
Abstract
Sirtuin-2 (Sirt2) is a member of the NAD (+)-dependent protein deacetylase family involved in neuroprotection, cellular metabolism, homeostasis, and stress responses after injury of the nervous system. So far, no data have been published describing the role of SIRT2 in motor functional recovery after damage. We found that SIRT2 expression and deacetylase activity were increased within motoneurons after axotomy. To shed light onto the biological relevance of this change, we combined in vitro and in vivo models with pharmacological and genetic ablation approaches. We found that SIRT2 KO (knockout) mice exhibited improved functional recovery after sciatic nerve crush. SIRT2 activity blockage, using AK7, increased neurite outgrowth and length in organotypic spinal cord cultures and human cell line models. SIRT2 blockage enhanced the acetyltransferase activity of p300, which in turn increased the levels of an acetylated form of p53 (Ac-p53 k373), histone 3 (Ac-H3K9), and expression of GAP43, a downstream marker of regeneration. Lastly, we verified that p300 acetyltransferase activity is essential for these effects. Our results suggest that bolstering an epigenetic shift that promotes SIRT2 inhibition can be an effective therapy to increase functional recovery after peripheral nerve injury.
Similar content being viewed by others


Introduction
Restoring the connectivity between the nervous system and muscle effectors is essential for optimal functional recovery after injury. In contrast to the central nervous system, nerves comprising the peripheral system retain the capability to regenerate [1]. Peripheral nerve injury (PNI) affects about 13–23 people per 100,000 in the USA and causes a substantial economic and social burden (i.e., $150 billion in US health care) [2,3,4]. Full recovery of lost motor function after PNI is fundamental and often difficult to achieve, particularly after severe PNI. Some authors have pointed out that boosting the intrinsic mechanisms triggered by motoneurons (MNs) to get a pro-regenerative mode can be an effective approach to improve functional recovery [5, 6].
Epigenetic modifications, such as histone acetylation or deacetylation, can shift the neuronal transcriptomic program leading to an enhanced axonal regrowth after nervous system injury [7, 8]. In this way, NAD+-dependent deacetylase sirtuins (SIRT1-7), which orchestrate a response to stress, metabolism, and neurodegeneration [9, 10], are also able to modify the epigenome. Among their known activities after PNI, SIRT1 exerts protection, while blockage of SIRT2 has a detrimental effect on MNs’ survival [11, 12]. SIRT2 also modulates cytoskeleton organization and acts as an anti-inflammatory mediator after PNI by modulating NF-κB [13,14,15]. However, the role of both deacetylases on axonal regrowth after nerve injury remains unclear. In agreement with previous studies [16], we have recently shown that SIRT1 accelerates axonal growth by autophagy induction [17]. On the other hand, SIRT2 knockdown rescues axonal transport and motor deficits in Parkinson’s disease animal models [18], although its genetic deletion provokes aging-related axonal degeneration by reactive oxygen species imbalance [19].
Seeking to unravel the role of SIRT2 in recovery after PNI, we used pharmacological and genetic manipulation to modulate its activity. Herein, we provide evidence that supports that the blockage of SIRT2 deacetylase activity improves motor functional recovery after PNI, and that it promotes an increase in the GAP43 levels within MNs, which is associated to a “pro-regenerative” neuronal phenotype. Furthermore, p300-dependent epigenetic mediators could facilitate this pro-regenerative state. These findings open a new therapeutic avenue to ameliorate functional recovery after injury to the nervous system by epigenetic modifications.
Material and Methods
In Vitro Cultures and Axonal Growth Analysis
Organotypic Culture on Collagen Matrix
We prepared collagen solution at 3 mg/mL mixing: rat tail collagen type I (Corning, Wiesbaden, Germany), phosphate-buffered saline (PBS) (Sigma-Aldrich), sodium bicarbonate at 0.3 mg/mL, and 10 × basal Eagle’s medium (Gibco). Twenty-four-well Petri dishes, pre-treated with poly-d-lysine (Sigma-Aldrich), were coated with 30-μL single drops of collagen and kept in the incubator for 1 h at 37 °C and 5% CO2 to induce collagen gel formation. Therefore, we extracted the lumbar spinal cord sections from 7-day-old Sprague-Dawley rats, placed them in 30% glucose cold Gey’s balanced salt solution (GBSS, Sigma-Aldrich), and removed the meninges and nerve roots. Spinal cords were cut into 350-μM-thickness slices and were placed onto collagen droplets (30 μL each). After 30 min in the incubator (5% CO2, 37 °C) slices were covered with 30 μL of the same collagen solution and after 30 s at 37 °C for collagen polymerization, we added Neurobasal cultured medium (Life Technologies) supplemented with B27 (Life Technologies), glutamine, and penicillin/streptomycin (Sigma-Aldrich). One day after culture, we replaced the media combined with DMSO (control) or AK7, a selective pharmacological inhibitor of SIRT2 activity [20]. We also changed the medium at 3 days of in vitro culture (DIV). The following day, we removed the media, post-fixed the sections with cold 4% paraformaldehyde (PFA) solution for 1 h, washed out excess fixative with Tris-buffered saline (TBS) twice, and incubated with primary antibodies at 4 °C during 48 h. For neurite growth analyses, we used the primary antibody RT-97 (1:200; Hybridoma Bank, USA). After three washes with 0.1% Tween-20 in TBS (TBST), we incubated the spinal cord sections with a secondary antibody conjugated to Alexa 594 (1:200; Life Technologies) overnight at 4 °C, and then counterstained with DAPI, and mounted with DPX (Sigma-Aldrich). We took sequential microphotographs with a fluorescence Olympus BX51 microscope (Olympus, Germany) attached to a DP73 camera and merged them with Adobe Photoshop CS3 (Adobe System, USA) to obtain whole spinal cord slice body with their neurites. To analyze neurite growth and length, whole culture images were analyzed using the Neurite-J plug-in of the ImageJ software [21]. The number of neurites for each intersection from the explants was calculated and compared between sets of cultures.
