
Abstract
A novel planctomycetal strain, designated Pla85_3_4T, was isolated from the surface of wood incubated at the discharge of a wastewater treatment plant in the Warnow river near Rostock, Germany. Cells of the novel strain have a cell envelope architecture resembling that of Gram-negative bacteria, are round to pear-shaped (length: 2.2 ± 0.4 µm, width: 1.2 ± 0.3 µm), form aggregates and divide by polar budding. Colonies have a cream colour. Strain Pla85_3_4T grows at ranges of 10–30 °C (optimum 26 °C) and at pH 6.5–10.0 (optimum 7.5), and has a doubling time of 26 h. Phylogenetically, strain Pla85_3_4T (DSM 103796T = LMG 29741T) is concluded to represent a novel species of a novel genus within the family Pirellulaceae, for which we propose the name Lignipirellula cremea gen. nov., sp. nov.
Similar content being viewed by others

Introduction
Planctomycetes is a phylum of bacteria which were once thought to have several exceptional eukaryote-like traits (Devos et al. 2013; Devos and Reynaud 2010; Fuerst and Sagulenko 2011; Fuerst and Webb 1991; König et al. 1984; Lindsay et al. 1997; Lonhienne et al. 2010). These have since been re-examined and re-interpreted (Acehan et al. 2013; Boedeker et al. 2017; Jeske et al. 2015; Jogler 2014; Jogler et al. 2011; Jogler and Jogler 2013; Neumann et al. 2014; Rast et al. 2017; Rivas-Marin et al. 2016b; Rivas-Marín and Devos 2018; Santarella-Mellwig et al. 2013; van Teeseling et al. 2015). The cell envelope architecture of Planctomycetes was shown to resemble that of Gram-negative bacteria (Boedeker et al. 2017; Devos 2014). The phylum Planctomycetes is part of the PVC superphylum, along with Verrucomicrobia, Chlamydiae and others (Wagner and Horn 2006). Members of the phylum can play major roles in biogeochemical cycles (Peeters and van Niftrik 2018; Strous et al. 1999; Wiegand et al. 2018), are ubiquitous and are found in high abundance on algal surfaces (Bengtsson and Øvreås 2010; Bengtsson et al. 2012; Bondoso et al. 2014, 2015, 2017; Lage and Bondoso 2014; Vollmers et al. 2017), on which they probably metabolise complex sugars (Frank et al. 2014; Jeske et al. 2013; Lachnit et al. 2013; Wiegand et al. 2018).
Major scientific interest is dedicated to the interesting cell biology of Planctomycetes. For example, they lack many otherwise essential cell division genes, including the canonical ftsZ (Jogler et al. 2012; Pilhofer et al. 2008; Wiegand et al. 2020). Even so, some Planctomycetes are capable of performing cell division by binary fission, while others divide by budding (Rivas-Marin et al. 2016a; Wiegand et al. 2020). Members of the orders Planctomycetales and Pirellulales also perform a lifestyle switch between sessile mother cells and motile daughter cells (Jogler et al. 2011). Most Planctomycetes feature unusual crateriform structures on their cell surfaces. Their periplasm can be enlarged to form invaginations into the cytoplasm. They are potential producers of small molecules with interesting bioactivities (Graça et al. 2016; Jeske et al. 2016; Wiegand et al. 2018), possess many giant genes (Kohn et al. 2016), and belong to the bacterial phylum with the highest number of predicted genes with unknown function (40–55% of the annotated proteins) (Bordin et al. 2018; Overmann et al. 2017; Wiegand et al. 2018).
In this study, we isolated strain Pla85_3_4T from wood particles floating in the estuary of the Warnow river next to a wastewater treatment plant discharge and close to the Baltic Sea. In phylogenetic analyses, the strain clusters within the family Pirellulaceae, which comprises most of the described members of the order Pirellulales. In the past, Planctomycetes have been found on decomposing wood in natural temperate forests by 16S rRNA gene analysis (Tlaskal et al. 2017), but to date only one other Planctomycete, Singulisphaera mucilagenosa, has been isolated from (degrading) wood (Zaicnikova et al. 2011).
Materials and methods
Isolation and cultivation of the strain
Strain Pla85_3_4T was isolated from submerged wood pellets suspended near the discharge of a wastewater treatment plant in the estuary of Warnow river next to the city Rostock located in Germany (exact location 54.106 N 12.096 E). This location is close to the Baltic Sea and has brackish water. Isolation was performed as described previously (Oberbeckmann et al. 2018; Wiegand et al. 2020). For further investigation, the strain was grown in M1H medium supplemented with N-acetyl glucosamine (NAG) and artificial seawater (ASW) (M1H NAG ASW medium) as described previously (Wiegand et al. 2020) and was cultivated at 28 °C under constant agitation at 110 rpm.
