sensitization for human nasopharyngeal carcinoma cells by modulating phosphorylated histone H2AX (γ-H2AX), phosphorylated CDC25C, phosphorylated cyclin-dependent kinase 1 (p-CDK1) and CDK1 (Ding et al. 2008; Wang et al. 2017c). In lung cancer cells, tetrandine decreased the proportion of cells blocked at G2 (Ding et al. 2016) and also enhanced the radiation sensitization of esophageal carcinoma cells (Bai et al. 2015). To date, tetrandine is the only alkaloid of S. tetrandra which has been reported to enhance radiation sensitization in various cancer cells. As these studies of mechanisms were only conducted in vitro, additional researches using in vivo models with appropriate controls and doses are required.
In summary, alkaloids in S. tetrandra have favorable anti-tumor effects. Furthermore, some alkaloids of S. tetrandra has the potential to be developed into novel anti-tumor drugs because of the mechanisms already defined and its pharmacological effects.
Central nervous system activity
Neuroprotective effects
Tetrandrine (9) and fangchinoline (10) are protective against the neurodegeneration occurring in Alzheimer's and Parkinson's disease. Rats were injected intraperitoneally with 10 or 30 mg/kg tetrandrine every other day for 4 weeks, tetrandrine has been shown to exert neuroprotective effects in chronic vascular dementia by reducing IL-1β expression, N-methyl-D-aspartate receptor 2B phosphorylation at tyrosine 1472 and neuronal necrosis (Lv et al. 2016). However, no positive control was included in these animal experiments and a dose–response effect was not observed. In a mouse model of middle cerebral artery occlusion (MCAO), two-dimensional difference gel electrophoresis (2-D DIGE) revealed that tetrandrine at the dose of 30 mg/kg attenuated cerebral ischemia/reperfusion injury and induced differential proteomic changes (Ruan et al. 2013). Nevertheless, the experiment had a number of limitations, including having no positive control and a single dose of drugs, resulting in a lower level of reliability and no possibility of observing a dose-dependent effect. Tetrandrine at the concentrations of 0.5 and 1.0 µM has also been shown to diminish the harmful effects of amyloid-β protein on cell survival by interfering with signals of necrosis relating to Ca2+ overload through L-type calcium channels (Chiou et al. 2006). Furthermore, tetrandrine (0.1–20 µM) also protects against injury induced by oxygen-glucose-serum deprivation/reoxygenation (OGSD/R) (Bao et al. 2016). Tetrandrine has been shown to facilitate the neuroprotective effects of glutathione in a 6-hydroxydopamine-lesioned rat model in Parkinson's disease at the dose of 15 mg/kg/day (Li et al. 2015c), reduce phenobarbital withdrawal symptoms and protect brain cells against apoptosis at the doses from 6.25 to 31.25 mg/kg/day (Han et al. 2015). Tetrandrine can also alleviate pathological nerve cell damage (Zhang et al. 2001) and ameliorate Aβ (1–42)-induced spatial learning and memory deficit in Alzheimer's disease at the dose of 40 mg/kg/day (He et al. 2011). Nonetheless, because the factors affecting nerve cell damage or Alzheimer's disease are numerous, the signaling pathways are extremely complex and the signaling molecules involved are in considerable abundance. Thus, the mechanism of tetrandrine in nerve cell protection and the treatment of Alzheimer's disease requires additional in-depth basic research including a search for factors that modulate the neuroprotective effects of tetrandrine, to explore related signaling molecules and pathways involved in the neuroprotective effects. Tetrandrine (9) and fangchinoline (10) over a concentration range of 0.1–10 µM have been shown to lessen the harmful effects of H2O2-induced neuronal cell death by interfering with increased Ca2+ concentrations, which inhibit glutamate release and generation of reactive oxygen species (ROS) (Koh et al. 2003). Yet this investigation was conducted without the use of a positive control. Furthermore, fangchinoline inhibited glutamate release from rat cortical synaptosomes by suppressing the activity of the voltage-dependent Ca2+ channels, with an IC50 values derived from a dose–response curve of about 93 µM, subsequently causing a reduction in the ingress of Ca2+ into nerve terminals (Lin et al. 2009; Kim et al. 2001). This investigation, too, was conducted without the use of a positive control. And the IC50 value (93 µM) was too high, which can not provide reliable experimental data for clinical application.
Butyrylcholinesterase and acetylcholinesterase (AChE) play important roles in Alzheimer's disease. N-methylcoclaurine (1), reticuline (5), dicentrine (39), dehydrodicentrine (51), roemerine (52), Stepharanine (54) and cyclanoline (64) have demonstrated inhibitory effects against AChE with IC50 value of 15.0 ± 1.4 µM, 33.6 ± 3.0 µM, 6.6 µM, 2.98 µM, 335.1 µM, 14.10 ± 0.81 µM, and 9.23 ± 3.47 µM, respectively (Hošt'álkováet al. 2015; Kongkiatpaiboon et al. 2016; Dong et al. 2015; Ingkaninan et al. 2006). Among them, roemerine showed the weakest acetylcholinesterase inhibitory activity. Compared with the positive control of tacrine, with IC50 value of 0.25 µM, the activities of these compounds are not strong. Investigations on potential mechanisms and in vivo study are required, since AChE inhibitory activity of these compounds was merely evaluated by analysis of their IC50 values. Stepholidine (57) at the doses of 3–10 mg/kg should be explored as a potential therapeutic drug for Alzheimer's disease because it acts through a mechanism that improves trafficking of the α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor and improves synaptic plasticity by activating the dopamine D1 receptor/protein kinase A (D1/PKA) signaling pathway (Hao et al. 2015). Nevertheless, this study also lacked a positive control and no analysis of dose-dependency has been conducted. These limitations put into question the reliability of this study.
Enhancement of sleep and hypnotic effects
Tetrandrine (9) is the sole constituent of S. tetrandra that has been studied for its effects on initiating sleep. Intragastric administration of tetrandrine (60 mg/kg/day) significantly decreased the c-Fos positive ratio of noradrenergic neurons in the locus coeruleus (LC) of spontaneously hypertensive rats. It also significantly decreased norepinephrine in endogenous pathways that regulate sites that modulate the sleep–wake cycle, such as the LC, hypothalamus and ventrolateral preoptic nucleus (Huang et al. 2016). Yet the authors did not explore dose-dependency, because only two doses were evaluated in this study. Tetrandrine (100 mg/kg, administered orally) has also been shown to markedly improve sleep efficiency by increasing the time of total sleep, rapid eye movement (REM) sleep and non-REM sleep, including deep sleep and light sleep (Zhang et al. 2014). Unfortunately, only a single dose of tetrandrine was used throughout the study, with no investigation of dose-dependency. Moreover, the dose of tetrandrine used was too high in the study, it can not provide reliable experimental data for clinical application. In mice, tetrandrine (30 or 60 mg/kg, administered orally) has also been shown to significantly potentiate pentobarbital-induced hypnosis by reducing sleep latency and increasing sleeping time in a dose-dependent manner, an effect potentiated by 5-hydroxytryptophan (Zhao et al. 2004). In general, future studies should attempt to identify the mechanism of action of tetrandrine.
Antinociceptive effects
Tetrandrine (9) (15, 30 or 45 mg/kg, administered intraperitoneally) exhibits antinociceptive effects against LPS-induced hyperalgesia in mice. These effects mediated by inhibition of inhibitor kappa B kinaseβ (IKKβ) phosphorylation. This has been shown to reduce the production of important pain mediators, such as PGE2 and COX-2 via the IKKβ/IκB/NF-κB pathway (Zhao et al. 2014). In this study, tetrandrine exhibited stronger antinociceptive effects than the positive control morphine (10 mg/kg). In the study, further research models are critically needed to confirm such activity in-vivo and in humans. Dicentrine (39) (100 mg/kg/day, administered orally) has demonstrated significant antinociceptive activity in a mouse model of acute visceral pain. It also reduced spontaneous nociception and attenuated mechanical and cold hypersensitivity, probably via a transient receptor potential ankyrin 1 (TRPA1)-dependent mechanism (Montrucchio et al. 2013). This investigation, too, was conducted without use of a positive control, and also lacked analysis of a dose–response, as well as the dose of dicentrine was used too high. Though research on the antinociceptive effects of S. tetrandra is limited, these results compliment the traditional use of this plant in the treatment of arthralgia.
Other central effects
Reticuline (5) possesses potent central nervous system depressant action. In vivo, reticuline (50–100 mg/kg, administered intraperitoneally) could produce alteration of behaviour pattern, prolongation of pentobarbital-induced sleep, reduction in motor coordination and d-amphetamine-induced hypermotility and suppression of the conditioned avoidance response (Morais et al. 1998). This study had the same shortage of lacking a positive control. Tetrandrine (9) (40 mg/kg, administered intraperitoneally) appears to be a potential adjuvant drug for the treatment of refractory epilepsy. It has demonstrated decreased cell resistance to phenytoin and valproic acid. It also reduced the frequency of seizures and increased the efficacy of antiepileptic drugs by reducing the expression of P-glycoprotein at both mRNA and protein levels in vivo (Chen et al. 2015). Anyhow, safety and efficacy data should be established, especially via a toxicity study prior to evaluation in a human study. Tetrandrine (30 mg/kg, administered intraperitoneally) also ameliorated sevoflurane-induced cognitive impairment by suppression of inflammation and apoptosis in aged rats. Thus, tetrandrine may potentially be a novel candidate to protect cognitive function against the effects of sevoflurane anesthesia (Ma et al. 2016). A limitation of this study was the single dose of tetrandrine used and not a positive control. Tetrandrine (10 mg/kg or 30 mg/kg, administered intraperitoneally) was also shown to reduce cognitive impairment in chronic cerebral hypo-perfused rats by inhibition of the activation of S100B and a decrease in the expression of IL-1β, TNF-α and iNOS in the hippocampal CA1 region (Chen et al. 2013). The limitations of this study were the single dose of tetrandrine used and the lack of a positive control.
Nantenine (44) (13.3, 20 and 30 mg/kg, administered intraperitoneally) has demonstrated inhibition of the head-twitch response induced by 5-hydroxytryptophan (5-HT) plus clorgyline by blocking 5-HT 2A (5-HT2A) receptors in the central nervous system. The positive control ketanserin at the doses of 0.0625, 0.25 and 1 mg/kg (administered intraperitoneally) also showed similar effects (Indra et al. 2002). Although nantenine had a similar effect with ketanserin, the doses of nantenine used were significantly larger than that of ketanserin. Additionally, nantenine (20–50 mg/kg, administered intraperitoneally) presented anticonvulsant effect in inhibiting pentylenotetrazol and maximal electroshock-induced seizures in rats, whereas nantenine at higher doses (75–105 mg/kg, administered intraperitoneally) has been shown to a convulsant activity appears to be related to inhibition of phosphatase at high doses and cause a decrease in Ca2+-influx into cells at low doses (Ribeiro and Leite 2003). Unfortunately, the authors of this study did not perform comparisons using a positive control. Magnoflorine (49) (20 mg/kg, administered intraperitoneally) alone enhanced cognition, and co-administration of magnoflorine with scopolamine blocked long-term, but not short-term, memory impairment (Kukula-Koch et al. 2017). However, no explanation of the active principle or mechanisms of action was provided and use of a positive control was lacking. Stepholidine (57) (2.5, 5.0 and 10 mg/kg, administered intraperitoneally) is a natural dopamine (D) receptor D1 agonist and D2 antagonist. It can significantly reduce cue-induced reinstatement of cocaine seeking, in a dose-dependent manner, and significantly decreased break points for cocaine reward (Manuszak et al. 2018). It is a pity that this trial lacked a positive control. The in vivo effects of stepholidine are largely mediated through blockade of the dopamine receptor with potential cross-talk to other receptors or signaling proteins (Meade et al. 2015). Stepholidine (1, 3 and 10 mg/kg, administered intraperitoneally) has been shown to dose-dependently attenuate reinstatement of a desire to seek 3, 4-methylenedioxypyrovalerone following priming by a cue plus 3, 4-methylenedioxypyrovalerone in an intravenous self-administration model (Hicks et al. 2018). Anyhow, it is important that an experimental animal model is also established to further verify the effects of stepholidine in vivo.
Cardiovascular activity
Antihypertensive effects
S. tetrandra has traditionally been used as an antihypertensive and antiarrhythmic treatment. In anesthetized rats, extracts of S. tetrandra (50 or 150 mg/kg/day, administered orally) reduced both frequency of arrhythmias and size of infarcts induced by myocardial ischemia and reperfusion. These effects were comparable to those of tetrandrine (50 mg/kg/day) and verapamil (Yu et al. 2004). In this study, phytochemical analysis of extracts of S. tetrandra was not evaluated. Moreover, only two doses of extracts of S. tetrandra were used throughout this study, limiting any data on dose-dependency. Intragastric administration of tetrandrine (9) (30 and 60 mg/kg/day) significantly lowered the blood pressure of spontaneously hypertensive rats by inhibition of the noradrenergic system, rather than by calcium channel blockade. The effects of tetrandrine at the dose of 60 mg/kg/day were similar to those treated with amlodipine (a positive control, 100 mg/kg/day) after one day. After treated with drugs on two and three days, the effects of tetrandrine (30 and 60 mg/kg/day) were weaker than those of amlodipine (100 mg/kg/day) (Huang et al. 2016). Nonetheless, only two doses of tetrandrine were used throughout this study, limiting any data on dose-dependency, as well as the trial lacked a positive control and toxicity assessment. A similar study also reported that intragastric administration of tetrandrine (100 mg/kg, administered orally) significantly lowered the blood pressure of spontaneously hypertensive rats over three days of treatment (Zhang et al. 2014). However, this investigation was also conducted without the use of a positive control and lacked any analysis of a dose-dependent relationship. And the dose of tetrandrine was used too high to provide reliable experimental data for clinical application. Tetrandrine (80 mg/kg/day, administered intraperitoneally) was shown to prevent monocrotaline-induced pulmonary arterial hypertension in rats by regulation of protein expression in inducible NOS and cyclic guanosine monophosphate (cGMP)-dependent protein kinase type 1. Compared with vardenafil (2.0 mg/kg/day, administered intraperitoneally), tetrandrine (80 mg/kg/day) exhibited similar effects against monocrotaline-induced pulmonary arterial hypertension in rats (Wang et al. 2016c). The flaws of the study lie in only a single dose of tetrandrine used throughout the study, limiting analysis of dose-dependency. Finally, tetrandrine substantially lowered intraocular pressure in a rat model of ocular hypertension, although no explanation of the active principle or mechanism of action was offered. More importantly, this study did not state the unit of tetrandrine dose, it was just indicated that 0.1%, 0.2%, 0.3% tetrandrine were administered in this study (Huang et al. 2011). Tetrandrine (2.0–20 mg/kg, administered intravenously) has been shown to lower portal venous pressure, mean arterial pressure and total peripheral resistance in cirrhotic rats (Huang et al. 1999). It is worth noting that this study also did not include a positive control or investigate the mechanism of action.