SH-SY5Y Culture and Transfection
SH-SY5Y cells were grown in high glucose Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 15% fetal bovine serum (Sigma-Aldrich), and 0.5 × penicillin/streptomycin solution (Sigma-Aldrich) and were kept in a humidified incubator at 37 °C under 5% CO2. Coated plastic plates (Thermo) with 10% collagen dissolved in Milli-Q water at 37 °C for 2 h were seeded with cells at a density of 2.5 × 105 per cm2. The differentiation medium used was Neurobasal supplemented with B27 (Life Technologies), 1 μM of retinoic acid and 0.5 × penicillin/streptomycin solution. After 3 DIV without changing the medium, SH-SY5Y cells presented a differentiated-like phenotype characterized by the presence of neurite extensions. At this time, we added the following drugs to the cells: DMOG, which stabilizes HIF1α by prolyl hydroxylase inhibition, AK7, and the selective inhibitor of p300, C646; drugs were dissolved in DMEM to the desired concentration and this medium was used to replace the culture medium. After 24 h, cells were fixed with 4% PFA, rinsed twice with PBS, and stored at 4 °C or were added blocking buffer, containing PBS plus 0.3% (v/v) Triton X-100 and 10% fetal bovine serum (FBS). Cells were then incubated with a mouse anti-β-tubulin (1:500, Covance/Biolegend, San Diego, CA, USA) antibody in 0.5 × blocking buffer in PBS, at 4 °C overnight. The following day, after several washes with PBS plus 0.05% Tween-20, coverslips were incubated with Cy3- or Cy2-conjugated secondary antibodies (Jackson Immunoresearch). Coverslips were counterstained with DAPI and mounted with Mowiol. Images were taken under the same exposure times, sensitivities, and resolutions for each marker analyzed with the aid of a digital camera (Olympus DP50) attached to the microscope (Olympus BX51).
For transfection experiments, we transfected 1 × 106 cells with 1 μg of shRNA GFP (CSHCTR001-CH1, Tebu-bio) or shRNA p300 (HSH008832-32-CH1, Tebu-bio) using the Amaxa Nucleofector II TM (Lonza) and the Nucleofactor V kit (Lonza), following the manufacturer’s recommendations.
To analyze neurite growth, we randomly took microphotographs at 20 × magnification (5 fields per slice) and analyzed them with the help of the ImageJ software. The growth of neurites was measured manually and compared between sets of cultures. Neurite length was calculated as the average of neurite length from all neurons analyzed, and the maximal neurite length was the maximal length obtained from 3 independent experiments. In total, 30–50 cells were analyzed per replicate.
Animals and Surgery
All procedures involving animals were approved by the Ethics Committee of Universitat Autònoma de Barcelona, and followed the European Community Council Directive 2010/63/EU.
The Sirt2−/− C57-BL6 (SIRT2 KO) mice used in our experiments were kindly provided by Dr. Alejandro Vaquero (Bellvitge Biomedical Research Institute (IDIBELL), Barcelona, Spain). These mice were generated in Dr. Tong’s lab using the original Sirt2−/− ES cells [22]. Wild-type C57BL/6 (Charles River) and SIRT2 KO mice aged 2 months old were kept under standard conditions of light and temperature, and fed with food and water ad libitum. Surgical procedures were performed under anesthesia after administrating an intramuscular injection of ketamine (90 mg/kg, i.m.) and xylazine (10 mg/kg, i.m.). We dissected the leg muscle and freed the right sciatic nerve from the connective tissue. We crushed the nerve with fine forceps (Dumont no. 5) in three different orientations for 10 s. Finally, the animals were allowed to recover in a warm environment, once we sutured the wound by planes and disinfected it with povidone iodine.
Electrophysiological and Functional Examination
All experiments were conducted in a blinded fashion. For electrophysiological evaluation, mice were anesthetized with ketamine/xylazine (100:10 mg/kg weight, i.m.) 1 week before surgery and at different times post-injury. The sciatic nerve was stimulated by transcutaneous electrodes placed at the sciatic notch by single pulses (20 μs), and the compound muscle action potential (CMAP) was recorded by placing electrodes on the tibialis anterior (TA), gastrocnemius (GA), and plantar interosseous. We gradually increased the stimulus intensity until reaching the supramaximal stimulus, which corresponded to the maximum CMAP amplitude. To measure the amplitude from baseline to peak and the latency, the evoked action potentials were displayed on a storage oscilloscope (Synergy Medelec, Viasys HealthCare) at settings appropriate after every stimulus (n = 6). After testing, animals were allowed to recover in a warm environment.
For the functional analysis of locomotion, we painted the plantar surface of mice hindpaws with acrylic paint and allowed them to walk along a corridor with white paper. Footprints from ipsi- and contralateral paws were analyzed by measuring the print length (PL) and the distance between 1st and 5th toes with a precision device. The three parameters were combined to obtain the Sciatic Functional Index (SFI) [23, 24], which quantifies the changes in the walking pattern (0 for uninjured; − 100 for maximally impaired gait).
Drugs
AK7 (Sigma-Aldrich, Saint Louis, MO), DMOG (Tocris), and C646 (Millipore) were diluted in DMSO and used in vitro at 25 μM, 1 mM and 20 μM, respectively.
Tissue Processing for Histology
We euthanized at 3 days post injury (dpi) or 30 dpi the animals with an intracardial dolethal injection (60 mg/kg i.p.) in a saline solution of heparin (10 U/mL) and we perfused with a transcardially infusion of 0.1 M PBS buffer solution of 4% PFA. We collected the L4-L5 spinal cord segments and the contra- and ipsilateral gastrocnemius and plantar interosseous muscles. The spinal cord was post-fixed 2 h with the same solution and introduced into a 30% sucrose solution for cryopreservation at 4 °C. Muscles were weighted and transferred into the same sucrose solution. We froze the spinal cords with Tissue-Tek O.C.T (Sakura Finetek) and cut them into serial slices of 15 μm (30 slices of 10 slides each) with the aid of a cryotome (Leica, Heidelberg, Germany) and kept them at − 20 °C until used. The same protocol was applied for plantar muscles, which were cut into serial slices of 15 μm (5 slices of 5 slices each) and kept them at − 20 °C until used.
Immunohistochemistry and Image Analysis
Immunohistochemistry of the sections to be compared were processed together on the same day and within the same slide. All images to be compared were analyzed the same day.