Light microscopy and electron microscopy
Phase contrast microscopy was performed with a Nikon Eclipse Ti inverted microscope with a Nikon DS-Ri2 camera. Cells were immobilised in MatTek glass bottom dishes (35 mm, No. 1.5) using a 1% (w/v) agarose cushion (Boedeker et al. 2017). ImageJ (Rueden et al. 2017) was used to examine cell size by sequentially applying an Otsu threshold, then the watershed function, and finally the count particles function, excluding particles smaller than 0.05 µm.
Field emission scanning electron microscopy was performed as described previously (Boersma et al. 2019). The bacteria were fixed in formaldehyde, washed and placed on poly-l-lysine-coated cover slips. Samples were then fixed in 1% (v/v) glutaraldehyde and washed twice before dehydrating in graded series of acetone [10, 30, 50, 70, 90, 100% (v/v)] on ice. Samples from the last acetone treatment step were brought to room temperature before placing them in fresh 100% acetone. Samples were then subjected to critical-point drying with liquid CO2 (CPD 300, Leica). Dried samples were covered with a gold/palladium (80/20) film by sputter coating (SCD 500, Bal-Tec), before examination in a field emission scanning electron microscope (Zeiss Merlin) using an Everhart–Thornley HESE2 detector and an inlens SE detector in a 25:75 ratio with an acceleration voltage of 5 kV.
Physiological and biochemical analyses
The pH optimum was determined at 28 °C, with buffering agents 100 mM 2-(N-morpholino)ethanesulfonic acid (MES) at pH 5 and 6, 100 mM (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) HEPES at pH 7, 7.5 and 8, and 100 mM N-cyclohexyl-2-aminoethanesulfonic acid (CHES) at pH 9 and 10. Temperature optimum determination was performed at pH 7.5 and temperatures ranging from 10 to 40 °C in steps of 5 °C. Cell densities were inferred from optical densities (λ = 600 nm).
Genome information and analysis of genome-encoded features
The genome and 16S rRNA gene sequences of strain Pla85_3_4T are available from GenBank (accession numbers MK559988 and CP036433, respectively). Numbers of carbohydrate-active enzymes were obtained from the CAZY database (Lombard et al. 2014). Gene clusters potentially involved in the production of secondary metabolites were determined using antiSMASH 4.0 (Blin et al. 2017).
Phylogenetic analysis
16S rRNA gene-based phylogeny was computed for strain Pla85_3_4T, the type strains of all described planctomycetal species (assessed in January 2020), all isolates recently published (Boersma et al. 2019; Kallscheuer et al. 2019a, b, c, d, 2020; Kohn et al. 2019; Peeters et al. 2019; Rensink et al. 2020) and with an outgroup of strains from outside the phylum Planctomycetes but part of the PVC superphylum. The alignment of 16S rRNA genes was made with SINA (Pruesse et al. 2012). Phylogenetic analysis was performed employing a maximum likelihood approach with 1000 bootstraps, the nucleotide substitution model GTR, gamma distribution, and estimation of proportion of invariable sites using GTRGAMMAI (Stamatakis 2014).
The genomes for the genome-based analyses were gathered from GenBank including the sequences for strain Pla85_3_4T recently published (Wiegand et al. 2020). Completeness and contamination of the genome was determined using CheckM v1.0.131 (Parks et al. 2015). The average nucleotide identity (ANI) was calculated using OrthoANI (Lee et al. 2016), the average amino acid identity (AAI) was computed with the aai.rb script from the enveomics collection (Rodriguez-R and Konstantinidis 2016) and the percentage of conserved proteins (POCP) was determined as previously described (Qin et al. 2014). The rpoB nucleotide sequences (encoding the β-subunit of RNA polymerase) were taken from the genome annotations and the sequence identities were determined as described (Bondoso et al. 2013). Upon extracting only those parts of the sequences that would have been sequenced with the primer set described by Bondoso et al. (2013), the alignment and matrix calculation was performed with Clustal Omega (Sievers et al. 2011).