Tetrandrine and fangchinoline inhibited high K+ (65.4 mM) and induced sustained contraction in the rat aorta smooth muscle strips, also inhibited 1.0 μM norepinephrine-stimulated Ca2+ influx in rat aorta strips. Meanwhlie, tetrandrine (3 mg/kg) and fangchinoline (30 mg/kg) administered by intravenous (i.v.) bolus injection lowered the mean arterial pressure significantly during the period of observation in conscious stroke-prone spontaneously hypertensive rats, respectively. These results showed that tetrandrine was more potent than fangchinoline in blocking calcium channels and antihypertensive activity (Kim et al. 1997). The main limitations of most of these studies lacked positive controls.
Dicentrine (39) (3–10 μM) was found to be a potent α1-adrenoceptor inhibitor in rat thoracic aortas, as revealed by competitive antagonism of norepinephrine- and phenylephrine-induced vasoconstriction (Teng et al. 1991). Anyhow, the mechanism by which this activity occurred should be investigated further. Oral administration of dicentrine (5 and 8 mg/kg) to conscious spontaneously hypertensive rats induced hypotension that lasted for more than 15 h. Additionally, administration of dicentrine (0.1, 0.5 or 1.0 mg/kg) and prazosin (0.01, 0.05 or 0.1 mg/kg) intravenously induced a dose-related reduction of mean arterial pressure (MAP) which reached a maximal effect 5–10 min after injection and persisted for 2 h. This suggests that dicentrine may have therapeutic potential as an oral or intravenous antihypertensive drug by acting as a blockade of the α-adrenoceptor (Yu et al. 1992a). Nonetheles, future studies should attempt to identify the mechanism of action and toxicity of dicentrine. These results reinforce the traditional uses of S. tetrandra as an antihypertensive and antiarrhythmic treatment.
Cardiovascular effects
The cardioprotective and antiarrhythmic effects of the S. tetrandra extract at the concentration of 16.8 μg/ml are equivalent to those of tetrandrine (18.7 μg/ml) alone and either may be a better treatment choice than classical calcium channel antagonists, verapamil, for the treatment of arrhythmia or myocardial infarction induced by ischemia and reperfusion injury (Yu et al. 2001; Wong et al. 2000). In these studies, phytochemical analysis of S. tetrandra extracts was not investigated. Moreover, only a single dose of S. tetrandra extract and tetrandrine were used throughout the studies, and thus limited the information on the dose-dependent effect. Tetrandrine (9) not only acts as an antihypertensive drug, it (50 mg/kg, administered orally) also reverses cardiac and vascular remodeling. The latter occurs through inhibition of vascular smooth muscle cell proliferation, through the inhibition of induction and sensitization of vascular smooth muscle cells to stimulate pro-apoptosis and by improving endothelial function (Rao 2002). Nevertheless, these results did not display a dose-dependent relationship and some investigations lacked positive controls. Renovascular hypertensive rats demonstrated hypertrophy and altered remodeling of the artery and arteriole walls in addition to the proliferation of vascular smooth muscle cells in renal arterioles. This vascular remodeling was shown to be suppressed by tetrandrine (50 mg/kg, administered orally), markedly attenuating these changes (Li et al. 2003), and inhibiting collagen synthesis and deposition in their arteries (Li et al. 2001). However, these studies had important limitations—only one dose of tetrandrine was used and thus no information about dose-dependency was investigated, as well as no appropriate positive controls were used. Tetrandrine (10, 20 or 40 μM) has been shown to antagonize angiotensin II-induced cardiomyocyte hypertrophy by decreasing angiotensin II-induced elevation of pulse rate, cell size, total protein content and rate of protein synthesis. It also inhibited the activity and expression of phosphorylated extracellular regulated kinase1/2 (p-ERK1/2) (Zhou et al. 2007). Unfortunately, the authors of this study did not perform comparisons using a positive control. Furthermore, tetrandrine (15 mg/kg) offered full protection against myocardial infarction that was experimentally induced by non-invasive treatment with isoproterenol in rabbits (Pinelli et al. 2010). The major limitation of this study was the lack of analysis of a dose-dependent relationship. Tetrandrine (0.1 and 1.0 mg/kg) has also been shown to ameliorate ischemic-reperfusion injury in rat myocardium by inhibition of neutrophil priming and activation (Shen et al. 1999). Nevertheless, only two doses of tetrandrine were used in these studies, and thus information regarding the dose-dependent effect is limited.
Dicentrine (39) (1, 3 or 9 μM), is equaliate to quinidine (from 0.5 to 4.5 μM), shows promise as an antiarrhythmic agent with type Ia and type III antiarrhythmic action (Young et al. 1994). Dicentrine has the potential to reduce two major cardiovascular risk factors, hypertension and hyperlipidemia. It does this via a decrease in mean arterial pressure and reduced vascular hyperreactivity to phenylephrine. Dicentrine (5 and 10 mg/kg, administered orally) also lowers blood concentrations of cholesterol, low density lipoprotein cholesterol, total plasma triglycerides and very low density lipoprotein cholesterol while increasing concentrations of high density lipoprotein cholesterol (Yu et al. 1993). These experiments had a number of shortcomings, including the absence of a positive control and only two doses of drug, resulting in the lack of reliability of results and absence of analysis of a dose-dependent effect. In anesthetized normotensive rats, acute intravenous administration of nantenine (44) (3–5 mg/kg, administered intravenously) produced a dose-dependent decrease in mean arterial pressure, accompanied by a significant decrease in heart rate (Orallo 2004). Nonetheless, future studies should attempt to replicate this effect under clinical conditions using a low dose suitable for human administration.
Stephenanthrine (65) (0.01–1.0 μM) is of potential therapeutic interest due to the control of leukocyte recruitment involving angiotensin-II that occurs in cardiovascular disease. Stephenanthrine inhibits angiotensin-II-induced endothelial P-selectin upregulation. It also inhibits ROS generation in endothelial cells stimulated by angiotensin-II in both N-formylmethionyl-leucyl-phenyl-alanine (fMLP)-stimulated human neutrophils and in the hypoxanthine-xanthine oxidase system (Estellés et al. 2003). However, it should be noted that this study also did not include a positive control.
Immunomodulatory effects
Extracts of S. tetrandra and its principal active components, tetrandrine (9) and fangchinoline (10), have been used to treat autoimmune diseases, such as rheumatoid arthritis, transplant rejection and systemic lupus erythematosus (Semwal et al. 2010; Lai 2002; Sekiya et al. 2004; Seow et al. 1988). Rheumatoid arthritis patients were administered 10 g extract of S. tetrandra orally for 12 weeks in a clinical trial. The clinical trial results showed that extract of S. tetrandra could significantly reduce the lipid peroxide level, the plasma human granulocyte elastase level, granulocyte count and proportion of granulocytes in peripheral blood of rheumatoid arthritis patients, and increase leukocyte/elastase ratio and granulocyte/elastase ratio (Sekiya et al. 2004). The limitations of this study were the lack of description of doses and a positive control. Tetrandrine at 0.1–100 μM inhibited neutrophil-monocyte chemotactic factor-1 upregulation and adhesion to fibrinogen induced by N-formyl-methionyl-leucylphenylalanine and phorbol 12-myristate 13-acetate, and reduced production of oxygen free radical, down-regulated synthesis and release of some proinflammatory cytokines (Semwal et al. 2010). In an in vitro study, tetrandrine at 16.0 μM had potent immunosuppressive activity by suppressing the antibody synthesis by B cells, and the mitogen-induced lymphoproliferative response (Seow et al. 1988). Nonetheless, the same shortages of this study as most of the aforementioned literature are lack of positive control and a single dose was used. The underlying mechanisms for these immunomodulatory effects have been studied extensively. These compounds inhibited activation of human peripheral blood T-cells by both phorbol 12-myristate 13-acetate (PMA) + cluster of differentiation 28 (CD28)-costimulation and CD3 + CD28-costimulation (Lai et al. 1999). They have also been shown to reverse the pathological process of collagen-induced arthritis by modulating metabolic pathway imbalances (Yun et al. 2018), markedly alleviating the severity of arthritis and reducing serum concentrations of pro-inflammatory cytokines. The compounds further restored the ratio of T helper 17 (Th17)/ regulatory T (Treg) cells. This has been demonstrated by measurement of serum concentrations of relevant cytokines (IL-17 and IL-10) and the proportion of each cell type (Yuan et al. 2016). They have also reduced expression of Rac family small GTPase 1 (Rac1), cell division cycle 42 (Cdc42) and Ras homolog family member A (RhoA), and inhibited Akt and JNK activation, resulting in the subsequent down-regulation of MMP-2/9, F-actin and FAK activation and/or expression (Lv et al. 2015). They have also been shown to reduce the concentrations of ROS, TNF-α, IL-6, prostaglandin-E2 and matrix metalloproteinases (Shan et al. 2018), and inhibit the secretion of IL-1α, TXB (2), ET-1 and E-selectin in LPS-induced endothelial cells (Hu et al. 2012).
It has also been reported that tetrandrine (20 mg/kg, administered orally of three times a day) exhibits synergistic effects towards rheumatoid arthritis when used in combination with leflunomide or methotrexate in two clinical trials (Zhang et al. 2018d; Zhang 2014). Although there are numerous studies of the immunomodulatory effects of tetrandrine and fangchinoline, future studies should attempt to replicate these immunomodulatory effects with appropriate postive controls, and under clinical conditions with low doses suitable for human administration. Neuromyelitis optica is a severe neurological demyelinating autoimmune disease that affects the optic nerves and the spinal cord. Isotetrandrine (27) at 100 μM has been shown to reduce astrocyte cytotoxicity in neuromyelitis optica by blocking the binding of neuromyelitis optica-immunoglobulin G (NMO-IgG) to aquaporin 4 (Sun et al. 2016). However, the authors of this study did not perform comparisons using a positive control, as well as conducted only one too high concentration.
Traditionally, S. tetrandra has the ability to treat rheumatoid arthritis. Although tetrandrine has been approved in China for its treatment, the immunosuppressive activity, mechanism of action and structure–function relationships of the other constituents in S. tetrandra require additional study.
Antifibrotic effects
Antifibrotic effects in pulmonary disease
Silicosis is a representative fibrotic pulmonary disease. Tetrandrine (9) modulates TNF and collagen gene expression and could be used to treat silicosis alone or in combination with other therapies (Idec-Sadkowska et al. 2006; Liu et al. 1994; Xv and Zhou 2015; Chen et al. 2018; Resit et al. 1993; Castranova et al. 1991). Tetrandrine (50 mg/kg, administered orally) was shown to inhibit the development of experimental silicosis of rats by reducing α1 (I) and α1(III) collagen mRNA levels of silicotic rat lung, but this study was conducted without the use of positive control, and also lacks analysis of dose-effects relationship of tetrandrine (Idec-Sadkowska et al. 2006; Liu et al. 1994). In a clinical trial, silicosis patients were administered 40 mg tetrandrine orally at three times a day for 6 month. The results of this trial showed that tetrandrine could significantly relieve the clinical symptoms and inhibit pulmonary fibrosis in the treatment of silicosis patients compared with the control group (Xv and Zhou 2015). In another clinical trial, tetrandrine (100 mg, administered orally) combined with ursolic acid was significantly superior to the simple medicine group (only tetrandrine group or only ursolic acid group) (Chen et al. 2018). Nevertheless, these clinical trials have the same shortages of no positive control and absence of dose-effects relationship. Moreover, these studies lacked the safety evaluation of tetrandrine on silicosis patients. The mechanisms governing the antifibrotic action of tetrandrine (2–10 μM) were shown to be inhibit the ability of fibroblasts to respond to stimulation by growth factors, such as platelet derived growth factor, fibroblast growth factor, tumor necrosis factor (Resit et al. 1993). Additionally, tetrandrine (96 μM) could inhibit particle-stimulated oxygen consumption, superoxide release, and hydrogen peroxide secretion by alveolar macrophages, and also exhibited substantial binding affinity for membrane lipids and alveolar macrophages (Castranova et al. 1991). Although tetrandrine has been approved in China for the treatment of silicosis, the antifibrotic effects, mechanisms of other alkaloids in S. tetrandra, and toxicity of these alkaloids require further investigation.
Antifibrotic effects in liver disease
Extracts of S. tetrandra (200 mg/kg) have been demonstrated to safely and effectively prevent and reverse hepatic fibrosis by activating hepatic stellate cell apoptosis in rats (Chor et al. 2009). Nevertheless, the experiment had some shortages, including no positive control and a single dose of drugs, resulting in lower reliability and absence of dose-dependent effect. More importantly, the dose of extracts of S. tetrandra used in the study was too high, and it cannot provide reliable experimental data for clinical application. Furthermore, the extracts tested were not chemically analyzed. Tetrandrine (9) (0.39–50 μM) exerted antifibrotic effects in hepatic stellate cells and in rats with hepatic fibrosis in a dose-dependent manner. It achieved this by regulating transforming growth factor-β-activated kinase 1 (TAK1), JNK and NF-κB. It also reduced fibrosis-related gene transcription, attenuated NF-κB-activated pathways and induced metallothionein gene transcription (Li et al. 2016; Hsu et al. 2007; Park et al. 2000). Yet some investigations lacked use of positive controls.
Other antifibrotic effects
Tetrandrine (9) at 1.0 or 5.0 μM was shown to reverse human cardiac myofibroblast activation and myocardial fibrosis through calcium channel blockade (Teng et al. 2015). However, this study lacked use of a positive control. Tetrandrine (8.0 μM) also exerted an antifibrotic effect on human hypertrophic scar fibroblasts by inducing the expression of microRNA, transforming growth factor-β1 (TGF-β1), SMAD family member 2 (Smad2) and SMAD family member 7 (Smad7) (Ning et al. 2016). Tetrandrine (4.0–20.0 μM) could inhibit human hypertrophic scar fibroblasts at least partially through induction of Smad7 and decrement of Smad2 resulting in inhibition of TGF-b1 transcription and its intracellular signaling, which led to reduction of type I and III collagen production and suppression of cell reproductive activity (Li et al. 2012). Nevertheless, the mechanism of the antifibrotic effect on human hypertrophic scar fibroblasts in vivo requires further study. Additionally, tetrandrine (5 mg/kg/day or 10 mg/kg/day or 20 mg/kg/day) exhibited a synergistic effect when used with prednisone (10 mg/kg/day) for renal fibrosis caused by adriamycin in rats. This was mediated by inhibition of fibrotropic factors and expression of connective tissue growth factor (CTGF) mediated by the promotion of anti-fibrosis factor MMP13 expression (Bai and Dong 2016). We believe that safety and efficacy data should be established next especially via toxicity study prior to entering the human study.