Spinal cord slices, or dorsal root explants, were washed with TBS, treated with TBS-glycine 0.1 mM and with a blocking solution of 10% donkey serum in TBS with 0.3% Triton X-100 for 1 h at room temperature (RT). Slices were incubated overnight at 4 °C with the following primary antibodies: rabbit-anti NAD-dependent deacetylase sirtuin-2 (SIRT2; 1:200, Sigma), rabbit anti-acetyl-histone H3 (Lys9) (Acetyl H3-K9; 1:50, Millipore), rabbit anti-acetyl-p53 (Lys373) (Acetyl p53-K373; 1:500, Millipore), rabbit anti-phosphoprotein kinase B/AKT (Ser-473) (pAKT; 1:500, Santa Cruz), rabbit anti-growth-associated protein 43 (GAP43) (1:1000; Millipore), rabbit anti-acetyl-p300 (Lys 1542) (1:200; Bionova), and mouse anti-p300 (1:200; Bionova). After several washes with TBS/0.1% Tween-20, sections were incubated with specific donkey-Cy3 or donkey-Alexa 488 (1:200; Jackson Immunoresearch) during 1 h at RT. The exceeding secondary antibodies were removed by washing with TBS/0.3% Triton X-100, and the slices were counterstained with fluorescent green or blue NeuroTracer Nissl Stain (Molecular Probes, Leiden, Netherlands) and DAPI (Sigma, St. Louis, MO, USA). The slices were washed with TBS and TB and were mounted with Fluoromount-G mounting medium (SouthernBiotech).
Spinal cord slices from different animals and experimental groups were examined with a confocal laser scanning microscope (Zeiss LSM 700; Zeiss, Jena, Germany). We systematically acquired confocal images using three separate photomultiplier channels with a 1.4 numerical aperture objective of 20 × under the same conditions of sensitivity, resolution, and exposure time for each analyzed marker. The obtained images were automatically separately projected and merged using a pseudocolor display. Signal intensity was analyzed using the ImageJ software (National Institutes of Health; available at http://rsb.info.nih.gov/ij/). For ROI (region of interest) delimitation, the Nissl or DAPI labeling was used to enclose the MN cytosol or nuclei, respectively, and the integrated density within the ROI was obtained for at least 15 MNs extracted from four different sections of L4-L5 spinal segments (separated 75 μm between each other) per animal for each marker.
For neuromuscular junction analysis, we took sequential confocal images from different sections of each animal. Bungarotoxin positive endplates were classified as positive or negative for double NF-200 and SYT1 labeling. Only motor endplates that were double positive with co-labeling were counted as reinnervated.
Western Blot
For immunoblotting studies, SH-SY5Y cells (n = 3–4 per experimental condition) were collected 24 h after treatment and were added lysis buffer (50 mM Tris, 2 mM EDTA, 0.5% Triton X-100, 10 mM nicotinamide, and a cocktail of protease [Sigma] and phosphatase inhibitors [Roche]; pH = 6.8). Cells were homogenized on ice with the aid of a Pellet pestle (Sigma-Aldrich), sonicated it with an ultrasonic homogenizer (Model 3000, Biologics Inc.), and were centrifuged at 13,000g during 10 min at 4 °C. The supernatant was harvested and quantified using the BCA assay, according to the manufacturer’s instructions (Pierce Chemical Co.; Rockford, IL, USA). An equal amount of protein from each animal (10 μg/well) was resolved in 10% or 15% SDS-PAGE gels and transferred to a nitrocellulose membrane in a BioRad cuvette system in 25 mM Tris, 192 mM glycine, 20% (v/v) methanol, and pH 8.4 transfer buffer. Proteins were blocked during 1 h at RT with 5% low fat milk in TBS/0.1% Tween-20 for 1 h at RT, and incubated overnight with different primary antibodies as follows: rabbit anti-acetyl-p53 (Lys373) (Acetyl p53-K373; 1:500, Millipore), rabbit GAP43 (1:1000; Millipore), mouse anti-p53 (1:500; Cell Signaling), mouse anti-p300 (1:500; Bionova), and mouse anti-β-actin (1:5000; Sigma-Aldrich). After several washes, we incubated the membrane for 1 h with an appropriate secondary antibody conjugated with horseradish peroxidase (1:5000, Vector). The proteins were visualized using a chemiluminescent method (ECL Clarity kit, Bio-Rad Laboratories, Berkeley, CA, USA) and the images were captured and quantified with Image Lab Software (Bio-Rad Laboratories).
Morphological Evaluation
To evaluate the macroscopic structure of the myelinated axons from contralateral and injured sides of the sciatic nerve, a 3 mm distal from the injury segment was sectioned in semithin sections (0.5-μm-thick) and stained with toluidine blue after EPON resin embedding. We counted the number of axons in the sciatic nerve by systematically choosing images of nerve sections at 100 × by light microscopy, representing at least 30% of the nerve cross-section area. The whole area of the sciatic nerve was calculated using a 4 × image and the total number of axons per section were counted. Axon area and diameter were calculated for 150–200 axons/animal using the ImageJ software (National Institutes of Health; available at http://rsb.info.nih.gov/ij/).
Motor Neuron Counting
Four slices covering the L4-L5 spinal segment (separated by 75 μm; n = 4 per group) of eight sections were randomly selected and stained with FluoroNissl green tracer (Life Technologies) following the manufacturer’s protocol. We took sequential microphotographs from the injured and contralateral ventral horns with the aid of a digital camera (Olympus DP76) attached to the microscope (Olympus BX51) at 10 ×. We counted those MNs located at the L4-L5 segment with an area bigger than 400 μm2, prominent nuclei, and a polygonal form. The percentage of survival for each animal was the number of MNs present at the injured site compared with the contralateral non-injured side.
Statistical Analysis
All values are presented as means ± standard error of the mean (SEM). All experiments were performed in triplicate unless specified. Statistical analyses were performed using GraphPad Prism 7 software by unpaired t tests (one or two-sided) or one or two-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison tests. These are specified in figure legends. In general, a P value ≤ 0.05 was taken to indicate a significant difference. CMAP apparition was analyzed using the Mantel-Cox test, and each CMAP was considered an event. All data was performed blinded to the experimental group.
Results
SIRT2 Activity Blockage Enhances Axonal Regeneration In Vitro
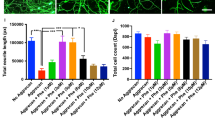
To test whether SIRT2 activity plays a role in MN axonal growth, we used an in vitro model based on spinal organotypic cultures (SOCs) that preserves spinal cord cytoarchitecture and allows MN neurite outgrowth. The correlation between MN neurite outgrowth in SOCs with motor nerve regeneration [25] has been established previously. We embedded SOCs on a collagen matrix and treated them with AK7, a selective SIRT2-inhibitor. We have recently described that AK7 inhibits SIRT2 deacetylase activity in MNs at SOCs [11]. Herein, we observed that SIRT2-activity blockage by this inhibitor promoted a significant increase in the number of neurites outgrowing from SOCs (Fig. 1(A)), although no change was observed in the maximum neurite length (Fig. 1(A)). Accordingly, AK7-treated MNs exhibited a significant increase in GAP43 immunofluorescence, a regeneration-associated marker (Fig. 1(B)). We performed a similar analysis to the above using dorsal root ganglion explants. In this case, there was no significant difference in the number of neurite outgrowths; however, the maximum neurite length was observed in the AK7-treated explants (Fig. S1).

