
Abstract
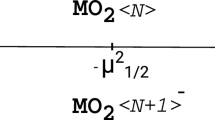
Hydrogen adsorption properties of the CN3Be3+ cluster have been studied using density functional theory and MP2 method with a 6–31++G** basis set. Five hydrogen molecules get adsorbed on the CN3Be3+ cluster with a hydrogen storage capacity of 10.98 wt%. Adsorption of three H2 molecules on one of the three Be atoms in a cluster is reported for the first time. It is due to the more positive charge on this Be atom than the remaining two. The average value for H2 adsorption energy in CN3Be3+ (5H2) complexes is 0.41 (0.43) eV/H2 at MP2 (wB97XD) level, which fits well within the ideal range. Adsorption energy from electronic structure calculations plays an important role in retaining the number of H2 molecules on a cluster during atom-centered density matrix propagation (ADMP) molecular dynamics (MD) simulations. According to ADMP-MD simulations, out of five H2 adsorbed molecules on CN3Be3+, four and two H2 molecules remain absorbed on CN3Be3+ cluster at 275 K and 350 K, respectively, during the simulation.






Similar content being viewed by others

References
Schlapbach L, Züttel A (2011) Hydrogen-storage materials for mobile applications. Mat Sust energy 4:265–270
Tabtimsai C, Rakrai W, Wanno B (2017) Hydrogen adsorption on graphene sheets doped with group 8B transition metal: a DFT investigation. Vacuum 139:101–108
Chaudhary A, Malakkal L, Siripurapu RK, Szpunar B, Szpunar J (2016) First principles calculations of hydrogen storage on cu and Pd-decorated graphene. Int J Hydrog Energy 41:17652–17656
Balat M (2008) Potential importance of hydrogen as a future solution to environmental and transportation problems. Int J Hydrog Energy 33:4013–4029
Si H, Peng LJ, James R, Morries JR, Pan BC (2011) Theoretical prediction of hydrogen storage on ZnO sheet. J Phys Chem C 115:9053–9058
Coontz R, Hanson B (2004) Not so simple. Science 305:957–957
Chen P, Zhu M (2008) Recent progress in hydrogen storage. Mater.Today 11:36–43
Principi G, Agresti F, Maddalena A, Russo SL (2009) The problem of solid state hydrogen storage. Energy 34:2087–2091
Zhang X, Tang C, Jiang Q (2016) Electric field induced enhancement of hydrogen storage capacity for Li atom decorated graphene with stone-Wales defects. Int J Hydrog Energy 41:10776–10785
Roszak R, Roszak S, Majumdar D, Kuchta B, Firlej L, Leszczynski J (2014) Unique bonding nature of carbon-substituted Be2 dimer inside the carbon (sp2) network. J Phys Chem A 118:5727–5733
Roszak R, Roszak S (2015) S-block metallabenzene: aromaticity and hydrogen adsorption. J Mol Model 21:28–46
Porter III WW, Wong-Foy A, Dailly A, Matzger AJ (2009) Beryllium benzene bicarboxylate: the first beryllium microporous coordination polymer. J Mater Chem 19:6489–6491
Sumida K, Hill MR, Horike S, Dailly A, Long JR (2009) Synthesis and hydrogen storage properties of be-12(OH)(12)(1,3,5benzenetribenzoate)(4). J Am Chem Soc 131:15120–15121
Han, S. S.; Deng, W. Q.; Goddard, W. A. (2007) Improved designs of metal-organic frameworks for hydrogen storage. Angew. Chem., Int Ed, 46: 6289–6292
Roszak R, Firlej L, Roszak S, Pfeifer P, Kuchta B (2016) Hydrogen storage by adsorption in porous materials: is it possible? Colloids Surf A Physicochem Eng Asp 496:69–76
Wu YB, Duan Y, Lu G, Lu HG, Yang P, Schleyer PVR, Merino G, Islas R, Wang ZX (2012) D3h CN3Be3+ and CO3Li3+: viable planar hexacoordinate carbon prototypes. Phys Chem Chem Phys 14:14760–14763
Pan S, Jalife S, Kumar R M, Subramanian V, Merino G, Chattaraj P K (2013) Structure and Stability of (NG)nCN3Be3+ Clusters and Comparison with (NG)BeY0/+ .Chem. Phys Chem 14: 2511–2517
Chai JD, Head-Gordon M (2008) Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys Chem Chem Phys 44:6615–6620
Schelegel HB, Iyengar SS, Li X, Millam JM, Voth GA, Scuseria GE, Frisch MJ (2002) Ab initio molecular dynamics: propagating the density matrix with Gaussian orbitals. III Comparison with Born–Oppenheimer dynamics J Chem Phys 117:8694–8704
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09. Gaussian, Inc., Wallingford
Gorelsky SI (2009). AOMix: program for molecular orbital analysis version:6.88
Gorelsky SI, Lever ABP (2001) Electronic structure and spectra of ruthenium diimine complexes by density functional theory and INDO/S, comparison of the two methods. J Organomet Chem 635:187–196
Reed AE, Weinstock RB, Weinhold F (1985) Natural population analysis. J Chem Phys 2:735–746
Deshmukh A, Konda R, Titus E, Chaudhari A (2017) Electronic structure calculations and molecular dynamics simulations of hydrogen adsorption on beryllium doped complexes. Int J Hydrog Energy 42:23708–23715
Konda RS, Titus E, Chaudhari A (2018) Adsorption of molecular hydrogen on inorganometallic complexes B2H4M (M=Li, be, Sc, Ti, V). Struct Chem 29:1–7
Zhou W, Yildirim T, Durgun E, Ceraci S (2007) Hydrogen absorption properties of metal-ethylene complexes. Phys Rev B 76:085434–008543
Lide DR (1994) Handbook of organic solvents. Press, CRC
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Solimannejad, M., Konda, R., Rahimi, R. et al. Ab initio calculations and molecular dynamics simulation of H2 adsorption on CN3Be3+ cluster. Struct Chem 31, 1757–1763 (2020). https://doi.org/10.1007/s11224-020-01532-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-020-01532-w