Antidiabetic effects
Tetrandrine (9) at 10 mg/kg or 20 mg/kgwas shown to greatly improve the pharmacokinetics and hypoglycemic effects of berberine by inhibiting P-glycoprotein (Shan et al. 2013). Nonetheless, only two doses of tetrandrine were used in this study and thus a full analysis of dose-dependent effects was not possible. Tetrandrine (1 mg/kg, administered intravenously) also activated nicotinic receptors in the adrenal medulla that enhanced the secretion of β-endorphin, and it stimulated opioid μ-receptors that increased glucose utilization and/or reduced hepatic gluconeogenesis, lowering plasma glucose concentrations in streptozotocin-induced diabetic rats (Hsu et al. 2004; Chen et al. 2004). A study design with only one dose of drug and an absence of a positive control were definite shortcomings in these studies. Fangchinoline (10) (0.3–3 mg/kg) decreased high blood glucose concentrations in a dose-dependent manner and also increased low blood insulin concentrations in streptozotocin-induced diabetic mice (Ma et al. 2007; Tsutsumi et al. 2003). It is unfortunate that these trials lacked proper positive controls, compromising the quality of the studies. Furthermore, fangchinoline (3.0 mg/kg) protected against nephron damage by attenuating changes in the p38 MAPK pathway that reduced oxidative stress and inflammation in rats with diabetic nephropathy (Jiang et al. 2018). Nonetheless, only a single dose of fangchinoline was used in the study, limiting the data on a dose response. Cepharanthine (25) (10 mg/kg/day, administered intraperitoneally) was shown to decrease blood and serum concentrations of glucose, urea nitrogen, malondialdehyde and creatinine. It also reduced proteinuria and the kidney to body weight ratio by inhibiting the activation of NF-κB and NLRP3 (Samra et al. 2016). Nevertheless, this investigation was conducted without the use of a positive control and also lacked analysis of the relationship with dose. Treatment with fangchinoline (10) (0.3–3.0 mg/kg), fangchinoline 2′-N-α-oxide (18) (1.0 mg/kg) and 2′-N-norfangchinoline (19) (1.0 mg/kg) reduced elevated blood glucose concentrations to almost half of their initial concentration. In addition, tetrandrine 2′-N-β-oxide (15) (1.0 mg/kg), tetrandrine 2′-N-α-oxide (16) (1.0 mg/kg), tetrandrine 2-N-β-oxide (17) (1.0 mg/kg) and fangchinoline 2′-N-α-oxide (18) (1.0 mg/kg) also decreased hyperglycemic blood concentrations by ~ 50% (Tsutsumi et al. 2003). However, this study lacked appropriate positive controls, compromising the quality of data. Furthermore, the relationship between the structure of those compounds and anti-hyperglycemic effects has not been investigated. Though various constituents in S. tetrandra have been evaluated for their anti-hyperglycemic activity, further research is required to verify the effectiveness of the constituents, the mechanisms involved and effective doses. Also, further investigation in both in vivo and in vitro models is required.
Antiplatelet effects
Tetrandrine (9) with IC50 value of 28.6 ± 3.24 μM and fanchinoline (10) with value of IC50 21.7 ± 2.61 μM are known to suppress thromboxane A2 formation, inhibiting platelet aggregation and thrombosis formation (Kim et al. 1999). Flaws in the studies lie in the lack of a positive control. N-methylcoclaurine (1) at 334.4 μM and coclaurine (3) at 350.8 μM have shown strong inhibition of arachidonic acid-induced platelet aggregation (Chen et al. 1996). Yet this investigation was also conducted without the use of a positive control and also lacked analysis of a dose–effect relationship of the two compounds. Moreover, no discussion of the principle of their activity or mechanism of action in vivo was provided, and the concentrations of N-methylcoclaurine and coclaurine used were too high to provide reliable experimental data for clinical application. Dicentrine (39) (30–150 μM) can inhibit thromboxane formation and increase cyclic AMP concentrations in rabbit platelets. Thus, it offers therapeutic potential as an antiplatelet agent (Yu et al. 1992b). Although the study reported on the effects of dicentrine on antiplatelet effects in vitro, they are insufficiently detailed. An experimental animal model should also be established to further verify the antiplatelet effects of dicentrine.
Miscellaneous uses
Tetrandrine (60 and 120 mg/kg) has shown great potential in the prevention and treatment of noise-induced hearing loss. It acts primarily by preventing outer hair cell damage and preventing synapse loss between inner hair cells and spiral ganglion neurons (Yu et al. 2018). Despite the dramatic effects of tetrandrine in protection against noise-induced hearing loss at the cellular level, molecular mechanisms underlying this protection are not well understood. Tetrandrine at 10 μM had strong anti-adipogenic effects on 3T3-L1 pre-adipocytes. These effects are possibly largely attributed to a reduction in the expression and/or phosphorylation of CCAAT/enhancer-binding protein-α (C/EBP-α), peroxisome proliferator-activated receptor-γ (PPAR-γ), fatty acid synthase (FAS), perilipin A and signal transducer and activator of transcription-3 (STAT-3) (Jang 2016). Nonetheless, important issues still require resolution, including the antiadipogenic effect of tetrandrine on obese animal models.
Tetrandrine (0.6, 2.0 or 6.0 mg/kg) prevented bone loss in sciatic-neurectomized mice by inhibiting receptor activation of NF-κB ligand-induced osteoclast differentiation (Takahashi et al. 2012). We believe that further work will be required to clarify that tetrandrine acts on the expression and/or function of transcription factors which form complexes with the nuclear factor of activated T cells c1 (NFATc1) in osteoclasts. Tetrandrine (10, 30 or 100 μM) was demonstrated to relax corpus cavernosum tissues in rabbits. This was attributed to the inhibition of extracellular Ca2+ influx and suppression of intracellular-stored Ca2+ release, but it was not mediated by NO, prostaglandin release or potassium channel activation (Chen et al. 2009b). However, further experiments are required to establish whether the cAMP or Rho-A/Rhokinase pathways contribute to tetrandrine-induced relaxation. Tetrandrine at the concentration of 3.2 μM has radioprotective effects against 10 Gy of ionizing irradiation and it was able to suppress irradiation-induced inflammatory processes (Chen et al. 1997). However, this study had some shortcomings, including lack of a positive control and exploration of only a single dose of drug, resulting in lower reliability of data and absence of any investigation of a dose–response effect.
Tetrandrine (15, 30 or 60 mg/kg, administered intraperitoneally) and fanchinoline (30 or 60 mg/kg, administered intraperitoneally) were shown to dose-dependently modulate serotonergic function and antagonize morphine-induced analgesia dependent on serotonergic mechanisms (Zhang and Fang 2001; Fang et al. 2005). Though these studies suggested that antagonism of morphine-induced antinociception by tetrandrine was dependent on serotonergic mechanisms, the precise domain of action on the serotonergic system requires further clarification. Reticuline (5) (IC50 474 μM) and coclaurine (3) (IC50 68.9 μM) have demonstrated antispasmodic activity related to either inhibition of extracellular calcium, an intracellular effect, or both (Martin et al. 1993). Yet the IC50 values of reticuline and coclaurine were too high to provide reliable experimental data for clinical application. Moreover, that study merely evaluated the antispasmodic activity of reticuline and coclaurine, their mechanisms of action in vivo requiring further investigation.
Tetrandrine is a classic calcium channel inhibitor involved in various activities (Wang and Lemos 1994; Wang et al. 2004; Leung et al. 1996). In rat phaeochromocytoma PC 12 cells, 100 μM tetrandrine abolished high K+ (30 mM)-induced sustained increase in cytoplasmic Ca2+ concentration, inhibited bombesin-induced inositol triphosphate accumulation in NIH/3T3 fibroblast and abolished Ca2+ entry (Takemura et al. 1996). The shortage of this study was that the dose of tetrandrine was too high and only one dose was used. Tetrandrine (3–100 μM) concentration and time dependently inhibited the amplitude of the [Ca2+]i transient without any significant effect on the resting level of [Ca2+]i in the isolated single rat cardiomyocyte (Wang et al. 1997). Tetrandrine (10–30 μM) significantly inhibited the increase of [Ca2+]i in induced by glutamate, serotonin and histamine in dissociated retina cells (Xuan et al. 2009). 30 μM tetrandrine inhibited NO production by the endothelial cells through blockade of Ca2+ release-activated Ca2+ channels (Kwan et al. 2001). Tetrandrine inhibited Ca2+-activated chloride channel in cultured human umbilical vein endothelial cells with an IC50 of 5.2 ± 0.4 μM (Fang et al. 2004). Fangchinoline (0.1–10 μM), a nonspecific Ca2+ channel blocker, mitigated the harmful effects of cyanide-induced neuronal cell death by interfering with [Ca2+]i influx by inhibiting glutam ate release and oxidants generation(Cho and Seong 2002). Some experimental animal models must be established to further verify these effects in vivo.
Tetrandrine (60–120 μmol/L) decreased Na+ current density in a concentration-dependent manner and made the voltage-dependent activation curve shift to more positive voltages in the atrial myocardium of patients in atrial fibrillation and sinus rhythm (Chen et al. 2009c). Large-conductance K+ channel is a target for therapeutic strategies aimed at modulating osteoblast proliferation and function. Tetrandrine (5–30 μM) could inhibit large-conductance K+ channel in MG63 cells and primary human osteoblasts (Henny et al. 2009). But these studies lacked proper positive controls.
Cepharanthine (25) (0.0625–2.0 μM) can inhibit receptor activation of osteoclast formation and bone-resorbing activity induced by either NF-κB ligand or estrogen deficiency. Thus, it could be potentially used as an alternative therapy for preventing or treating osteolytic diseases (Lin et al. 2019; Zhou et al. 2018). However, these authors did not compare the efficacy of cepharanthine with a positive control. Isotetrandrine (27) (8, 16 or 32 μM) ameliorates tert-butyl hydrogen peroxide-induced oxidative damage through the upregulation of heme oxygenase-1 expression. This occurred through the dissociation of nuclear translocation of factor-erythroid 2 p45-related factor 2 (Nrf2) from the Nrf2-Keap1 complex by activation of extracellular signal-regulated protein kinase and c-Jun NH2-terminal kinase, and by inactivation of Keap1 (Wang et al. 2016b). The major limitation of this study was the lack of a positive control. Dicentrine (39) (0.03–0.3 μM) causes a blockade of the α1-adrenoceptor. It has therapeutic potential for relieving bladder outlet obstruction caused by benign prostatic hyperplasia (Yu et al. 1994). Since only in vitro isometric tension experiments were studied, an experimental animal model should also be established to further verify the effectiveness of relief of bladder outlet obstruction.
In addition, at the concentration of 10−4 M, tetrandrine (9), cepharanthine (25), fangchinoline (10) and N-methylcoclaurine (1) have demonstrated potential anti-inflammatory effects through inhibition of the release of histamine. Of these compounds, N-methylcoclaurine exhibited an inhibitory effect similar to that of fangchinoline (Nakamura et al. 1992). However, this study evaluated the anti-inflammatory effects of these compounds by inhibition of histamine release only, their mechanisms of action requiring additional investigation. Nantenine (44) (0.3 mg/kg, administered intraperitoneally) was shown to block and rapidly reverse hyperthermia induced by 3,4-methylenedioxymethamphetamine (MDMA), attenuate lethality and reduce MDMA-induced locomotor stimulation and head twitches in mice (Legendre et al. 2010). However, although the study was conducted in an animal model, only one dose was investigated and so additional research is required to further verify its effects. Magnoflorine (49) at 53.5 μM in combination with hyaluronic acid gel promoted subchondral bone regeneration. It also exhibited a protective effect by stimulating the recovery of subchondral bone integrity and attenuating cartilage degradation, preventing the progression of osteoarthritis (Cai et al. 2018). However, this study evaluated the potential benefits of hyaluronic acid-gel combined with magnoflorine in promoting subchondral bone regeneration and the protective effects of stimulating recovery of subchondral bone integrity on attenuating cartilage degradation. The effect of magnoflorine without hyaluronic acid-gel should also be evaluated. Magnoflorine (0–10 μM) was also found to inhibit copper-mediated oxidation of low and high density lipoproteins. It also inhibited glycated and glycoxidated low density lipoproteins by increasing the lag time of conjugated diene formation and prevented the generation of thiobarbituric acid reactive substances (Hung et al. 2007a, b). As above, this trial lacked a positive control.
Pharmacokinetics, including metabolism
Oral administration of 100 mg of tetrandrine (9) in six healthy male volunteers (28–41 years of age) resulted in an area under the curve of plasma concentrations over 0 to infinity (AUC0→∞) of 1554.16 ± 25.07 μg.h/L. The time to reach maximum plasma concentration (Tmax) was 14.00 ± 10.02 h with a maximum plasma concentration (Cmax) of 67.26 ± 23.22 μg/L. The elimination half-life (t1/2) was 22.34 ± 21.22 h (Yang et al. 2017). In a Sprague Dawley (SD) rat model, there was no statistical difference between males and females in tetrandrine plasma concentrations at identical time points, indicating that no sex differences existed in the pharmacokinetics of tetrandrine in SD rats. The principal pharmacokinetic parameters were as follows: AUC0→72 6279.2 ± 2411.5 μg h/L, AUC0→∞ 7002.7 ± 2528.0 μg h/L, Cmax 237.1 ± 95.9 μg/L, Tmax 6.0 ± 1.8 h, t1/2 20.6 ± 3.7 h and an elimination rate constant (Ke) of 0.034 ± 0.006 h−1 (Song et al. 2008). After single intravenous dose of 4 mg/kg tetrandrine in rabbits, a biphasic concentration–time curve was observed, essentially conforming to a two-compartment pharmacokinetic model. The t1/2 of the two-compartments was short at approximately 7.20 min and 92.68 min (Lou and Zhang 1993). A two-compartment pharmacokinetic model and similar pharmacokinetic parameters were also obtained after single intravenous doses of 13 mg/kg tetrandrine to five anesthetized dogs (Zeng et al. 1985).
Although fangchinoline (10) has been shown to have various pharmacological properties, there have been few pharmacokinetic studies on this compound. To compare the different pharmacokinetic parameters of tetrandrine (9) and fangchinoline, 9.3 g/kg tetrandrine and 3.1 g/kg fangchinoline were administered orally to two groups of rats. With the exception of clearance, the majority of the pharmacokinetic parameters were greater for tetrandrine than for fangchinoline. For example, the t1/2 of tetrandrine was 49.6 h and 12.273 h for fangchinoline (Li et al. 2009b).
The pharmacokinetic parameters of cepharanthine (25) in human trials have been reviewed (Desgrouas et al. 2014a). After a single oral dose of 50 mg in 12 healthy Chinese volunteers, cepharanthine was rapidly distributed to tissues before being gradually eliminated. It had the pharmacokinetic parameters Cmax: 136 ± 67 µg/L; Tmax: 0.75 ± 0.21 h and t1/2: 132 ± 48 h (Hao et al. 2010). A Japanese study indicated a linear relationship between oral administration of doses in the range 25 to 100 mg and measured plasma AUC. Absolute oral availability was low at 6–9%, suggesting extensive pre-systemic liver metabolism. No side effects were reported during the study period (Yasuda et al. 1989). In addition, a recent pharmacokinetic study in rats (Deng et al. 2017) demonstrated poor oral availability of cepharanthine at only 5.65 ± 0.35%. In that study, a single intravenous injection of 1 mg/kg cepharanthine produced a Cmax of 153.17 ± 16.18 µg/L and t1/2 of 6.8 ± 1.2 h. Oral administration of 10 mg/kg cepharanthine demonstrated poor oral availability, with a Cmax of 46.89 ± 5.25 µg/L of approximately 2.67 h. The t1/2 was 11.02 ± 1.32 h (Deng et al. 2017).