(A) Top, representative microphotographs of vehicle- (Veh) and AK7-treated spinal organotypic cultures (SOCs) embedded in collagen and immunolabeled to reveal neurofilaments using the RT97 antibody. Bottom, graphs histogram showing the number of neurites per intersection, and bar graph indicating the maximum neurite length (n = 11–12, ANOVA, post hoc Bonferroni, *P < 0.05 vs. Veh). Scale bar = 250 μm and 50 μm (images with high magnification). (B) Representative confocal images of motoneurons (MNs) (arrows) from SOCs of different conditions immunostained against GAP43 (green) and counterstained with RT-97 (red). Scale bar = 25 μm. Bar graph of the mean (± SEM) intensity of GAP43 immunoreactivity inside the cytoplasm of MNs (n = 4 SOCs slices, t test *P < 0.05)
Peripheral Nerve Injury Modifies SIRT2 Expression and Activity
Considering these initial results in vitro and that sirtuins are sensors of cellular stress, we first aimed to characterize SIRT2 abundance and activity within MNs after axotomy by sciatic nerve crush, as we recently performed for SIRT1 [17]. Axotomized MNs at the ipsilateral ventral horn showed a significant increase in cytosolic SIRT2 immunolabeling (Fig. 2(A)). We explored two SIRT2 substrates, and, consistently with its deacetylase activity, the abundance of the acetylated forms of histone 4 (H4K16) and of α-tubulin were found reduced compared to that observed in MNs from sham-operated animals (Fig. 2(B)). These results showed that both the SIRT2 expression and its deacetylase activity increase after nerve crush, which suggests a role of SIRT2 for motor regeneration.

(A) Representative confocal images of MNs (arrows) stained with SIRT2 (magenta) and FluoroNissl (green) from sham and injury animals at 3 days post injury (dpi), and the associated bar graph of the mean (± SEM) intensity for each marker inside the cytoplasm of MNs (n = 3–4 animals per group, t test, *P < 0.05 vs. Sham). Scale bar = 100 μm. (B) Top, confocal images of MNs (arrows) immunolabeled against α-tubulin (α-Tub), acetylated-α-tubulin (Ac-α-Tub) in red, and Ac-H4k16 in magenta, and counterstained with FluoroNissl (green) from Sham and crush animals at 3 dpi. Down, bar graph of the mean of the immunofluorescence intensity of Ac-α-Tub related to total α-Tub (%Ac-α-Tub)—or of Ac-H4k16 inside the cytoplasm or nuclei of L4-L5 MNs, respectively (n = 3–4 animals per group, t test, *P < 0.05 vs. Sham). Scale bar = 100 μm
Electrophysiological Characterization of SIRT2 KO Mice
SIRT2 has been shown as an essential regulator of myelin production in the peripheral nervous system (PNS) during development [26]. Moreover, the SIRT2 KO mice exhibit axonal degeneration during aging due to redox and energetic imbalance [19]. For these reasons, we first tested whether SIRT2 genetic depletion would impair motor function, by analyzing the compound muscle action potential at hindlimb muscles. SIRT2 KO mice did not show any significant difference of CMAP amplitude or latency at tibialis anterior, gastrocnemius, or plantar interosseous compared to wild-type (WT) littermates (Fig. 3(A, B)). In agreement with this finding, the number of EPON-stained axons across ipsilateral sciatic nerve sections from injured SIRT2 KO mice was similar to that of injured WTs (Fig. 3(C)). In addition, the myelin sheath from SIRT2 KO mice had normal morphology and thickness compared to that of WT littermates. Thus, we concluded that the genetic deletion of SIRT2 had no detrimental effects on motor behavior, at least until 2 months of age.

(A) Mean values of the amplitudes and latencies (± SEM) from CMAP recordings obtained at tibialis anterior (TA), gastrocnemius (GA), and plantar muscles in wild type (WT) or SIRT2 KO mice (n = 6 animals per group). (B) High-magnification images of semithin sections of the contralateral sciatic nerve from the WT and SIRT2 KO mice. Scale bar = 20 μm
SIRT2 Genetic Deletion Improves Motor Functional Recovery
We analyzed motor functional recovery in the SIRT2 KO mice after PNI. We crushed the right sciatic nerve of WT and SIRT2 KO mice to perform a reproducible PNI, and periodically tested the grade of muscle reinnervation. Experimental design is depicted in Fig. 4(A). Early, at 7 dpi, 100% of SIRT2 KO animals presented CMAP response at TA, in contrast to 33.33% of WT littermates, suggesting a better CMAP recovery promoted by SIRT2 genetic ablation (Fig. 4(B)). CMAP latencies of SIRT2 KO were found similar to WT mice in gastrocnemius while significantly lower at the more distal plantar interosseous muscle at 21 dpi (Fig. 4(C)) (Table 1). These observations indicated that remyelination might occur earlier than in the control when SIRT2 is absent. On the other hand, SIRT2 KO mice presented a significant increase in the CMAP amplitude at any of the muscles tested at 28 dpi suggesting an improved reconnection (Fig. 4(D)) (Table 1). Altogether, these results indicate that the lack of SIRT2 improved functional reconnection after injury.

(A) Experimental design. (B) Graph showing the percentages of animals that presented electrophysiological evidence of reinnervation at the tibialis anterior (TA) muscle at different days post injury (dpi) (n = 6 animals per group, Mantel-Cox test). (C, D) Mean of the latencies and amplitudes (± SEM) of CMAP from ipsilateral studied muscles after sciatic nerve crush from wild type (WT) or SIRT2 KO at different dpi (n = 6 animals per group, ANOVA, post hoc Bonferroni, *P < 0.05 vs. WT)
Improved Motor Functional Recovery by Genetic Deletion of SIRT2
Several reports suggest that, although nerve regeneration is achieved, this does not correlate with a better functional recovery [27]. We analyzed the reinnervation of plantaris muscle endplates by double co-labeling of axons with neurofilament (NF200) and synaptotagmin (SYT1), an essential protein of vesicle liberation at pre-synaptic level, and α-bungarotoxin, which binds to the nicotinic acetylcholine receptor (AChR) at the post-synaptic side. SIRT2 KO animals exhibited a non-significant increase in the number of functional reinnervation of neuromuscular junctions (p = 0.073) (Fig. 5(A)). To test whether SIRT2 blockage increased motor axon recovery is converted into a better motor recovery, we used the walking track technique. SIRT2 KO animals exhibited a significant reduction in the sciatic functional index compared to WT littermates at 25 dpi, indicating better motor performance (Fig. 5(B)).