For the multi-locus sequence analysis (MLSA), the unique single-copy core genome of all analysed genomes was determined with proteinortho5 (Lechner et al. 2011) with the ‘selfblast’ option enabled. The protein sequences of the resulting orthologous groups were aligned using MUSCLE v.3.8.31 (Edgar 2004). After clipping, partially aligned C- and N-terminal regions and poorly aligned internal regions were filtered using Gblocks (Castresana 2000). The final alignment was concatenated and clustered using the maximum likelihood method implemented by RaxML (Stamatakis 2014) with the ‘rapid bootstrap’ method and 500 bootstrap replicates. The outgroup consisted of concatenated gene sets of strains from the order Planctomycetales.
Results and discussion
Phylogenetic inference
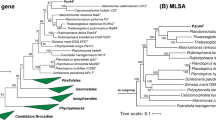
Based on 16S rRNA sequence analysis and MLSA, the isolated strain Pla85_3_4T clusters within the family Pirellulaceae (Fig. 1). 16S rRNA gene sequence identities to other current genera of this family range from 87.7 to 89.2% (Fig. 2). These values fall below the 94.5% cut-off value for delineating genera (Yarza et al. 2014), indicating that this species is part of a novel genus. To substantiate this claim, other phylogenetic markers such as RNA polymerase β-subunit (rpoB) gene similarity (Bondoso et al. 2013), AAI (Konstantinidis and Tiedje 2005) and POCP (Qin et al. 2014) were employed. Comparison of POCP values of strain Pla85_3_4T and the other genera in the family Pirellulaceae yielded maximum values between 27.8% and 45.7% (Fig. 2) which are all below the 50% cut-off value for delineation of genera (Qin et al. 2014). Comparison of AAI values of strain Pla85_3_4T yielded maximal similarities ranging from 48.1 to 53.8% (Fig. 2), which also fall below the cut-off range of 60–80% for defining genera (Luo et al. 2014). The maximal similarities of a 1200 base pair region of the rpoB gene between strain Pla85_3_4T were found to be in a range of 66.9–72.3% (Fig. 2), which again fall below the cut-off range of 75.5–78% for delineating genera (Bondoso et al. 2013; Kallscheuer et al. 2019d). Taken together, all four parameters support the conclusion that strain Pla85_3_4T belongs to a novel genus. A most closely related genus could not be clearly identified, although this strain formed a branch with Pirellula staleyi in the MLSA-based tree.

a 16S rRNA gene-based phylogenetic tree of described planctomycetal species and the novel isolate Pla85_3_4T indicated in blue. Bootstrap values indicated as a proportion of 1000 re-samplings (in %). The outgroup consisted of three 16S rRNA genes from the PVC superphylum outside the phylum Planctomycetes. b Whole genome-based MLSA phylogeny, with bootstrap values based on 500 re-samplings, indicated at the nodes (in %). The outgroup consisted of several representatives of the order Planctomycetales

Delineation of strain Pla85_3_4T from known genera in the family Pirellulaceae. Methods used: 16S rRNA gene identity (16S), average amino acid identity (AAI), average nucleotide identity (ANI), rpoB gene (partial) identity and percentage of conserved proteins (POCP)
Morphological, physiological and biochemical analyses
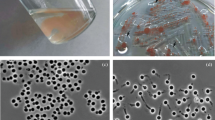
Morphology, physiology and life cycle of strain Pla85_3_4T were found to be similar to those of many other Planctomycetes. Adult cells were attached by loose fimbriae, enabling the cells to grow in aggregates (Fig. 3a). In other Planctomycetes, these fimbriae originate from crateriform structures, but these structures were either absent or difficult to observe in this strain. When examined with phase contrast microscopy, the cells appeared round to pear-shaped and 2.2 ± 0.4 by 1.2 ± 0.3 µm in length and width, respectively (Table 1). As typical for members of the family Pirellulaceae, the cells divide by polar budding. Colonies of strain Pla85_3_4T have a cream colour, indicating a lack of carotenoid production. The novel strain might be interesting for future studies on pigmentation of Planctomycetes since its phylogenetic position is between genera with mostly pigmented species (Rhodopirellula, Rubripirellula, Novipirellula, Crateriforma, Roseimaritima) and unpigmented species of the genera Pirellula, Blastopirellula and Bremerella (Fig. 1).

Phase contrast micrographs of strain Pla85_3_4T (a) and cell size, pH optimum and temperature optimum (b). The strain grows in aggregates or rosettes and divides by polar budding, as can be observed in the overview and close up, respectively. Asterisks indicate budding cells. Scale bar is 1 µm
Strain Pla85_3_4T is able to grow in medium containing artificial seawater, consistent with the observation that the section of the river from which this strain was isolated is quite brackish due to the influx of water from the Baltic Sea. Strain Pla85_3_4T grows chemoorganotrophically, aerobically and at temperatures ranging from 10 to 30 °C, with the optimum at 26 °C (Fig. 3b). The strain grows in the neutral to alkaline pH range from 6.5 to 8.5, with the optimum at 7.5. The growth rate of this strain was calculated to be 0.027 h−1, which corresponds to a doubling time of 26 h.