Two mature female Langyu small-ear miniature pigs were fed 51.3 mg/kg dicentrine (39) mesylate (equivalent to dicentrine 40.0 mg/kg). Twenty-four metabolites were identified in the urine collected from the two pigs. These included nine phase I metabolites, including N-demethylated dicentrine, N-oxidated dicentrine and O-demethylated dicentrine. There were also 15 phase II metabolites, O-glucuronidation and O-glucosylation derivatives of the phenolic groups of the phase I metabolites (Lai et al. 2010). After intravenous administration of 10 mg/kg dicentrine to rats, the detailed pharmacokinetic parameters of dicentrine were measured as: volume of distribution (Vd): 12.58 ± 1.11 L/kg; clearance (CL): 0.51 ± 0.046 L/kg/min; AUC0→∞: 20.95 ± 2.28 μg.min/ml; area under the first moment curve (AUMC): 1262 ± 354 μg min2/ml; mean residence time (MRT): 55.18 ± 8.52 min (Tsai et al. 1996).
An intravenous dose of 5 mg/kg or an oral dose of 15 mg/kg magnoflorine (49) was administered to two groups of male SD rats. The resultant pharmacokinetic evaluation indicated that magnoflorine had a low oral availability of 22.6%. Assessment of the concentration–time curves of magnoflorine in plasma and various tissues (heart, liver, spleen, lung, kidney and brain) after oral administration of 40 mg/kg revealed that it underwent rapid and wide tissue distribution, with the highest concentrations found in the liver, followed by the heart, spleen and lungs. The concentration was least in the brain, indicating that it has difficulty crossing the blood–brain barrier (Bao et al. 2015). After intraperitoneal and oral administration of magnoflorine to rats, the 12 phase I metabolites already identified that were found in various biological matrices (e.g. urine, feces, plasma and organ tissues) were products of demethylation, dehydrogenation, hydroxylation, methylene to ketone transformation, N-ring opening and dihydroxylation. The 11 phase II metabolites consisted of products of methylation, acetylation, glucuronidation and N-acetylcysteine conjugation (Tian et al. 2016a).
The plasma pharmacokinetics and tissue distribution of roemerine (52) in rats was evaluated after oral administration of 20 mg/kg or intravenous administration of 6 mg/kg. Significant roemerine concentrations were detected in all tissues examined 5 min after oral administration, with peak concentrations observed after 20 min in all tissues except the liver, where the highest tissue concentrations were found, in addition to in the lungs, then in decreasing order in the kidneys, spleen and heart. This implies that the liver and lungs might be the target organs of roemerine. As roemerine was also detected in the brain, this suggests that it was able to cross the blood–brain barrier (Liu et al. 2014).
Orally administered stepholidine (57) was rapidly absorbed in the gastrointestinal tract of SD rats, although the oral availability was poor due to extensive pre-systemic metabolism via glucuronidation and sulfation. There were two concentration peaks of stepholidine in plasma, the second peak potentially resulting from enterohepatic circulation. A total of 17 metabolites of glucuronidated stepholidine and sulphated stepholidine were detected in different tissues. Of note, stepholidine was efficiently transported across the blood–brain barrier (Sun et al. 2009).
After oral administration of 60 mg/kg corydalmine (60) to SD rats, the tissue distribution of corydalmine was measured after 0, 0.5, 1, 2, 4, 6, 8, 12, 24 and 48 h post-dose in the brain, heart, kidneys, liver, lungs, spleen, spinal cord and muscle. The results revealed that corydalmine was distributed widely in tissues, although the time to peak concentration varied. Peak concentrations of corydalamine were observed after less than 1 h in the heart, spleen and brain, but after 2–6 h in the kidneys, lungs, spinal cord and muscle. The time to peak concentration occurred after approximately 6 h in the liver, the principle site of corydalamine metabolism by hepatic microsomes. These data indicate that corydalmine was absorbed after oral administration and then distributed quickly in SD rats in vivo. Furthermore, high concentrations in the brain and spinal cord may be related to the pharmacological activity of this compound in the central nervous system (Zhang et al. 2018c). Metabolism of L-corydalmine was evaluated in vitro in liver microsomes from five species: mice, rats, monkeys, dogs and humans. Six metabolites, including three O-demethylated corydalmines and three hydroxylated corydalmines were identified in all five species (Tang et al. 2016).
Isotetrandrine (27) (100 mg/L) was incubated with a male rat hepatic S9 fraction in the presence of a nicotinamide adenine dinucleotide phosphate-generating system. Five metabolites identified were N-desmethyl-isotetrandrine, hydroxyl-isotetrandrine, oxo-isotetrandrine, oxohydroxy-isotetrandrine and N, N-didesmethyl-isotetrandrine (Wu et al. 2004).
The pharmacokinetics and metabolites were evaluated in SD rats administered 30 mg/kg isoboldine (47) by oral gavage. Isoboldine was found to have extremely low oral availability due to a strong first-pass effect. The principle metabolic routes for isoboldine were glucuronidation and sulfonation (Li et al. 2015d).
Toxicity
Recently, the toxicity of traditional medicines, including TCM, have attracted attention, especially those related to nephrotoxicity caused by aristolochic acids and a number of alkaloids (Xu et al. 2016b). To date, no aristolochic acids have been found in S. tetrandra. However, confusion surrounding the use and misidentification of TCMs has increased the risk of toxicity. A. fangchi can cause nephrotoxicity. Misidentification often occurs between S. tetrandra and A. fangchi, resulting in inappropriate use and possible toxic effects (Vanherweghem 1994). In addition to nephrotoxicity, traditional herbs may cause toxicity to other organs, including the liver and lungs, in addition to genotoxicity.
Tetrandrine (9) has been shown to induce oxidative stress and mitochondrial dysfunction in hepatocytes (Qi et al. 2013; Cai et al. 2006). Tetrandrine has been shown to cause acute pulmonary toxicity. This may be due to its biotransformation to a quinone methide-derived metabolite mediated by cytochrome P450 3A (CYP3A) enzymes (Tian et al. 2016b). Tetrandrine has the potential for genotoxicity and carcinogenicity (Rosenkranz and Klopman 1990; Whong et al. 1989; Xing et al. 1989). After intravenous administration of a single dose of 150 mg/kg tetrandrine to mice, tetrandrine induced transient toxicity to the liver, lungs and kidneys. Withdrawal of tetrandrine was associated with reversal of these pathological changes (Shi et al. 2016b). Although no nephrotoxicity caused by aristolochic acids has been reported in S. tetrandra, it may have the potential to cause nephrotoxicity. After S. tetrandra was chronically administered to SD male rats for more than 4 weeks, it damaged tubulointerstitial tissue and damaged the structure of glomeruli (Sun and Wu 2012; Cai et al. 2005). However, as described above, tetrandrine used in combination with prednisone caused a synergistic effect on renal fibrosis caused by adriamycin in rats (Bai and Dong 2016). Therefore, a safe and effective dose of tetrandrine still requires evaluation.
Of the compounds in S. tetrandra, only tetrandrine has been reported to have the potential for hepatotoxicity, pulmonary toxicity, genotoxicity or nephrotoxicity. The toxicity of other structurally similar compounds in S. tetrandra requires further evaluation.
Conclusions, discussion and future perspectives
The present review summarized the research progress in the traditional use, phytochemistry, pharmacology and toxicology of S. tetrandra and its extracts and constituents. The research achievements and gaps were discussed. Although there have been some achievements in the traditional use, phytochemistry, pharmacology and toxicology of S. tetrandra, there are still some scientific gaps. Theses scientific gaps are as follows: (1) the plant origin of “Fang Ji” and the strating time of traditional uses of S. tetrandra is needed to further investigation. (2) Lack of phytochemistry studies on the aerial part of S. tetrandra. (3) Anti-inflammatory and anticancer effects are the most studied in the wide range of pharmacological properties of S. tetrandra. However, other pharmacological properties need to further in-depth investigated S. tetrandra. (4) Toxicological studies on S. tetrandra are limited. To date, only tetrandrine from S. tetrandra has been identified as causing potential toxicity to the liver, lungs, genes and kidneys. Due to a lack of data assessing other alkaloids, their toxicity requires further evaluation. (5) There are many alkaloids that have only been tentatively identified by mass spectrometry. Their precise structures need to be determined by an in-depth exploration using nuclear magnetic spectroscopy (NMR), infrared spectroscopy (IR), ultraviolet spectroscopy (UV), circular dichroism (CD) and mass spectrometry (MS). (6) Alkaloids are traditionally considered the major bioactive compounds in S. tetrandra, which leads to the result that other constituents of S. tetrandra such as bisflavone have not yet been investigated. It severely limited the diversity of research and applications of S. tetrandra. (7) Analyses of the structure–activity relationships of these alkaloids are insufficient. (8) In vitro and in vivo pharmacological studies have increasingly confirmed the traditional use of S. tetrandra as an analgesic, antiphlogistic and immunoregulatory medicine for arthralgia caused by rheumatism and inflamed sores. However, it’s antinociceptive and immunomodulatory effects have been insufficiently studied. (9) Studies on the pharmacology, especially anti-tumor effects, have critical limitations, with insufficient numbers, replications and a lack of in-depth or systematic study at the molecular and cellular levels. (10) S. tetrandra has been shown to possess a number of distinct pharmacological activities at both the cellular level and that of the whole organism. However, the mechanisms of action of component molecules are poorly described and require further investigation.
Future perspectives
- 1.
Based on the traditional uses S. tetrandra to treat arthralgia caused by rheumatism and inflamed sores, a greater number of studies of the antinociceptive and immunomodulatory effects should be conducted in future.
- 2.
Most of the compounds studied are those identified in the roots of S. tetrandra, and so more studies are required of the chemical constituents of the aerial parts of S. tetrandra.
- 3.
The definitive structures of the majority of the alkaloids should be confirmed using comprehensive spectroscopy techniques.
- 4.
The anticancer effects of alkaloids in S. tetrandra should be further verified in vivo. Thus, S. tetrandra has the potential to be explored as a new anticancer drug.
- 5.
Further in vitro and in vivo animal studies are required for toxicological evaluation prior to future clinical studies.
References
Agnihotri VK, ElSohly HN, Khan SI et al (2008) Constituents of Nelumbo nucifera leaves and their antimalarial and antifungal activity. Phytochem Lett 1(2):89–93
Angerhofer CK, Guinaudeau H, Wongpanich V et al (1999) Antiplasmodial and cytotoxic activity of natural bisbenzylisoquinoline alkaloids. J Nat Prod 62(1):59–66
Aota K, Yamanoi T, Kani K et al (2018) Cepharanthine inhibits IFN-γ induced CXCL10 by suppressing the JAK2/STAT1 signal pathway in human salivary gland ductal cells. Inflammation 41(1):50–58
Avci FG, Sayar NA, Sariyar Akbulut B (2018) An OMIC approach to elaborate the antibacterial mechanisms of different alkaloids. Phytochemistry 149:123–131
Ayyildiz D, Arga KY, Avci FG et al (2017) Transcriptomic analysis displays the effect of (-)-roemerine on the motility and nutrient uptake in Escherichia coli. Curr Genet 63(4):709–722
Baghdikian B, Mahiou-Leddet V, Bory S et al (2013) New antiplasmodial alkaloids from Stephania rotunda. J Ethnopharmacol 145(1):381–385
Bai SS, Dong C (2016) The protective effect and mechanism of tetrandrine combined with prednisone for renal fibrosis rats caused by Adriamycin. Jilin Med J 37(8):1845–1848
Bai HM, Liu H, Zhou BQ et al (2015) Radiosensitization of tetrandrine in the esophageal carcinoma. J Basic Clin Oncol 28(2):129–131
Bai XY, Liu YG, Song W et al (2018) Anticancer activity of tetrandrine by inducing pro-death apoptosis and autophagy in human gastric cancer cells. J Pharm Pharmacol 70(8):1048–1058
Bailly C (2019) Cepharanthine: an update of its mode of action, pharmacological properties and medical applications. Phytomedicine 62:152956
Bao S, Geng P, Wang S et al (2015) Pharmacokinetics in rats and tissue distribution in mouse of magnoflorine by ultra performanceliquid chromatography-tandem mass spectrometry. Int J Clin Exp Med 8:20168–20177
Bao G, Li C, Qi L et al (2016) Tetrandrine protects against oxygen-glucose serum deprivation/reoxygenation-induced injury via PI3K/AKT/NF-κB signaling pathway in rat spinal cord astrocytes. Biomed Pharmacother 84:925–930
Benhamou RI, Steinbuch KB, Fridman M (2016) Antifungal imidazole-decorated cationic amphiphiles with markedly low hemolytic activity. Chemistry 22(32):11148–11151
Bhagya N, Chandrashekar KR (2016) Tetrandrine-a molecule of wide bioactivity. Phytochemsitry 125:5–13
Bhagya N, Chandrashekar KR (2018) Tetrandrine and cancer-an overview on the molecular approach. Biomed Pharmacother 97:624–632