(A) Representative microphotographs with immunolabeling against syntaxin 1 (SYT1, magenta) and neurofilament (NF, red) in α-bungarotoxin-labeled (a-bunga, green) neuromuscular junctions (NMJs), and a bar graph of the average percentage of reinnervated NMJs (red, green, and magenta overlapping signal) at 30 days post injury (dpi) in both experimental groups. The dotted line indicates the mean number of reinnervated NMJs in wild type (WT) uninjured animals. (n = 4, t test, *P < 0.05 vs. WT). Scale bar = 10 μm. (B) Left: plot of the sciatic functional index (SFI) obtained with walking track analysis after sciatic nerve crush in WT or SIRT2 KO animals (n = 6 animals per group, ANOVA, post hoc Bonferroni, *P < 0.05 vs. WT). Right: representative footprints from ipsi- and contralateral paws at 30 dpi
SIRT2 Blockage Triggers p300/H3K9 Pro-regenerative Molecular Axis In Vivo
We recently reported that SIRT2 blockage has detrimental effects on MN survival after hypoglossal axotomy due to a possible microglia-cytotoxic phenotype [11]. Therefore, we analyzed MN survival and gliotic response on L4-L5 spinal cord segments after crush injury, which represents a more distal axotomy. However, we did not find any particular significant loss of lumbar MNs after crush injury due to SIRT2 genetic ablation (Fig. 6(A)).

(A) Representative microphotographs of ventral horns from crushed wild-type (WT) and SIRT2 KO animals at 30 days post injury (dpi) stained with FluoroNissl (green), and the bar graphs showing the average percentage of surviving MNs (arrows) (± SEM) on the injured side with respect to the contralateral side. Scale bar = 100 μm. (B) Confocal microphotographs of MNs immunolabeled for GAP43 and pAKT (Ser-473) (in red), and counterstained with FluoroNissl Blue and DAPI within the ventral horn of WT and SIRT2 KO animals at 30 dpi. Scale bar = 20 μm. Bar graphs of the mean of the immunofluorescence intensity for each marker inside the cytoplasm of injured MNs (n = 4, t test, *P < 0.05 vs. WT). (C) High magnification of representative images of semithin sections from regenerating distal sciatic nerves of the WT and SIRT2 KO mice, respectively, at 30 dpi, and the bar graphs showing the quantification of the number of myelinated axons, the mean of the axon area, and the axon diameter within the injured nerves. Scale bar = 20 μm
To check the pro-regenerative phenotype of crushed MNs, we immunohistochemically labeled GAP43 and analyzed its presence in the cytosol. SIRT2 genetic deletion led to an increase in GAP43 immunolabeling within MNs (Fig. 6(B)). Moreover, to understand which are the molecular axes involved in this pro-regenerative phenotype, we firstly analyzed the activation of AKT, which has been shown to increase axonal growth [28, 29] and to induce GAP43 [28]. In particular, we immunohistochemically labeled phosphorylated AKT at residue S473 (pAKT), which correlates with axonal regrowth [30, 31]. In this case, we observed a reduction in the level of pAKT in injured MNs of SIRT2 KO mice compared to that of injured WT mice (Fig. 6(B)). This result indicates that this pathway is attenuated after SIRT2 deletion; thus, it may not be involved in the observed pro-regenerative phenotype.
We also analyzed the myelin sheath of regenerating nerves. Semithin sections of distal regenerating nerves showed a tendency of higher number of axons in SIRT2 KO mice than in WT littermates, although the difference did not reach significance levels (p = 0.061, Fig. 6(C)). In addition, the axon area and diameter were similar in both groups, corroborating the data obtained from the electrophysiological results.
GAP43 expression is a tightly regulated process, which depends on the acetylation level of histone 3 (Ac-H3K9) at its promoter, and is also p53-dependent (Ac-p53k373) [32]. The acetylation/deacetylation cycle of both factors depends on the histone acetyltransferase p300/PCAF and SIRT2, respectively. Aiming to determine the acetylation state of p300/PCAF, we immunohistochemically labeled both its acetylated form at Lys1542, which is also a SIRT2-target, and the total form of p300 [32]. We observed that the levels of the acetylated p300/PCAF form as well as Ac-H3K9 and Ac-p53k373 were increased in SIRT2 KO mice compared to WT littermates (Fig. 7(A)) [30]. Altogether, these data suggest that the genetic deletion of SIRT2 confers an increase in the acetyltransferase activity of p300, which in turn increased Ac-H3k9 or Ac-p53k373 levels and downstream GAP43 expression [31].

(A) Top: Confocal images of motoneurons (MNs) immunolabeled with antibodies against Ac-H3K9, Ac-p53 (k373), Ac-p300 (k1542), and p300 in red and counterstained with FluoroNissl (green) and DAPI (blue) from MNs of wild-type (WT) and SIRT2 KO animals at 30 dpi. Scale bar = 100 μm and 20 μm. Down: Bar graphs of the mean of the Ac-H3k9, Ac-p53, and the ratio of Ac-p300/total p300 immunofluorescence intensity inside the cytoplasm or nuclei from injured MNs (n = 4, t test, *P < 0.05 vs. WT). (B) Representative microphotographs and bar graphs showing the mean (± SEM) values of neurite elongation and maximum neurite length per cell from SH-SY5Y cells after 24 h of treatment with vehicle (Veh), DMOG, AK7, C646, or AK7+C646, or eGFP, eGFP+AK7, shRNA/p300, and shRNA/p300+AK7. Insets are a black and white magnification of a single cell at each condition (n = 3–4, ANOVA, post hoc Bonferroni *P < 0.05 vs. Veh. Scale bar = 100 μm; inset, 20 = μm). (C) Western blots and histogram showing the analysis of p53, Ac-p53, and GAP43, in different experimental groups (n = 3–4, ANOVA, post hoc Bonferroni, *P < 0.05 vs. Veh, #P < 0.05 vs. AK7)
p300 Orchestrates the pro-Regenerative Effect of SIRT2 Blockage
To confirm whether the SIRT2 pro-regenerative effect was mediated by p300, we used another in vitro model based on a SH-S5Y5 human cell culture, which we recently used to describe that the pro-regenerative effect of SIRT1 is mediated through HIF1α stabilization [17]. We pharmacologically treated the cells with DMOG, a positive control of enhanced neuritogenesis [17], and with the SIRT2 inhibitor AK7, alone or in combination with the selective p300 inhibitor C646 [33](Fig. 7(B)). We observed that SIRT2 inhibition with AK7 significantly increased the average number and maximum neurite length outgrowing from these cells in a similar way as DMOG did. Moreover, this effect was blocked when we inhibited p300 using C646.
In parallel, we nucleofected the cells to silence p300 using shRNA in combination with AK7 (Fig. 7(B)). We confirmed that the use of shRNA effectively reduced p300 levels by immunoblotting (Fig. S2), and observed that p300 shRNA blocked the positive effects of AK7, including neurite elongation and Ac-p53 and GAP43 increase (Fig. 7(B, C)). Altogether, these results suggest that neurite outgrowth promoted by SIRT2 activity blockage depends on modulation of p53 by p300 acetyltransferase activity.
Discussion
Here, we propose that the inhibition of SIRT2-deacetylase activity can be an effective treatment to boost functional recovery after peripheral nerve injury. We provide evidence that SIRT2 inhibition favors neurite outgrowth that depends on p300 activity, which turns on epigenetic changes via the acetylation of H3K9 and p53, leading to a downstream increase of GAP43, a pro-regenerative marker.
As far as we know, this is the first evidence showing that SIRT2 blockage improves functional recovery after injury. This is in agreement with previous reports showing that SIRT2 knockdown reduces locomotor deficits in a Drosophila model of Parkinson’s disease [18] and that SIRT2 overexpression blocks neurite outgrowth [34]. However, our results are contradictory to those of previous studies that showed that SIRT2 tubulin-deacetylase activity prevented neurite shortening in a Parkinson’s disease in vitro model [35] and its genetic deletion led to axonal degeneration in aging [19]. Considering that epigenetic changes that may be promoted by SIRT2 largely depend on the type of neuron, state of maturity, its physiology, and its pathology, different outcomes may be expected in different models.
Herein, we have demonstrated that modulation of the SIRT2/p300 epigenetic axis is involved in promoting a pro-regenerative phenotype and neuritogenesis. Epigenetic modifications govern several aspects of cell response and strongly determine the outcome after injuries to the nervous system. They range from changes in the Schwann cell (SC) proliferative capacity by excessive deacetylation promoted by HDAC3 [36] to the appearance of neuropathic pain via hypomethylation of CpG sites in dorsal root ganglia (DRG) [37].
In this sense, although some authors claim that p300 acts coordinately with HDAC3 to activate myelination inhibitory programs, and knockdown of SIRT2 exclusively on SCs leads to an altered myelin sheath [26], we did not observe major alterations in myelin in the injured whole SIRT2 KO mice. These apparent discrepancies indicate that the outcome of epigenetic changes highly depends on the tissular environment, the cross-talk among cells in the tissue, and the physiological or pathological context. Epigenetic changes have been shown to be central elements for the promotion of axonal growth, at least in the PNS [26]. In fact, either AAV-mediated overexpression of HDAC3 deacetylase-dead mutant or pharmacological HDAC3 inhibition have been shown to be sufficient to promote axonal growth of sensory axons [26]. Importantly, these changes in HDACs and HATs do not occur after axotomy in the CNS.
Further studies are needed to decipher if SIRT2 deacetylase activity has the same role promoting regeneration in the central nervous system (CNS) or in sensory axons of peripheral nerves. SIRT2 is downregulated after nerve constrictive injuries and in DRG neurons of neuropathic pain models [37, 38]. Therefore, it seems that levels of this enzyme are differently regulated when the injury affects MNs or sensory neurons. SIRT2 knockdown by lentiviruses reduces axonal growth in sensory neurons, while its overexpression increases axonal growth in vitro [39]. We also observed that SIRT2 inhibition increases the neurite length in collagen-cultured DRGs, while it has no effect on the number of projected neurites (Fig. S2); this possibly indicates differential effects on motor or sensory axon regeneration. Besides, it would also be interesting to know the details on how SIRT2 inhibition promotes this improvement in functional recovery since we have not observed a significant increase in the number of regenerating axons. Recovery of function after nerve injury depends on different aspects aside from an accelerated axonal regeneration; on newly axons elongating, on branching/sprouting from the injury site or close to the denervated territory, on modulating membrane conductive properties [40], on changes in the motor unit size, and also on axonal fusion events [41], on accelerated maturation of NMJ, regenerated or surviving axons. Further research would clarify whether motor units are enlarged or axons are reinforced after nerve damage in SIRT2 KOs. Finally, the role of SIRT2 after CNS injury has not been addressed yet, and our results may open avenues to advance investigation in this field as well.
We have found that a determinant factor downstream SIRT2 inhibition, leading to an increase in neurite length in a human cell line, was mediated through the activity of p300. Viral overexpression of p300/CBP-associated factor (P/CAF) promotes axonal regeneration after injury by reactivating a regenerative gene expression program [30]. In the PNS, inhibition of p300 by C646 has detrimental effects after crush injury in hindlimb fine movements, supporting our data [42]. In the CNS, although p300 expression is not modified, its acetyltransferase activity promotes axonal regeneration after spinal cord injury [31, 43]. p300/CBP and PCAF can mediate axonal regeneration also via acetylation of p53 on K373/320 and C/EBP [30, 44]. The downstream activity of p300 favors GAP43 expression, which is a determinant of nerve regeneration [1]. Thus, the fact that SIRT2 blockage increases GAP43 expression within the CNS, opening new therapeutic windows by imprinting cortical motor neurons to engage a pro-regenerative mode that favors functional recovery.
In addition, we observed that SIRT2 blockage did not increase the phosphorylation state of AKT at Ser473, which directly depends on PTEN. In fact, we observed a non-significant reduction of pAKT levels within the MNs in SIRT2 KO mice, suggesting that SIRT2 activity is essential for an optimal AKT activation in MNs as it is in PC12 cells [45], although this depends on the biological state of the cells [46]. Hence, we describe a novel intrinsic molecular axis for nerve regeneration that does not depend on AKT/mTOR, raising therapeutic interest due to the pro-oncogenic role of this axis [47].
In summary, we provide evidence that SIRT2 blockage enhances functional recovery after PNI that may be via epigenome changes. Because SIRT2 inhibition has demonstrated beneficial effects in neurodegenerative disease [48, 53], the inhibition of SIRT2 activity may constitute an effective therapeutic avenue for treating those PNS injuries, in which functional recovery is not obtained spontaneously. Nonetheless, due to the anti-inflammatory role of SIRT2, it is important to remark that caution should be taken since its inhibition could exacerbate the neuropathic pain that appears after PNI, similar to the observed in chronic constriction injury [38].