Cai ZY, Zhou JM, Ge YR (2005) Influence of high-dose tetrandra on rat renal function and tissue morphology. Chin J Hosp Pharm 25:1200–1201
Cai Y, Qi XH, Gong LK et al (2006) Tetrandrine-induced apoptosis in rat primary hepatocytes is initiated from mitochondria: caspases and endonuclease G (Endo G) pathway. Toxicology 218(1):1–12
Cai XH, Wang S, Chen BA (2011) Research advances on the pharmacological effects of tetrandrine. Chin J Nat Med 9:473–480
Cai Z, Feng Y, Li C et al (2018) Magnoflorine with hyaluronic acid gel promotes subchondral bone regeneration and attenuates cartilage degeneration in early osteoarthritis. Bone 116:266–278
Castranova V, Kang JH, Ma JK et al (1991) Effects of bisbenzylisoquinoline alkaloids on alveolar macrophages: correlation between binding affinity, inhibitory potency, and antifibrotic potential. Toxicol Appl Pharmacol 108(2):242–252
Chea A, Bun SS, Azas N et al (2010) Antiplasmodial activity of three bisbenzylisoquinoline alkaloids from the tuber of Stephania rotunda. Nat Prod Res 24:1766–1770
Chen W (2006) Research on herbal literuatures of Fangji and its adverse drug reaction. Chengdu University of Traditional Chinese Medicine
Chen KS, Ko FN, Teng CM et al (1996) Antiplatelet of vaso relaxing actions of some benzylisoquinoline and phenanthrene alkaloids. J Nat Prod 59:531–534
Chen YJ, Tu ML, Kuo HC et al (1997) Protective effect of tetrandrine on normal human mononuclear cells against ionizing irradiation. Biol Pharm Bull 20(11):1160–1164
Chen G, Shi DZ, Li WK (2002) Effect of tetrandrine and albendazole on Echinococcus multilocularis infection in mice. Chin J Zoonoses 18(6):69–72
Chen G, Shi DZ, Li WK (2003) An approach to mechanism of tetrandrine and albendazole against Echinococcus multilocularis infection in mice. Endemic Dis Bull 18(2):18–21
Chen WC, Hayakawa S, Yamamoto T et al (2004) The plasma glucose lowering action of tetrandrine in streptozotocin-induced diabetic rats. J Pharm Pharmacol 56:643–648
Chen JH, Du ZZ, Shen YM et al (2009a) Aporphine alkaloids from Clematis parviloba and their antifungal activity. Arc Pharm Res 32(1):3–5
Chen J, Liu J, Wang T et al (2009b) The relaxation mechanisms of tetrandrine on the rabbit corpus cavernosum tissue in vitro. Nat Prod Res 23:112–121
Chen L, Li QY, Li ZW et al (2009c) Inhibitory effects of tetrandrine on the Na+ channel of human atrial fibrillation myocardium. Acta Pharmcol Sin 30:166–174
Chen L, Chen L, Lv Y et al (2013) Tetrandrine ameliorates cognitive impairment via inhibiting astrocyte-derived S100B activation in a rat model of chronic cerebral hypoperfusion. Neurol Res 35(6):614–621
Chen T, Ji B, Chen Y (2014) Tetrandrine triggers apoptosis and cell cycle arrest in human renal cell carcinoma cells. J Nat Med 68:46–52
Chen Y, Xiao X, Wang C et al (2015) Beneficial effect of tetrandrine on refractory epilepsy via suppressing P-glycoprotein. Int J Neurosci 125(9):703–710
Chen S, Liu W, Wang K et al (2017a) Tetrandrine inhibits migration and invasion of human renal cell carcinoma by regulating Akt/NF-κB/MMP-9 signaling. PLoS ONE 12:e0173725
Chen Z, He T, Zhao K et al (2017b) Anti-metastatic activity of fangchinoline in human gastric cancer AGS cells. Oncol Lett 13:655–660
Chen N, Rong XH, Zhao J (2018) Clinical observation on the therapeutic effect of tetrandrine combined with ursolic acid in the treatment of silicosis. Ind Health Occup Dis 44(6):454–456
Chinese Botany Editorial Committee (1996) Flora of China. Science Press, Beijing, p 31
Chinese Pharmacopoeia Commission (2015) Pharmacopoeia of People’s Republic of China, Part 1. China Medical Science Press, Beijing, p 148
Chiou WF, Lee WS, Yeh PH (2006) Tetrandrine selectively protects against amyloid-beta protein -but not against MPTP-induced cytotoxicity in SK-N-SH neuroblastoma cells. Plant Med 72(14):1300–1304
Cho SO, Seong YH (2002) Protective effect of fangchinoline on cyanide-induced neurotoxicity in cultured rat cerebellar granule cells. Arch Pharm Res 25:349–356
Cho HS, Chang SH, Chung YS et al (2009) Synergistic effect of ERK inhibition on tetrandrine-induced apoptosis in A549 human lung carcinoma cells. J Vet Sci 10:23–95
Choi SU, Park SH, Kim KH et al (1998) The bisbenzylisoquinoline alkaloids, tetrandine and fangchinoline, enhance the cytotoxicity of multidrug resistance-related drugs via modulation of P-glycoprotein. Anticancer Drug 9:255–261
Choi HS, Kim HS, Min KR et al (2000) Anti-inflammatory effects of fangchinoline and tetrandrine. J Ethnopharmacol 69(2):173–179
Chor JS, Yu J, Chan KK et al (2009) Stephania tetrandra prevents and regresses liver fibrosis induced by carbon tetrachloride in rats. J Gastroenterol Hepatol 24(5):853–859
Chow LWC, Cheng KS, Leong F et al (2018) Enhancing tetrandrine cytotoxicity in human lung carcinoma A549 cells by suppressing mitochondrial ATP production. Naunyn Schmiedebergs Arch Pharmacol 392(4):427–436
Cos P, Vlietinck AJ, Berghe DV (2006) Anti-infective potential of natural products: how to develop a stronger in vitro ‘proof-of-concept’. J Ethnopharmacol 106:209–302
Deng JZ, Zhao SX, Lou FC (1990) A new monquaternary bisbenzylisoquinoline alkaloid from Stephania tetrandra. J Nat Prod 53:993–994
Deng Y, Wu W, Ye S et al (2017) Determination of cepharanthine in rat plasma by LC-MS/MS and its application to a pharmacokinetic study. Pharma Biol 55(1):1775–1779
Desgrouas C, Taudon N, Bun SS et al (2014a) Ethnobotany, phytochemistry and pharmacology of Stephania rotunda Lour. J Ethnopharmacol 154:537–563
Desgrouas C, Dormoi J, Chapus C et al (2014b) In vitro and in vivo combination of cepharanthine with anti-malarial drugs. Malar J 13:90
Desgrouas C, Chapus C, Desplans J et al (2014c) In vitro antiplasmodial activity of cepharanthine. Malar J 13:327
Ding W, Zhang HB, Zhang DF et al (2008) Clinical study on radiotherapy sensitization of nasopharyngeal carcinoma by tetrandrine. J Benbu Med Coll 33:339–341
Ding W, Yang AZ, Xu HJ et al (2016) Effects of tetrandrine on radiosensitvity of lung adenocarcinoma cell line and its mechanism. Chin Clin Oncol 21:409–412
Dong JW, Cai L, Fang YS et al (2015) Proaporphine and aporphine alkaloids with acetylcholinesterase inhibitory activity from Stephania epigaea. Fitoterapia 104:102–107
Estellés R, López-Martín J, Milian L et al (2003) Effect of two phenanthrene alkaloids on angiotensin II-induced leukocyte-endothelial cell interactions in vivo. Br J Pharmacol 140(6):1057–1067
Fadaeinasab M, Taha H, Fauzi PN et al (2015) Anti-malarial activity of isoquinoline alkaloids from the stem bark of Actinodaphne macrophylla. Nat Prod Commun 10(9):1541–1542
Fan B, Zhang X, Ma Y et al (2017) Fangchinoline induces apoptosis, autophagy and energetic impairment in bladder cancer. Cell Physiol Biochem 43(3):1003–1011
Fang QZ, Zhong N, Zhang Y et al (2004) Tetrandrine inhibits Ca2+-activated chloride channel in cultured human umbilical vein endothelial cells. Acta Pharmacol Sin 25:327–333
Fang LH, Zhang YH, Ku BS (2005) Fangchinoline inhibited the antinociceptive effect of morphine in mice. Phytomedicine 12:183–188
Feng YX, Zhu ZY, Chen H (1983) Comparison of pharmacognosy morphology and histological structure of the medicinal plants of the genus Stephania (Menispermaceae). Acta Pharm Sin 18:849–861
Gao KY (2017) Clinical experience of YU Junsheng in treating renal edema with modified FangjiHuangqi decoction. Shandong J Trad Chin Med 36:786–790
Gao LN, Feng QS, Zhang XF et al (2016) Tetrandrine suppresses articular inflammatory response by inhibiting pro-inflammatory factors via NF-κB inactivation. J Orthop Res 34(9):1557–1568
Gao S, Li X, Ding X et al (2017) Cepharanthine induces autophagy, apoptosis and cell cycle arrest in breast cancer cells. Cell Physiol Biochem 41(4):1633–1648
García Díaz J, Tuenter E, Escalona Arranz JC et al (2019) Antimicrobial activity of leaf extracts and isolated constituents of Croton linearis. J Ethnopharmacol 236:250–257
Gertsch J (2009) How scientific is the science in ethnopharmacology? Historical perspectives and epistemological problems. J Ethnopharmacol 122:177–183
Gokgoz NB, Akbulut BS (2015) Proteomics evidence for the activity of the putative antibacterial plant alkaloid (-)-roemerine: mainstreaming omics-guided drug discovery. OMICS 19(8):478–489
González-Coloma A, Reina M, Sáenz C et al (2012) Antileishmanial, antitrypanosomal, and cytotoxic screening of ethnopharmacologically selected Peruvian plants. Parasitol Res 110(4):1381–1392
Guo Y, Pei X (2019) Tetrandrine-induced autophagy in MDA-MB-231 triple-negative breast cancer cell through the inhibition of PI3K/AKT/mTOR signaling. Evid based Complement Altern Med 2019:7517431
Guo C, Wang M, Li J et al (2015) Effect of inflammatory cytokines in the LPS-induced RAW264.7 cells by the decoction and its split components from Stephania tetrandra S.Moore. Acta Chin Med Pharmacol 43:33–36
Guo B, Xie P, Su J et al (2016) Fangchinoline suppresses the growth and invasion of human glioblastoma cells by inhibiting the kinase activity of Akt and Akt-mediated signaling cascades. Tumour Biol 37(2):2709–2719
Guo S, Jiang K, Wu H et al (2018) Magnoflorine ameliorates lipopolysaccharide-induced acute lung injury via suppressing NF-κB and MAPK activation. Front Pharmacol 30:982
Han B, Fu P, Ye Y et al (2015) Protective effects of tetrandrine on brain cells in phenobarbital dependent and -withdrawn rats. Mol Med Rep 11:1939–1944
Hao G, Liang H, Li Y et al (2010) Simple, sensitive and rapid HPLC-MS/MS method for the determination of cepharanthine in human plasma. J Chromatogr B Anal Technol Biomed Life Sci 878(28):2923–2927
Hao JR, Sun N, Lei L et al (2015) L-Stepholidine rescues memory deficit and synaptic plasticity in models of Alzheimer's diseasevia activating dopamine D1 receptor/PKA signaling pathway. Cell Death Dis 6(11):e1965
Haque MA, Jantan I, Harikrishnan H et al (2018) Magnoflorine enhances LPS-activated pro inflammatory responses via MyD88-dependent pathways in U937 macrophages. Plant Med 84:1255–1264
He FQ, Qiu BY, Zhang XH et al (2011) Tetrandrine attenuates spatial memory impairment and hippocampal neuroinfla-mmation viainhibiting NF-κB activation in a rat model of Alzheimer's disease induced by amyloid-β(1–42). Brain Res 1384:89–96
Henny NC, Li B, Elford C et al (2009) A large-conductance (BK) potassium channel subtype affects both growth and mineralization of human osteoblasts. Am J Physiol Cell Physiol 297:C1397–C1408
Hicks C, Huang P, Ramos L et al (2018) Dopamine D1-like receptor agonist and D2-like receptor antagonist (–)-stepholidine reduces reinstatement of drug-seeking behavior for 3,4-methylenedioxypyrovalerone (MDPV) in rats. ACS Chem Neurosci 9(6):1327–1337
Hošt'álková A, Opletal L, Kuneš J et al (2015) Alkaloids from Peumus boldus and their acetylcholinesterase, butyrylcholinesterase and prolyloligopeptidase inhibition activity. Nat Prod Commun 10:577–580
Hsu JH, Wu YC, Liou SS et al (2004) Mediation of endogenous beta endorphin by tetrandrine to lower plasma glucose in streptozotocin-induced diabetic Rats. Evid based Complement Altern Med 1(2):193–201
Hsu YC, Chiu YT, Cheng CC et al (2007) Antifibrotic effects of tetrandrine on hepatic stellate cells and rats with liver fibrosis. J Gastroenterol Hepatol 22(1):99–111
Hu SL (2009) Textual research on herbal medicine of Fang Ji. Modern Pharm Clin 24:286–288
Hu S, Dutt J, Zhao T et al (1997) Tetrandrine potently inhibits herpes simplex virus type-1 induced keratitis in BALB/c mice. Ocul Immunol and Inflamm 5(3):173–180
Hu YY, He KW, Guo RL (2012) Six alkaloids inhibit secretion of IL-1α, TXB (2), ET-1 and E-selectin in LPS-induced endothelial cells. Immunol Invest 41(3):261–274
Hua P, Sun M, Zhang G et al (2015) Cepharanthine induces apoptosis through reactive oxygen species and mitochondrial dysfunction in human non-small-cell lung cancer cells. Biochem Biophys Res Commun 460(2):136–142
Huang RL, Chen CC, Huang YL et al (1998) Anti-tumor effects of d-dicentrine from the root of Lindera megaphylla. Plant Med 64:212–215
Huang YT, Chang FC, Chen KJ et al (1999) Acute hemodynamic effects of tetramethylpyrazine and tetrandrine on cirrhotic rats. Plant Med 65:130–134
Huang P, Xu Y, Wei R et al (2011) Efficacy of tetrandrine on lowering intraocular pressure in animal model with ocular hypertension. J Glaucoma 20(3):183–188
Huang AC, Lien JC, Lin MW et al (2013) Tetrandrine induces cell death in SAS human oral cancer cells through caspase activation-dependent apoptosis and LC3-I and LC3-II activation-dependent autophagy. Int J Oncol 43(2):485–494
Huang HP, Peng HS, Wang DL et al (2015) Textual research on historical evolution of Traditional Chinese medicine of Fang Ji. J Chin Med Mater 38:1533–1535
Huang YL, Cui SY, Cui XY et al (2016) Tetrandrine, an alkaloid from S. tetrandra exhibits anti-hypertensive and sleep-enhancing effects in SHR via different mechanisms. Phytomedicine 23:1821–1829
Hung TM, Na M, Min BS et al (2007a) Protective effect of magnoflorine isolated from coptidis rhizoma on Cu2+-induced oxidation of human low density lipoprotein. Plant Med 73:1281–1284
Hung TM, Lee JP, Min BS et al (2007b) Magnoflorine from Coptidis Rhizoma protects high density lipoprotein during oxidant stress. Biol Pharm Bull 30:1157–1160
Idec-Sadkowska I, Andrzejak R, Antonowicz-Juchniewicz J et al (2006) Trials of casual treatment of silicosis. Med Pr 57:271–280
Indra B, Tadano T, Nakagawasai O et al (2002) Suppressive effect of nantenine, isolated from Nandina domestica Thunberg, on the 5-hydroxy-L-tryptophan plus clorgyline-induced head-twitch response in mice. Life Sci 70:2647–2656