Abbreviations
- Ac-H3K9:
-
Histone H3 acetylated at Lys9
- Ac-p53:
-
p53 acetylated at lys 373
- AKT:
-
Protein kinase B
- CMAP:
-
Compound muscle action potential
- DRG:
-
Dorsal root ganglia
- GAP43:
-
Growth-associated protein 43
- SIRT2 KO:
-
Sirt2−/− C57-BL6
- MN:
-
Motoneuron
- NF:
-
Neurofilament
- PBS:
-
Phosphate-buffered saline
- PNI:
-
Peripheral nerve injury
- SFI:
-
Sciatic functional index
- SIRT:
-
Sirtuin
- SOC:
-
Spinal organotypic culture
- SYT1:
-
Synaptotagmin
- TBS:
-
Tris-buffered
References
Fagoe ND, van Heest J, Verhaagen J. Spinal Cord Injury and the Neuron-Intrinsic Regeneration-Associated Gene Program. NeuroMolecular Med 2014;16(4):799–813.
Noble J, Munro CA, Prasad VS, Midha R. Analysis of upper and lower extremity peripheral nerve injuries in a population of patients with multiple injuries. J Trauma 1998;45(1):116–22.
Taylor CA, Braza D, Rice JB, Dillingham T. The Incidence of Peripheral Nerve Injury in Extremity Trauma. Am J Phys Med Rehabil 2008;87(5):381–5.
Grinsell D, Keating CP. Peripheral nerve reconstruction after injury: a review of clinical and experimental therapies. Biomed Res Int 2014;2014:1–13.
Romeo-Guitart D, Casas C. Network-centric medicine for peripheral nerve injury: Treating the whole to boost endogenous mechanisms of neuroprotection and regeneration. Neural Regen Res 2019 Jul;14(7):1122–8.
Cho Y, Shin JE, Ewan EE, Oh YM, Pita-Thomas W, Cavalli V. Activating Injury-Responsive Genes with Hypoxia Enhances Axon Regeneration through Neuronal HIF-1α. Neuron. 2015;88(4):720–34.
Hutson TH, Kathe C, Palmisano I, Bartholdi K, Hervera A, De Virgiliis F, et al. Cbp-dependent histone acetylation mediates axon regeneration induced by environmental enrichment in rodent spinal cord injury models. Sci Transl Med. 2019;11(487):eaaw2064.
Venkatesh I, Mehra V, Wang Z, Califf B, Blackmore MG. Developmental Chromatin Restriction of Pro-Growth Gene Networks Acts as an Epigenetic Barrier to Axon Regeneration in Cortical Neurons. Dev Neurobiol 2018 Oct;78(10):960–77.
Haigis MC, Guarente LP. Mammalian sirtuins - Emerging roles in physiology, aging, and calorie restriction. Vol. 20, Genes Dev 2006;20:2913–21.
Langley B, Sauve A. Sirtuin Deacetylases as Therapeutic Targets in the Nervous System. Neurotherapeutics. 2013;10(4):605–20.
Romeo-Guitart D, Leiva-Rodriguez T, Espinosa-Alcantud M, Sima N, Vaquero A, Dominguez-Martin H, et al. SIRT1 activation with neuroheal is neuroprotective but SIRT2 inhibition with AK7 is detrimental for disconnected motoneurons. Cell Death Dis 2018 May;9(5):531.
Romeo-Guitart D, Forés J, Herrando-Grabulosa M, Valls R, Leiva-Rodríguez T, Galea E, et al. Neuroprotective Drug for Nerve Trauma Revealed Using Artificial Intelligence. Sci Rep 2018 Dec;8(1):1879.
Rothgiesser KM, Erener S, Waibel S, Luscher B, Hottiger MO. SIRT2 regulates NF-kappaB dependent gene expression through deacetylation of p65 Lys310. J Cell Sci 2010 Dec;123(Pt 24):4251–8.
Lin J, Sun B, Jiang C, Hong H, Zheng Y. Sirt2 suppresses inflammatory responses in collagen-induced arthritis. Biochem Biophys Res Commun 2013;441(4):897–903.
Pais TF, Szego ÉM, Marques O, Miller-Fleming L, Antas P, Guerreiro P, et al. The NAD-dependent deacetylase sirtuin 2 is a suppressor of microglial activation and brain inflammation. EMBO J 2013;32(19):2603–16.
Liu CM, Wang RY, Saijilafu, Jiao ZX, Zhang BY, Zhou FQ. MicroRNA-138 and SIRT1 form a mutual negative feedback loop to regulate mammalian axon regeneration. Genes Dev 2013;27(13):1473–83.
Romeo-Guitart D, Leiva-Rodriguez T, Forés J, Casas C. Improved Motor Nerve Regeneration by SIRT1/Hif1a-Mediated Autophagy. Cells. 2019 Oct 30;8(11):1354.
Godena VK, Brookes-Hocking N, Moller A, Shaw G, Oswald M, Sancho RM, et al. Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat Commun 2014 Oct;5:5245.
Fourcade S, Morato L, Parameswaran J, Ruiz M, Ruiz-Cortes T, Jove M, et al. Loss of SIRT2 leads to axonal degeneration and locomotor disability associated with redox and energy imbalance. Aging Cell 2017 Dec;16(6):1404–13.
Taylor DM, Balabadra U, Xiang Z, Woodman B, Meade S, Amore A, et al. A brain-permeable small molecule reduces neuronal cholesterol by inhibiting activity of sirtuin 2 deacetylase. ACS Chem Biol 2011;6(6):540–6.
Torres-Espín A, Santos D, González-Pérez F, del Valle J, Navarro X. Neurite-J: An Image-J plug-in for axonal growth analysis in organotypic cultures. J Neurosci Methods 2014;236:26–39.
Serrano L, Martínez-Redondo P, Marazuela-Duque A, Vazquez BN, Dooley SJ, Voigt P, et al. The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev 2013;27(6):639–53.
Navarro X. Functional evaluation of peripheral nerve regeneration and target reinnervation in animal models: A critical overview. Eur J Neurosci 2016;43:271–86.
de Medinaceli L. Functional consequences of experimental nerve lesions: Effects of reinnervation blend. Exp Neurol 1988;100(1):166–78.
Allodi I, Mecollari V, Gonzalez-Perez F, Eggers R, Hoyng S, Verhaagen J, et al. Schwann cells transduced with a lentiviral vector encoding Fgf-2 promote motor neuron regeneration following sciatic nerve injury. Glia. 2014 Oct;62(10):1736–46.
Beirowski B, Gustin J, Armour SM, Yamamoto H, Viader A, North BJ, et al. Sir-two-homolog 2 (Sirt2) modulates peripheral myelination through polarity protein Par-3/atypical protein kinase C (aPKC) signaling. Proc Natl Acad Sci U S A 2011 Oct;108(43):E952–61.
Sakuma M, Gorski G, Sheu SH, Lee S, Barrett LB, Singh B, et al. Lack of motor recovery after prolonged denervation of the neuromuscular junction is not due to regenerative failure. Eur J Neurosci 2016;43(3):451–62.
Liu Z, Cai H, Zhang P, Li H, Liu H, Li Z. Activation of ERK1/2 and PI3K/Akt by IGF-1 on GAP-43 expression in DRG neurons with excitotoxicity induced by glutamate in vitro. Cell Mol Neurobiol 2012 Mar;32(2):191–200.
Agostinone J, Alarcon-martinez L, Gamlin C, Yu W, Wong ROL, Polo A Di. Insulin signalling promotes dendrite and synapse regeneration and restores circuit function after axonal injury. Brain. 2018;141(June):1963–80.
Puttagunta R, Tedeschi A, Soria MG, Hervera A, Lindner R, Rathore KI, et al. PCAF-dependent epigenetic changes promote axonal regeneration in the central nervous system. Nat Commun 2014 Apr;5:3527.
Gaub P, Joshi Y, Wuttke A, Naumann U, Schnichels S, Heiduschka P, et al. The histone acetyltransferase p300 promotes intrinsic axonal regeneration. Brain. 2011 Jul;134(Pt 7):2134–48.
Black JC, Mosley A, Kitada T, Washburn M, Carey M. The SIRT2 deacetylase regulates autoacetylation of p300. Mol Cell 2008 Nov;32(3):449–55.
Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: Identification of a selective small molecule inhibitor. Chem Biol 2010;17(5):471–82.
Pandithage R, Lilischkis R, Harting K, Wolf A, Jedamzik B, Luscher-Firzlaff J, et al. The regulation of SIRT2 function by cyclin-dependent kinases affects cell motility. J Cell Biol 2008 Mar;180(5):915–29.
Patel VP, Chu CT. Decreased SIRT2 activity leads to altered microtubule dynamics in oxidatively-stressed neuronal cells: implications for Parkinson’s disease. Exp Neurol 2014 Jul;257:170–81.
He X, Zhang L, Queme LF, Liu X, Lu A, Waclaw RR, et al. A histone deacetylase 3-dependent pathway delimits peripheral myelin growth and functional regeneration. Nat Med 2018 Mar;24(3):338–51.
Garriga J, Laumet G, Chen S-R, Zhang Y, Madzo J, Issa J-PJ, et al. Nerve Injury-Induced Chronic Pain Is Associated with Persistent DNA Methylation Reprogramming in Dorsal Root Ganglion. J Neurosci 2018 Jul;38(27):6090–101.
Zhang Y, Chi D. Overexpression of SIRT2 Alleviates Neuropathic Pain and Neuroinflammation Through Deacetylation of Transcription Factor Nuclear Factor-Kappa B. Inflammation. 2018;41(2):569–78.
Schartner E, Saleh A, Da Silva RV, Chowdhury SR, Smith D, Fernyhough P. SIRT2 is Required for Axon Regeneration in Adult Sensory Neurons and High Glucose Concentration Reduces its Expression in Diabetic Neuropathy. Can J Diabetes 2014 Oct 1;38(5):S62.
Moldovan M, Alvarez S, Rosberg MR, Krarup C. Persistent alterations in active and passive electrical membrane properties of regenerated nerve fibers of man and mice. Eur J Neurosci 2016;43(3):388–403.
Neumann B, Linton C, Giordano-Santini R, Hilliard MA. Axonal fusion: An alternative and efficient mechanism of nerve repair. Prog Neurobiol 2019;173:88–101.
Ding Z, Cao J, Shen Y, Zou Y, Yang X, Zhou W, et al. Resveratrol Promotes Nerve Regeneration via Activation of p300 Acetyltransferase-Mediated VEGF Signaling in a Rat Model of Sciatic Nerve Crush Injury. Vol. 12, Front Neurosci. 2018 May 23:341.
Chen J, Laramore C, Shifman MI. Differential expression of HDACs and KATs in high and low regeneration capacity neurons during spinal cord regeneration. Exp Neurol 2016 Jun 1;280:50–9.
Tedeschi A, Nguyen T, Puttagunta R, Gaub P, Di Giovanni S. A p53-CBP/p300 transcription module is required for GAP-43 expression, axon outgrowth, and regeneration. Cell Death Differ 2009 Apr 5;16(4):543–54.
Cao W, Hong Y, Chen H, Wu F, Wei X, Ying W. SIRT2 mediates NADH-induced increases in Nrf2, GCL, and glutathione by modulating Akt phosphorylation in PC12 cells. FEBS Lett 2016;590:2241–55.
Ramakrishnan G, Davaakhuu G, Kaplun L, Chung W-C, Rana A, Atfi A, et al. Sirt2 deacetylase is a novel AKT binding partner critical for AKT activation by insulin. J Biol Chem 2014 Feb;289(9):6054–66.
Tolkacheva T, Chan a M. Inhibition of H-Ras transformation by the PTEN/MMAC1/TEP1 tumor suppressor gene. Oncogene. 2000;19(5):680–9.
Chopra V, Quinti L, Kim J, Vollor L, Narayanan KL, Edgerly C, et al. The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington’s disease mouse models. Cell Rep 2012 Dec 27;2(6):1492–7.
Acknowledgements
We are sincerely grateful to Dr. Alejandro Vaquero for providing the SIRT2 transgenic mice and to Jessica Jaramillo for the nerve histology. This work was supported by the Spanish Ministerio de Economía y Competitividad (no. SAF 2014-59701). We are also grateful for the support from CIBERNED. The RT-97 antibodies were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biology. We thank Vassiliki Fotaki for editing service.
Author information
Authors and Affiliations
Contributions
DRG conceived research, performed experiments, analyzed results, and wrote the paper. TLR performed the SH-SY5Y experiments and analyzed results. CC conceived research, supervised experiments and the analysis, and wrote the paper.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Romeo-Guitart, D., Leiva-Rodríguez, T. & Casas, C. SIRT2 Inhibition Improves Functional Motor Recovery After Peripheral Nerve Injury. Neurotherapeutics 17, 1197–1211 (2020). https://doi.org/10.1007/s13311-020-00860-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-020-00860-3