Ingkaninan K, Phengpa P, Yuenyongsawad S et al (2006) Acetylcholinesterase inhibitors from Stephania venosa tuber. J Pharm Pharmacol 58:695–700
Jang BC (2016) Tetrandrine has anti-adipogenic effect on 3T3-L1 preadipocytes through the reduced expressionand/or phosphorylation levels of C/EBP-α, PPAR-γ, FAS, perilipin A, and STAT-3. Biochem Biophys Res Commun 476:481–486
Jiang Y, Liu J, Zhou Z et al (2018) Fangchinoline protects against renal injury in diabetic nephropathy by modulating the MAPK signaling pathway. Exp Clin Endocrinol Diabetes. https://doi.org/10.1055/a-0636-3883
Jiang YW, Cheng HY, Kuo CL et al (2019) Tetrandrine inhibits human brain glioblastoma multiforme GBM 8401 cancer cell migration and invasion in vitro. Environ Toxicol 34(4):364–374
Jiangsu New Medical College (2006) Dictionary of traditional Chinese medicine. Shanghai Science and Technology Publishing House, Shanghai, pp 1182–1185
Joshi VC, AvulaIkhlas B, Khan IA (2008) Authentication of Stephania tetrandra S. Moore (Fang Ji) and differentiation of its common adulterants using microscopy and HPLC analysis. J Nat Med 62:117–121
Kang HS, Kim YH, Lee CS et al (1996) Anti-inflammatory effects of Stephania tetrandra S Moore on interleukin 6 production and experimental inflammatory disease models. Mediat Inflamm 5(4):280–291
Kang OH, An HJ, Kim SB et al (2014) Tetrandrine suppresses pro-inflammatory mediators in PMA plus A23187-induced HMC-1 cells. Int J Mol Med 33(5):1335–1340
Kashiwada Y, Aoshima A, Ikeshiro Y et al (2005) Anti-HIV benzylisoquinoline alkaloids and flavonoids from the leaves of Nelumbo nucifera, and structure-activity correlations with related alkaloids. Bioorg Med Chem 13(2):443–448
Kim HS, Zhang YH, Oh KW et al (1997) Vasodilating and hypotensive effects of fangchinoline and tetrandrine on the rat aorta and the stroke-prone spontaneously hypertensive rat. J Ethnopharmacol 58:117–123
Kim HS, Zhang YH, Fang LH et al (1999) Effects of tetrandrine and fangchinoline on human platelet aggregation and thromboxane B2 formation. J Ethnopharmacol 66:241–246
Kim SD, Oh SK, Kim HS et al (2001) Inhibitory effect of fangchinoline on excitatory amino acids-induced neurotoxicity in cultured ratcerebellar granule cells. Arch Pharm Res 24(2):164–170
Kim J, Ha Q, Bao T et al (2018) Antifungal activity of magnoflorine against Candida strains. World J Microbiol Biotechnol 34(11):167
Koh SB, Ban JY, Lee BY et al (2003) Protective effects of fangchinoline and tetrandrine on hydrogen peroxide-induced oxidative neuronal cell damage in cultured rat cerebellar granule cells. Plant Med 69:506–512
Kondo Y, Takano F, Hojo H (1993) Inhibitory effect of bisbenzylisoquinoline alkaloids on nitric oxide production in activated macrophages. Biochem Pharmacol 46:1887–1892
Kongkiatpaiboon S, Duangdee N, Prateeptongkum S et al (2016) Acetylcholinesterase inhibitory activity of alkaloids isolated from Stephania venosa. Nat Prod Commun 11:1805–1806
Konkimalla VB, Efferth T (2010) Inhibition of epidermal growth factor receptor over-expressing cancer cells by the aphorphine-type isoquinoline alkaloid, dicentrine. Biochem Pharmacol 79(8):1092–1099
Kukula-Koch W, Kruk-Słomka M, Stępnik K et al (2017) The evaluation of pro-cognitive and antiamnestic properties of berberine and magnoflorine isolated from Barberry species by centrifugal partition chromatography (CPC), in relation to QSAR modelling. Int J Mol Sci 18(12):E2511
Kwan CY, Leung YM, Kwan TK et al (2001) Tetrandrine inhibits Ca2+ release-activated Ca2+ channels in vascular endothelial cells. Life Sci 68:841–847
Lai JH (2002) Immunomodulatory effects and mechanisms of plant alkaloid tetrandrine in autoimmune diseases. Acta Pharmacol Sin 23:1093–1101
Lai JH, Ho LJ, Kwan CY et al (1999) Plant alkaloid tetrandrine and its analog block CD28-costimulated activities of human peripheral blood T cells: potential immunosuppressants in transplantation immunology. Transplantation 68:1383–1392
Lai YC, Kuo TF, Chen CK et al (2010) Metabolism of dicentrine: identification of the phase I and phase II metabolites in miniature pig urine. Drug Metab Dispos 38(10):1714–1722
Lan J, Huang L, Lou H et al (2018) Design and synthesis of novel C14-urea-tetrandrine derivatives with potent anti-cancer activity. Eur J Med Chem 143:1968–1980
Lee YS, Han SH, Lee SH et al (2011) Synergistic effect of tetrandrine and ethidium bro-mide against methicillin-resistant staphylococcus aureus. J Toxicol Sci 36:645–651
Lee YS, Han SH, Lee SH et al (2012) The mechanism of antibacterial activity of tetrandrine against Staphylococcus aureus. Foodborne Pathog Dis 9:686–691
Lee HS, Safe S, Lee SO et al (2017) Inactivation of the orphan nuclear receptor NR4A1 contributes to apoptosis induction by fangchinoline in pancreatic cancer cells. Toxicol Appl Pharmacol 332:32–39
Legendre O, Pecic S, Chaudhary S et al (2010) Synthetic studies and pharmacological evaluations on the MDMA ('Ecstasy') antagonist nantenine. Bioorg Med Chem Lett 20(2):628–631
Leung YM, Kwan CY, Loh TT et al (1996) Capacitative Ca2+ entry in HL-60 cells: tetrandrine and SK & F 96365 as probes. China J Chin Mater Med 17(2):97–101
Li FX, Zhang H (2006) In vitro study of the synergistic effect of tetrandrine and fluconazole against Candida albicans. Chin J Dermatol 39:454–456
Li CX, Li L, Lou J et al (1998) The protective effects of traditional Chinese medicine prescription, han-dan-gan-le, on CCl4-induced liver fibrosis in rats. Am J Chin Med 26(3–4):325–332
Li QP, Lu ZA, Rao MR (2001) Depressive effect of tetrandrine on collagen synthesis in vascular smooth muscle cells. Acta Pharm Sin 36(7):481–484
Li QP, Leng J, Peng T et al (2003) Regression of vascular remodeling in renovascular hypertensive rats by tetrandrine and enalapril. Acta Pharm Sin 38(5):328–332
Li XN, Yan HX, Sha N et al (2009a) Isolation and identification of alkaloids from the root of Stephania tetrandra. J Shenyang Pharm Univ 26:430–433
Li ZH, Fan XL, Cai MM et al (2009b) Pharmacokinetics of fangchinoline and tetrandrine in rats. China J Chin Mater Med 34(23):3110–3113
Li ZW, Zhong SZ, Liu DW et al (2012) Effect of tetrandrine on the TGF-β-induced smad signal transduction pathway in human hypertrophic scar fibroblasts in vitro. Burns 38(3):404–413
Li SX, Song YJ, Zhang LL et al (2015a) An in vitro and in vivo study on the synergistic effect and mechanism of itraconazole or voriconazole alone and in combination with tetrandrine against Aspergillus fumigatus. J Med Microbiol 64:1008–1020
Li D, Lu Y, Sun P et al (2015b) Inhibition on proteasome β1 subunit might contribute to the anti-cancer effects of fangchinoline in human prostate cancer cells. PLoS ONE 10:e0141681
Li XY, Mei GH, Dong Q et al (2015c) Enhanced neuroprotective effects of coadministration of tetrandrine with Glutathione in preclinical model of parkinson's disease. Parkinsons Dis 2015:931058
Li Y, Zeng RJ, Chen JZ et al (2015d) Pharmacokinetics and metabolism study of isoboldine, a major bioactive component from Radix Linderae in male rats by UPLC-MS/MS. J Ethnopharmacol 171:154–160
Li X, Jin Q, Wu YL et al (2016) Tetrandrine regulates hepatic stellate cell activation via TAK1 and NF-κB signaling. Int Immunopharmacol 36:263–270
Li SX, Song YJ, Jiang L et al (2017a) Synergistic effects of tetrandrine with posaconazole against Aspergillus fumigatus. Microb Drug Resist 23(6):674–681
Li X, Yang Z, Han W et al (2017b) Fangchinoline suppresses the proliferation, invasion and tumorigenesis of human osteosarcomacells through the inhibition of PI3K and downstream signaling pathways. Int J Mol Med 40(2):311–318
Li HJ, Wang GM, Li Y et al (2018a) Comparison of therapeutic effect of Fangji Fuling decoction formula granule and conventional Fangji Fuli decoction on post-thrombotic syndrome. Shandong J Trad Chin Med 37(10):821–823
Li X, Wu Z, He B et al (2018b) Tetrandrine alleviates symptoms of rheumatoid arthritis in rats by regulating the expression of cyclooxygenase-2 and inflammatory factors. Exp Ther Med 16(3):2670–2676
Li JN, Wang QH, Wang ZB et al (2019) Tetrandrine inhibits colon carcinoma HT-29 cells growth via the Bcl-2/Caspase 3/PARP pathway and G1/S phase. Biosci Rep 39(5):BSR20182109
Liang Q, Ni C, Yan XZ et al (2010) Comparative study on metabonomics and on liver and kidney toxicity of Aristolochia fangchi and Stephania tetrandra. China J Chin Mater Med 35(21):2882–2888
Lien JC, Lin MW, Chang SJ et al (2017) Tetrandrine induces programmed cell death in human oral cancer CAL 27 cells through the reactive oxygen species production and caspase-dependent pathways and associated with beclin-1-induced cell autophagy. EnvironToxicol 32(1):329–343
Lin TY, Lu CW, Tien LT et al (2009) Fangchinoline inhibits glutamate release from rat cerebral cortex nerve terminals (synaptosomes). Neurochem Int 54(8):506–512
Lin YC, Chang CW, Wu CR (2015) Anti-nociceptive, anti-inflammatory and toxicological evaluation of Fang-Ji-Huang-Qi-Tang in rodents. BMC Complement Altern Med 15:10
Lin YJ, Peng SF, Lin ML et al (2016) Tetrandrine induces apoptosis of human nasopharyngeal carcinoma NPC-TW 076 cells through reactive oxygen species accompanied by an endoplasmic reticulum stress signaling pathway. Molecules 21:1353
Lin X, Song F, Zhou L et al (2019) Cepharanthine suppresses osteoclast formation by modulating the nuclear factor-κB and nuclear factor of activated T-cell signaling pathways. J Cell Biochem 120:1990–1996
Liou JT, Chen ZY, Ho LJ et al (2008) Differential effects of triptolide and tetrandrine on activation of COX-2, NF-κB, and AP-1 and virus production in dengue virus-infected human lung cells. Eur J Pharmacol 589(1–3):288–298
Liu T (2017) Studies on the antitumor differentiation therapy of tetrandrine, Wuhan University
Liu BC, He YX, Miao Q et al (1994) The effects of tetrandrine (TT) and polyvinylpyridine-N-oxide (PVNO) on gene expression of type I and type III collagens during experimental silicosis. Biomed Environ Sci 7(3):199–204
Liu XJ, Wang YF, Zhang MY et al (2004) Study on the inhibitory effect of cepharanthine on herpes simplex type-1 virus (HSV-1) in vitro. J Chin Med Mater 27(2):107–109
Liu YQ, He GH, Li HL et al (2014) Plasma pharmacokinetics and tissue distribution study of roemerine in rats by liquidchromatography with tandem mass spectrometry (LC-MS/MS). J Chromatogr B Anal Technol Biomed Life Sci 969:249–255
Liu T, Liu X, Li WH (2016) Tetrandrine, a Chinese plant-derived alkaloid, is a potential candidate for cancer chemotherapy. Oncotarget 7:40800–40815
Liu J, Zhang Y, Zhao W et al (2017a) Advances in chemical constituents and pharmacology of Stephania tetrandra. Acta Chin Med Pharm 45:100–103
Liu KC, Lin YJ, Hsiao YT et al (2017b) Tetrandrine induces apoptosis in human nasopharyngeal carcinoma NPC-TW 039 cells by endoplasmic reticulum stress and Ca2+/Calpain pathways. Anticancer Res 37(11):6107–6118
Liu T, Zhang Z, Yu C et al (2017c) Tetrandrine antagonizes acute megakaryoblastic leukaemia growth by forcing autophagy-mediated differentiation. Br J Pharmacol 174(23):4308–4328
Lohombo-Ekomba ML, Okusa PN, Penge O et al (2004) Antibacterial, antifungal, antiplasmodial, and cytotoxic activities of Albertisia villosa. J Ethnopharmacol 93(2–3):331–335
Lou JS, Zhang CL (1993) Pharmcokinetic studies studies on drug interaction between tetrandrine and propranolol in rabbits. China Trad Herb Drugs 24:24–26
Lu Y, Li F, Xu T et al (2017) Tetrandrine prevents multidrug resistance in the osteosarcoma cell line, U-2OS, by preventing Pgp overexpression through the inhibition of NF-κB signaling. Int J Mol Med 39(4):993–1000
Luo XR (1982) A systematic notes on the genus Stephania of China. Bull Botan Res 2:33–59
Luo X, Peng JM, Su LD et al (2016) Fangchinoline inhibits the proliferation of SPC-A-1 lung cancer cells by blocking cell cycle progression. Exp Ther Med 11(2):613–618
Lv XL, Zhang H, Song YJ et al (2014) A preliminary research of tetrandrine and fluconazole affecting the cell cycle in Candida albicans. Chin J Mycol 9:215–217
Lv Q, Zhu XY, Xia YF et al (2015) Tetrandrine inhibits migration and invasion of rheumatoid arthritis fibroblast-like synoviocytesthrough down-regulating the expressions of Rac1, Cdc42, and RhoA GTPases and activation of the PI3K/Akt and JNK signaling pathways. Chin J Nat Med 13(11):831–841
Lv YL, Wu ZZ, Chen LX et al (2016) Neuroprotective effects of tetrandrine against vascular dementia. Neural Regen Res 11(3):454–459
Ma W, Nomura M, Takahashi-Nishioka T et al (2007) Combined effects of fangchinoline from Stephania tetrandra Radix and formononetin and calycosin from Astragalus membranaceus Radix on hyperglycemia and hypoinsulinemia in streptozotocin-diabetic mice. Biol Pharm Bull 30(11):2079–2083
Ma C, Du F, Yan L et al (2015) Potent activities of roemerine against Candida albicans and the underlying mechanisms. Molecules 20:17913–17928
Ma H, Yao L, Pang L et al (2016) Tetrandrine ameliorates sevoflurane-induced cognitive impairment via the suppression of inflammation and apoptosis in aged rats. Mol Med Rep 13(6):4814–4820
Ma HB, Tian ZS, Gui SL et al (2017) Anti-prostate cancer effect of roemerine: an experimental study. Natl J Androl 23(1):27–33
Makarasen A, Sirithana W, Mogkhuntod S et al (2011) Cytotoxic and antimicrobial activities of aporphine alkaloids isolated from Stephania venosa (Blume) Spreng. Plant Med 77:1519–1524
Manuszak M, Harding W, Gadhiya S et al (2018) (–)-Stepholidine reduces cue induced reinstatement of cocaine seeking and cocaine self-administration in rats. Drug Alcohol Depend 189:49–54
Martin ML, Diaz MT, Montero MJ et al (1993) Antispasmodic activity of benzylisoquinoline alkaloids analogous to papaverine. Plant Med 59:63–67
Meade JA, Free RB, Miller NR et al (2015) (–)-Stepholidine is a potent pan-dopamine receptor antagonist of both G protein- and β-arrestin-mediated signaling. Psychopharmacology 232:917–930
Meng LH, Zhang H, Hayward L et al (2004) Tetrandrine induces early G1 arrest in human colon carcinoma cells by down-regulating the activity and inducing the degradation of G1-S-specific cyclin-dependent kinases and by inducing p53 and p21Cip1. Cancer Res 64:9086–9092
Meyer MM, Chen TP, William M et al (2000) Chinese herb nephropathy. Proc (Bayl Univ Med Cent) 13(4):334–337
Montrucchio DP, Córdova MM, Santos AR et al (2013) Plant derived aporphinic alkaloid S-(+) dicentrine induces antinociceptive effect in both acute and chronic inflammatory pain models: evidence for a role of TRPA1 channels. PLoS ONE 28:e67730
Morais LC, Barbosa-Filho JM, Almeida RN (1998) Central depressant effects of reticuline extracted from Ocotea duckei in rats and mice. J Ethnopharmacol 62(1):57–61
Mullaicharam AR (2011) Counterfeit herbal medicine. Int J Nutr Pharm Neurol Dis 1:97–102
Nakamura K, Tsuchiya S, Sugimoto Y et al (1992) Histamine release inhibition activity of bisbenzylisoquinoline alkaloids. Plant Med 58:505–508
Naman CB, Gupta G, Varikuti S et al (2015) Northalrugosidine is a bisbenzyltetrahydroisoquinoline alkaloid from Thalictrum alpinum with in vivo antileishmanial activity. J Nat Prod 78:552–556
Ning P, Peng Y, Liu DW et al (2016) Tetrandrine induces microRNA differential expression in human hypertrophic scar fibroblasts in vitro. Genet Mol Res 15(1):gmr7288
Nortier J, Pozdzik A, Roumeguere T et al (2015) Aristolochic acid nephropathy ("Chinese herb nephropathy"). Nephrol Ther 11(7):574–588
Ogino T, Katsuhara T, Sato T et al (1998) New alkaloids from the root of Stephania tetrandra (Fen-Fang-Ji). Heterocycles 48:311–317
Okada M (1999) Chinese-herb nephropathy. Lancet 354:1732
Orallo F (2004) Acute cardiovascular effects of (+)-nantenine, an alkaloid isolated from Platycapnos spicata, in an aesthetised normotensive rats. Plant Med 70:117–126
Otshudi AL, Apers S, Pieters L et al (2005) Biologically active bisbenzylisoquinoline alkaloids from the root bark of Epinetrum villosum. J Ethnopharmacol 102(1):89–94
Park PH, Nan JX, Park EJ et al (2000) Effect of tetrandrine on experimental hepatic fibrosis induced by bile duct ligation and scission in rats. Pharmacol Toxicol 87(6):261–268
Payon V, Kongsaden C, Ketchart W et al (2019) Mechanism of cepharanthine cytotoxicity in human ovarian cancer cells. Plant Med 85:41–47
Pinelli A, Trivulzio S, Brenna S et al (2010) Pretreatment with tetrandrine has protective effects against isoproterenol-induced myocardialinfarction in rabbits. In Vivo 24(3):265–270
Qi XM, Miao LL, Cai Y et al (2013) ROS generated by CYP450, especially CYP2E1, mediate mitochondrial dysfunction induced by tetrandrine in rat hepatocytes. Acta Pharmacol Sin 34(9):1229–1236
Qian QW, Zhu YM, Zhang MY et al (2010) Study on the anti-HSV-2 effect of cepharanthine hydrochloride in vitro. Chin J Health LabTechnol 20:2082–2083
Qin R, Shen H, Cao Y et al (2013) Tetrandrine induces mitochondria-mediated apoptosis in human gastric cancer BGC-823 cells. PLoS ONE 8:e76486
Rao MR (2002) Effects of tetrandrine on cardiac and vascular remodeling. Acta Pharmacol Sin 23:1075–1085
Rattanawong A, Payon V, Limpanasittikul W et al (2018) Cepharanthine exhibits a potent anticancer activity in p53-mutated colorectal cancer cells through upregulation of p21Waf1/Cip1. Oncol Rep 39:227–238
Reist RH, Dey RD, Durham JP et al (1993) Inhibition of proliferative activity of pulmonary fibroblasts by tetrandrine. Toxicol App Pharm 122:70–76
Rhein BA, Maury WJ (2015) Ebola virus entry into host cells: identifying therapeutic strategies. Curr Clin Microbiol Rep 2:115–124
Ribeiro RA, Leite JR (2003) Nantenine alkaloid presents anticonvulsant effect on two classical animal models. Phytomedicine 10:563–568
Rosenkranz HS, Klopman G (1990) Novel structural concepts in elucidating the potential genotoxicity and carcinogenicity of tetrandrine, a traditional herbal drug. Mutat Res 244(4):265–271
Ruan L, Huang HS, Jin WX et al (2013) Tetrandrine attenuated cerebral ischemia/reperfusion injury and induced differential proteomic changes in a MCAO mice model using 2-D DIGE. Neurochem Res 38(9):1871–1879
Sakurai Y, Kolokoltsov AA, Chen CC et al (2015) Two pore channels control Ebolavirus host cell entry and are drug targets for disease treatment. Science 347:995–998
Samra YA, Said HS, Elsherbiny NM et al (2016) Cepharanthine and piperine ameliorate diabetic nephropathy in rats: role of NF-κB and NLRP3 inflammasome. Life Sci 157:187–199
Schafer A, Cheng H, Lee C et al (2018) Development of potential small molecule therapeutics for treatment of Ebola virus disease. Curr Med Chem 25:5177–5190
Sekiya N, Shimada Y, Niizawa A et al (2004) Suppressive effects of Stephania tetrandra on the neutrophil function in patients with rheumatoid arthritis. Phytother Res 18:247–249
Semwal DK, Badoni R, Semwal R et al (2010) The genus Stephania (Menispermaceae): chemical and pharmacological perspectives. J Ethnopharmacol 132(2):369–383
Seow WK, Ferrante A, Goh DB et al (1988) In vitro immunosuppressive properties of the plant alkaloid tetrandrine. Int Arch Allergy Appl Immunol 85(4):410–415
Shan YQ, Zhu YP, Pang J et al (2013) Tetrandrine potentiates the hypoglycemic efficacy of berberine by inhibiting P-glycoprotein function. Biol Pharm Bull 36(10):1562–1569
Shan L, Tong L, Hang L et al (2018) Fangchinoline supplementation attenuates inflammatory markers in experimental rheumatoidarthritis-induced rats. Biomed Pharmacother 111:142–150
Shen YC, Chen CF, Sung YJ (1999) Tetrandrine ameliorates ischaemia-reperfusion injury of rat myocardium through inhibition of neutrophil priming and activation. Br J Pharmacol 128(7):1593–1601
Shen YC, Chou CJ, Chiou WF et al (2001) Anti-inflammatory effects of the partially purified extract of Radix Stephaniae tetrandrae: comparative studies of its active principles tetrandrine and fangchinoline on human polymorphonuclear leukocyte functions. Mol Pharmacol 60(5):1083–1090
Shi JP, Li SX, Zhu KJ et al (2016a) Tetrandrine on the dynamic changes of cytokines and antifungal effects in mice with Candida albicans. Chin J Zoonoses 32:689–695
Shi JP, Li SX, Ma ZL et al (2016b) Acute and sub-chronic toxicity of tetrandrine in intravenously exposed female BALB/c mice. Chin J Integr Med 22:925–931
Shi J, Guo B, Hui Q et al (2017) Fangchinoline suppresses growth and metastasis of melanoma cells by inhibiting the phosphorylation of FAK. Oncol Rep 38:63–70
Si DY, Zhao SX (1991) Aporphine alkaloidal components from the aerial part of Stephania tetrandra. J Jiling Med Coll 14:1–6
Si DY, Zhao SX (1993) Non-alkaloidal components from the aerial part of Stephania tetrandra. J Jiling Med Coll 16:1–5
Si DY, Zhao SX, Deng JZ et al (1992) A 4, 5-dioxoaporphine from the aerial parts of Stephania tetrandra. J Nat Prod 55:828–829
Si DY, Zhong DF, Sha Y et al (2001) Biflavonoids from the aerial part of Stephania tetrandra. Phytochemistry 58:563–566
Sim HJ, Kim JH, Lee KR et al (2013) Simultaneous determination of structurally diverse compounds in different Fangchi species by UHPLC-DAD and UHPLC-ESI-MS/MS. Molecules 18:5235–5250
Sim HJ, Yoon SH, Kim MS et al (2015) Identification of alkaloid constituents from Fangchi species using pH control liquid-liquid extraction and liquid chromatography coupled to quadrupole time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 29(9):837–854
Singh K, Dong Q, Timiri PS et al (2018) Tetrandrine inhibits deregulated cell cycle in pancreatic cancer cells: Differential regulation of p21Cip1/Waf1, p27Kip1 and cyclin D1. Cancer Lett 425:164–173
Song N, Zhang S, Li Q et al (2008) Establishment of a liquid chromatographic/mass spectrometry method for quantification of tetrandrine in rat plasma and its application to pharmacokinetic study. J Pharm Biomed Anal 48(3):974–979
Sun YF, Wink M (2014) Tetrandrine and fangchinoline, bisbenzylisoquinoline alkaloids from Stephania tetrandra can reverse multidrug resistance by inhibiting P-glycoprotein activity in multidrug resistant humancancer cells. Phytomedicine 21:1110–1119
Sun K, Wu JH (2012) Modern research development of fangchi. Chin Foreign Med Res 10:157–158
Sun SA, Tang YP, Wang H et al (1999) Clinical and experimental study on the treatment of hepatic fibrosis by Stephania tetrandra. Chin J Hepatol 7:62–63
Sun Y, Dai J, Hu Z et al (2009) Oral bioavailability and brain penetration of (–)-stepholidine, a tetrahydroprotoberberine agonistat dopamine D(1) and antagonist at D(2) receptors, in rats. Br J Pharmacol 158(5):1302–1312
Sun M, Wang J, Zhou Y et al (2016) Isotetrandrine reduces astrocyte cytotoxicity in neuromyelitis optica by blocking the binding of NMO-IgG to aquaporin 4. NeuroImmunomodulation 23(2):98–108
Takahashi T, Tonami Y, Tachibana M et al (2012) Tetrandrine prevents bone loss in sciatic-neurectomized mice and inhibits receptor activator of nuclear factor κB ligand-induced osteoclast differentiation. Biol Pharm Bull 35(10):1765–1774
Takemura H, Imoto K, Ohshika H et al (1996) Tetrandrine as a calcium antagonist. Clin Exp Pharmacol Physiol 23(8):751–753
Tang X, Di X, Zhong Z et al (2016) In vitro metabolism of l-corydalmine, a potent analgesic drug, in human, cynomolgus monkey, beagle dog, rat and mouse liver microsomes. J Pharm Biomed Anal 128:98–105
Tankeu S, Vermaak L, Chen W et al (2016) Differentiation between two “fang ji” herbal medicines, Stephania tetrandra and the nephrotoxic Aristolochia fangchi, using hyperspectral imaging. Phytochemistry 122:213–222
Teng CM, Yu SM, Ko FN et al (1991) Dicentrine, a natural vascular alpha 1-adrenoceptor antagonist, isolated from Lindera megaphylla. Br J Pharmacol 104:651–656
Teng G, Svystonyuk D, Mewhort HE et al (2015) Tetrandrine reverses human cardiac myofibroblast activation and myocardial fibrosis. Am J Physiol Heart Circ Physiol 308(12):H1564–H1574
Tian XQ (2014) Study on the constituents of Brucea Javanica and Stephania Tetrandra. Donghua University
Tian X, Zhang Y, Li Z et al (2016a) Systematic and comprehensive strategy for metabolite profiling in bioanalysis using software-assisted HPLC-Q-TOF: magnoflorine as an example. Anal Bioanal Chem 408(9):2239–2254
Tian Y, Shen S, Jiang Y et al (2016) CYP3A5 mediates bioactivation and cytotoxicity of tetrandrine. Arch Toxicol 90:1737–1748
Tian DD, Zhang RX, Wu N et al (2017) Tetrandrine inhibits the proliferation of human osteosarcoma cells by upregulating the PTEN pathway. Oncol Rep 237:2795–2802
Tsai TH, Tsai TR, Chou CJ et al (1996) Determination of dicentrine in rat plasma by high-performance liquid chromatography and its application to pharmacokinetics. J Chromatogr B 681:277–281
Tsutsumi T, Kobayashi S, Liu YY et al (2003) Anti-hyperglycemic effect of fangchinoline isolated from Stephania Tetrandra Radix in streptozotocin-diabetic mice. Biol Pharm Bull 26(3):313–317
Tzeng CC, Wu YC, Su TL et al (1990) Inhibitory effects of isoquinoline type alkaloids on leukemic cell growth and macromolecule biosynthesis. Kaohsiung J Med Sci 6:58–65
Uche FI, Drijfhout FP, McCullagh J et al (2016) Cytotoxicity effects and apoptosis induction by bisbenzylisoquinoline alkaloids from Triclisia subcordata. Phytother Res 30:1533–1539
Vanherweghem JL (1994) A new form of nephropathy secondary to the absorption of Chinese herbs. Bulletin et Memoires de I′ Academie Royale de Medecine Belgique 149:128–135
Vichkanova SA, Makarova LV, Solov'eva LF (1973) Chemotherapeutic properties of the alkaloid tetrandrine in experimental tuberculosis. Farmakologiya i Toksikologiya (Moscow) 36:74–78
Wan Z, Lu Y, Liao Q et al (2012) Fangchinoline inhibits human immunodeficiency virus type 1 replication by interfering with gp160 proteolytic processing. PLoS ONE 7:e39225
Wang G, Lemos JR (1994) Tetrandrine: a new ligand to block voltage-dependent Ca2+ and Ca(+)-activated K+ channels. Life Sci 56:295–306
Wang M, Lu S (2015) Clinical observation of Fangji Fuling decoction on chronic heart failure. J Emerg Trad Chin Med 24:355–357
Wang HX, Kwan CY, Wong TM (1997) Tetrandrine inhibits electrically induced [Ca2+]i transient in the isolated single rat cardiomyocyte. Eur J Pharmacol 319:115–122
Wang G, Lemos JR, Ladecola C (2004) Herbal alkaloid tetrandrine: from an ion channel blocker to inhibitor of tumor proliferation. TRENDS Pharmacol Sci 25(3):120–123
Wang N, Pan W, Zhu M et al (2011) Fangchinoline induces autophagic cell death via p53/sestrin2/AMPK signalling in human hepatocellular carcinoma cells. Br J Pharmacol 164(2b):731–742
Wang TH, Wan JY, Gong X et al (2012) Tetrandrine enhances cytotoxicity of cisplatin in human drug-resistant esophageal squamous carcinoma cells by inhibition of multidrug resistance-associated protein 1. Oncol Rep 28:1681–1686
Wang Y, Chen J, Wang L et al (2013) Fangchinoline induces G0/G1 arrest by modulating the expression of CDKN1A and CCND2 in K562 human chronic myelogenous leukemia cells. Exp Ther Med 5:1105–1112
Wang QS, Cui YL, Gao LN et al (2014a) Reduction of the pro-inflammatory response by tetrandrine-loading poly(l-lactic acid) films in vitro and in vivo. J Biomed Mater Res 102:4098–4107
Wang CD, Yuan CF, Bu YQ et al (2014b) Fangchinoline inhibits cell proliferation via Akt/GSK-3beta/ cyclin D1 signaling and induces apoptosis in MDA-MB-231 breast cancer cells. Asian Pac J Cancer Prev 15(2):769–773
Wang M, Li J, Wei Q et al (2016a) Study on analgesic and anti-inflammatory effects and mechanism of total alkaloids from Stephania tetrandra S.Moore decoction. Lishizhen Med Mater Med Res 27:335–338
Wang L, Ci X, Lv H et al (2016b) Isotetrandrine ameliorates tert-butyl hydroperoxide-induced oxidative stress through upregulation of heme oxygenase-1 expression. Exp Biol Med (Maywood) 241(14):1568–1576
Wang X, Yang Y, Yang D et al (2016c) Tetrandrine prevents monocrotaline-induced pulmonary arterial hypertension in rats through regulation of the protein expression of inducible nitric oxide synthase and cyclic guanosinemonophosphate-dependent protein kinase type 1. J Vasc Surg 64(5):1468–1477
Wang R, Ma TM, Liu F et al (2017a) Research progress on pharmacological action and clinical application of Stephania Tetrandrae Radix. China J Chin Mater Med 42:634–639
Wang B, Xing Z, Wang F et al (2017b) Fangchinoline inhibits migration and causes apoptosis of human breast cancer MDA-MB-231 cells. Oncol Lett 14:5307–5312
Wang J, Chang LH, Li X et al (2017c) Effect of tetrandrine on radiosensitivity of nasopharyngeal carcinoma cells. Chin J Pathophysiol 33:1611–1618
Wang J, Chang LH, Lai XP et al (2018) Tetrandrine enhances radiosensitivity through the CDC25C/CDK1/cyclin B1 pathway in nasopharyngeal carcinoma cells. Cell Cycle 17:671–680
Whong WZ, Lu CH, Stewart JD et al (1989) Genotoxicity and genotoxic enhancing effect of tetrandrine in Salmonella typhimurium. Mutat Res 222(3):237–244
Wino T, Sato T, Sasaki H et al (1988) Four new bisbenzylisoquionoline alkaloids from the root of Stephania tetrandra. Heterocycles 27:1149–1154
Wong TM, Wu S, Yu XC et al (2000) Cardiovascular actions of Radix Stephaniae tetrandrae: a comparison with its main component, tetrandrine. Acta Pharm Sin 21:1083–1088
Wu SJ, Ng LT (2007) Tetrandrine inhibits proinflammatory cytokines, iNOS and COX2 expression in human monocytic cells. Biol Pharm Bull 30(1):59–62
Wu J, Suzuki H, Zhou YW et al (2001) Cepharanthine activates caspases and induces apoptosis in Jurkat and K562 human leukemia cell lines. J Cell Biochem 82:200–214
Wu WN, McKown LA, Gopaul VS (2004) In-vitro metabolism of isotetrandrine, a bisbenzylisoquinoline alkaloid, in rat hepatic S9 fractionby high-performance liquid chromatography-atmospheric pressure ionization mass spectrometry. J Pharm Pharmacol 56:749–755
Wu CJ, Wang YH, Lin CJ et al (2011) Tetrandrine down-regulates ERK/NF-κB signaling and inhibits activation of mesangial cells. Toxicol In Vitro 25:1834–1840
Wu XL, Li JX, Li ZD et al (2015) Protective effect of tetrandrine on sodium taurocholate-induced severe acute pancreatitis. Evid Based Complement Altern Med 2015:129103
Xiao J, Song N, Lu T et al (2018) Rapid characterization of TCM qianjinteng by UPLC-QTOF-MS and its application in the evaluation of three species of Stephania. J Pharm Biomed Anal 156:284–296
Xie DT (2014) A taxonomic study of the genus Stephania Lour. from China. Fudan University
Xing SG, Shi XC, Wu ZL (1989) Effect of tetrandrine on micronucleus formation and sister-chromatid exchange in both in vitro and in vivo assays. Mutat Res 224:5–10
Xing ZB, Wang FM, Wang CP et al (2014) Advance on study of chemical components and pharmacological effect of Stephania tetrandra. Chin J Exp Trad Med Form 20:241–246
Xu J, Liu D, Yin Q et al (2016a) Tetrandrine suppresses β-glucan-induced macrophage activation via inhibiting NF-κB, ERK and STAT3 signaling pathways. Mol Med Rep 13:5177–5184
Xu XL, Yang LJ, Jiang JG et al (2016b) Renal toxic ingredients and their toxicology from traditional Chinese medicine. Expert Opin Drug Metab Toxicol 12(2):149–159
Xuan B, Liu F, Zhang MY et al (2009) Inhibitory effects of tetrandrine on intracellular free Ca2+ increase induced by glutamate, serotonin and histamine in dissociated retina cells. J Ocul Pharmacol Ther 12(3):331–336
Xue Y, Wang Y, Feng DC et al (2008) Tetrandrine suppresses lipopolysaccharide-induced microglial activation by inhibiting NF-kappaB pathway. Acta Pharmacol Sin 29:245–251
Xv J, Zhou ZH (2015) Effect of tetrandrine on anti-lung fibrosis of patients with silicosis. J Clin Pulm Med 20:1658–1660
Yang G, Zhang C, Hu P et al (2017) An UPLC-MS/MS method for quantifying tetrandrine and its metabolite berbamine in human blood: Application to a human pharmacokinetic study. J Chromatogr B Anal Technol Biomed Life Sci 1070:92–96
Yang HY, Wang YH, Du GH (2018a) Tetrandrine, in natural small molecule drugs from Plants, (G.-H. Du, editor), p 161
Yang X, Gao X, Cao Y et al (2018b) Anti-inflammatory effects of boldine and reticuline isolated from Litsea cubeba through JAK2/STAT3 and NF-κB signaling pathways. Plant Med 84:20–25
Yao M, Yuan B, Wang X et al (2017) Synergistic cytotoxic effects of arsenite and tetrandrine in human breast cancer cell line MCF-7. Int J Oncol 51:587–598
Yasuda K, Moro M, Akasu M et al (1989) Pharmacokinetic disposition of cepharanthin following single and multiple intravenous doses in healthy subjects. Jpn J Clin Pharmacol Ther 20:741–748
Ye Z, Dyke KV (1989) Selective antimalarial activity of tetrandrine against chloroquine resistant Plasmodium falciparum. Biochem Biophys Res Commun 159(1):242–248
Ye Z, Dyke KV, Castranova V (1989) The potentiating action of tetrandrine in combination with chloroquine or qinghaosu against chloroquine sensitive and resistant falciparum malaria. Biochem Biophys Res Commun 165(2):758–765
Ye Z, Dyke KV, Rossan RN (2013) Effective treatment with a tetrandrine/ chloroquine combination for chloroquine-resistant falciparum malaria in Aotusmonkeys. Malar J 12:117
Yin S, Rao G, Wang J et al (2015) Roemerine improves the survival rate of septicemic BALB/c mice by increasing the cell membrane permeability of staphylococcus aureus. PLoS ONE 10:e0143863
Yodkeeree S, Ooppachai C, Pompimon W et al (2018) O-methylbulbocapnine and dicentrine suppress LPS-induced inflammatory response by blocking NF-κB and AP-1 activation through inhibiting MAPKs and Akt signaling in RAW264.7 macrophages. Biol Pharm Bull 41(8):1219–1227
Young ML, Su MJ, Wu MH et al (1994) The electrophysiological effects of dicentrine on the conduction system of rabbit heart. Br J Pharmacol 113(1):69–76
Yu VW, Ho WS (2013) Tetrandrine inhibits hepatocellular carcinoma cell growth through the caspase pathway and G2/M phase. Oncol Rep 29:2205–2210
Yu SM, Hsu SY, Ko FN et al (1992a) Haemodynamic effects of dicentrine, a novel alpha 1-adrenoceptor antagonist: comparison with prazosin in spontaneously hypertensive and normotensive Wistar-Kyoto rats. Br J Pharmacol 106(4):797–801
Yu SM, Chen CC, Ko FN et al (1992b) Dicentrine, a novel antiplatelet agent inhibiting thromboxane formation and increasing the cyclicAMP level of rabbit platelets. Biochem Pharmacol 43:323–329
Yu SM, Kang YF, Chen CC et al (1993) Effects of dicentrine on haemodynamic, plasma lipid, lipoprotein level and vascular reactivity in hyperlipidaemic rats. Br J Pharmacol 108(4):1055–1061
Yu SM, Ko FN, Chueh SC et al (1994) Effects of dicentrine, a novel alpha 1-adrenoceptor antagonist, on human hyperplastic prostates. Eur J Pharmacol 252(1):29–34
Yu XC, Wu S, Wang GY et al (2001) Cardiac effects of the extract and active components of radix stephaniae tetrandrae. II. Myocardial infarct, arrhythmias, coronary arterial flow and heart rate in the isolated perfused rat heart. Life Sci 68:2863–2872
Yu XC, Wu S, Chen CF et al (2004) Antihypertensive and anti-arrhythmic effects of an extract of Radix Stephaniae Tetrandrae in the rat. J Pharm Pharmacol 56:115–122
Yu Y, Hu B, Bao J et al (2018) Otoprotective effects of stephania tetrandra S. Moore herb isolate against Acoustic Trauma. J Assoc Res Otolaryngol 19(6):653–668
Yuan X, Tong B, Dou Y et al (2016) Tetrandrine ameliorates collagen-induced arthritis in mice by restoring the balance between Th17and Treg cells via the aryl hydrocarbon receptor. Biochem Pharmacol 101:87–99
Yun X, Dong S, Hu Q et al (2018) 1H NMR-based metabolomics approach to investigate the urine samples of collagen-inducedarthritis rats and the intervention of tetrandrine. J Pharm Biomed Anal 154:302–311
Zeng FD, Shaw DH, Ogilvie RI (1985) Kinetic disposition and hemodynamic effects of tetrandrine in anesthetized dogs. J Cardiovasc Pharmacol 7:1034–1039
Zhang YZ (2014) Clinical efficacy and safety evaluation of tetrandrine combined with methotrexate in the treatment of rheumatoid arthritis. Chin J Clin 42:55–56
Zhang YH, Fang LH (2001) Antagonism of morphine-induced antinociception by tetrandrine is dependent on serotonergic mechanisms. Life Sci 69:1429–1439
Zhang X, Huang HJ, Liu YM et al (2001) Effects of tetrandrine on intracelluar free Ca2+ and lipid peroxidation of hippocampus in rat cerebral ischemic damage. J Tongji Med Univ 30:53–55
Zhang CH, Wang YF, Liu XJ et al (2005) Antiviral activity of cepharanthine against severe acute respiratory syndrome coronavirus in vitro. Chin Med J 118:493–496
Zhang H, Gao A, Li F et al (2009a) Mechanism of action of tetrandrine, a natural inhibitor of Candida albicans drug efflux pumps. Yakugaku Zasshi 129:623–630
Zhang DK, Cheng LN, Huang XL et al (2009b) Tetrandrine ameliorates dextran-sulfate-sodium-induced colitis in mice through inhibition of nuclear factor -kappaB activation. Int J Colorectal Dis 24(1):5–12
Zhang H, Wang K, Zhang G et al (2010) Synergistic anti-candidal activity of tetrandrine on ketoconazole: an experimental study. Plant Med 76:53–61
Zhang X, Guo H, Gao L et al (2013) Molecular mechanisms underlying the tetrandrine-mediated reversal of the fluconazole resistance of Candida albicans. Pharm Biol 51(6):749–752
Zhang J, Yu B, Zhang XQ et al (2014) Tetrandrine, an antihypertensive alkaloid, improves the sleep state of spontaneously hypertensive rats (SHRs). J Ethnopharmacol 151(1):729–732
Zhang Z, Yan J, Xu KJ et al (2015) Tetrandrine reverses drug resistance in isoniazid and ethambutol dual drug-resistant Mycobacterium tuberculosis clinical isolates. BMC Infect Dis 15:153
Zhang MF, Ding XD, Fei ZW et al (2017a) Growth inhibition and apoptosis induction of tetrandrine in human thyroid cancer cells (B-CPAP). Mod Oncol 25:3410–3414
Zhang Y, Wen YL, Ma JW et al (2017a) Tetrandrine inhibits glioma stem-like cells by repressing β-catenin expression. Int J Oncol 50:101–110
Zhang Z, Liu T, Yu M et al (2018a) The plant alkaloid tetrandrine inhibits metastasis via autophagy-dependent Wnt/β-catenin and metastatic tumor antigen 1 signaling in human liver cancer cells. J Exp Clin Cancer Res 37:7
Zhang H, Xie B, Zhang Z et al (2018b) Tetrandrine suppresses cervical cancer growth by inducing apoptosis in vitro and in vivo. Drug Des Dev Ther 13:119–127
Zhang H, Gao L, Shu M et al (2018c) Development of a highly sensitive and specific ELISA method for the determination of l-corydalmine in SD rats with monoclonal antibody. J Chromatogr B Anal Technol Biomed Life Sci 1073:163–169
Zhang L, Cui MY, Ding LL et al (2018d) Tetrandrine combined with leflunomide in the treatment of rheumatoid arthritis. Prog Anat Sci 24:69–71
Zhao TX (2018) Fang-Ji-Di-Huang decoction combined with methotrexate in the treatment of 40 cases of early active rheumatoid arthritis. Zhejiang J Trad Chin Med 53:444–445
Zhao WH, Hu ZQ, Okubo S et al (2001) Mechanism of synergy between epigallocatechin gallate and beta-lactams against methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 45(6):1737–1742
Zhao X, Cui XY, Chen BQ et al (2004) Tetrandrine, a bisbenzylisoquinoline alkaloid from Chinese herb Radix, augmented the hypnotic effect of pentobarbital through serotonergic system. Eur J Pharmacol 506(2):101–105
Zhao H, Luo F, Li H et al (2014) Antinociceptive effect of tetrandrine on LPS-induced hyperalgesia via the inhibition of IKKβ phosphorylation and the COX-2/PGE2 pathway in mice. PLoS ONE 10:e94586
Zhao YJ, Liu WD, Shen YN et al (2019) The efflux pump inhibitor tetrandrine exhibits synergism with fluconazole or voriconazole against Candida parapsilosis. Mol Biol Rep 46(4):5867–5874
Zhou DX, Yang GT, He XX et al (2007) Effects of tetrandrine on Ang II-induced cardiomyocyte hypertrophy and p-ERK1/2 expression. China J Chin Mater Med 32:1921–1924
Zhou YB, Wang YF, Zhang Y et al (2012) In vitro activity of cepharanthine hydrochloride against clinical wild-type and lamivudine-resistant hepatitis B virus isolates. Eur J Pharmacol 683(1–3):10–15
Zhou CH, Meng JH, Yang YT et al (2018) Cepharanthine prevents estrogen deficiency-induced bone loss by inhibiting bone resorption. Front Pharmacol 9:210
Zhu ZY, Feng YX, He LY et al (1983) Studies on the utilization of medicinal plant resources of the genus Stephania (Menispermaceae) in China. Acta Pharm Sin 18:460–467
Zhu R, Liu T, Tan Z et al (2014) Tetrandrine induces apoptosis in gallbladder carcinoma in vitro. Int J Clin Pharmacol ther 52:900–905
Zhu Q, Guo B, Chen L et al (2017) Cepharanthine exerts antitumor activity on choroidal melanoma by reactive oxygen species production and c-Jun N-terminal kinase activation. Oncol Lett 13:3760–3766
Acknowledgements
This research did not receive any funding from public, commercial or non-profit sectors.
Author information
Authors and Affiliations
Contributions
YJ retrieved the relevant literature and drafted the manuscript. SL originated the work, led the discussions, provided helpful comments and revised the manuscript. ML and HL provided helpful comments and revised the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jiang, Y., Liu, M., Liu, H. et al. A critical review: traditional uses, phytochemistry, pharmacology and toxicology of Stephania tetrandra S. Moore (Fen Fang Ji). Phytochem Rev 19, 449–489 (2020). https://doi.org/10.1007/s11101-020-09673-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11101-020-09673-w