Genomic characteristics
The genome of strain Pla85_3_4T has a 61.4% G + C content and is 9,565,229 bp in length (Table 1). This distinguishes the strain from other members of the family Pirellulaceae as these species have smaller genomes of between 6.1 and 8.0 Mb, and G + C contents between 53.4 and 57.8%. The strain has a larger genome and a larger number of genes, while the coding density and number of proteins per Mb is lower than in its relatives from the family Pirellulaceae (Table 1). The genome of strain Pla85_3_4T contains both a large number of transposable elements (27) and a large number of tRNAs (144) in comparison to those of other members of the family Pirellulaceae (Table 1). The genome of strain Pla85_3_4T contains a singular 16S rRNA gene.
Genome-based analysis of metabolic features
Based on the genome of strain Pla85_3_4T and of species of closely related genera, we analysed the numbers of putative carbohydrate-active enzymes and of gene clusters putatively involved in the synthesis of secondary metabolites (Table 2). These numbers can give a first impression on the metabolic capabilities of the strain, e.g. in competitive environments, in which complex polysaccharides (e.g. derived from macroscopic phototrophs) function as a major source of carbon and energy. The observed number of 124 putative carbohydrate-active enzymes of strain Pla85_3_4T is in the lower to middle range compared to its relatives, which harbour between 87 and more than 200 of such enzymes. Higher numbers of putative carbohydrate-active enzymes are not reflected by larger genomes. Strain Pla85_3_4T has the largest genome of the compared strains, but is the strain with the second-lowest number of carbohydrate-active enzymes. Although the difference in the genome size of Crateriforma conspicua and P. staleyi is only around 1 Mb, the number of carbohydrate-active enzymes is nearly 2.5-fold different in a direct comparison of these two species. The numbers of proteins belonging to such classes are more likely a reflection of the complexity of the natural environment and probably do not depend on the genome size.
In contrast, numbers of gene clusters putatively involved in the production of secondary metabolites clearly correlated with the genome size. Strain Pla85_3_4T has both the largest genome and the highest number of predicted clusters (Table 2). The lowest numbers were found in Mariniblastus fucicola and P. staleyi, both with genomes below 7 Mb. In strain Pla85_3_4T, three clusters putatively involved in the biosynthesis of bacteriocin, one cluster for ectoine production and one cluster related to resorcinol production were identified. These appear to be restricted to strain Pla85_3_4T (of the other strains examined only P. staleyi harbours one putative cluster related to bacteriocin production). Three terpenoid biosynthetic gene clusters are present in Pla85_3_4T, however, since the strain lacks pigmentation, they are likely not exclusively involved in carotenoid biosynthesis, but relevant for synthesis of other terpenoids. Putative polyketide synthases of the types I and III are present in all strains, except for the two above-mentioned species with genome sizes smaller than 7 Mb. Type II polyketide synthases appear to be absent from all compared strains.
Conclusion
Taken together, based on the phylogenetic inference and supported by differences in morphology as well as genomic characteristics, we conclude that strain Pla85_3_4T represents a novel species of a new genus, for which we propose the name Lignipirellula cremea gen. nov., sp. nov.
Lignipirellula gen. nov.
Lignipirellula (Lig.ni.pi.rel’lu.la. L. neut. n. lignum wood; N.L. fem. n. Pirellula name of a bacterial genus; N.L. fem. n. Lignipirellula a Pirellula isolated from wood).
Members of the genus have a Gram-negative cell envelope architecture, are aerobic, mesophilic, neutrophilic and heterotrophic. Cells are round to pear-shaped and divide by polar budding. The genus is part of the family Pirellulaceae, order Pirellulales, class Planctomycetia, phylum Planctomycetes.
Lignipirellula cremea sp. nov.
Lignipirellula cremea (cre’me.a. N.L. fem. adj. cremea of creme; corresponding to the creamy colour of the cells).
In addition to the characteristics described for the genus, cells have a length of 2.2 ± 0.4 µm and width of 1.2 ± 0.3 µm, form aggregates and lack pigmentation. Crateriform structures are not observed. Daughter cells have the same shape as the mother cell. Grows aerobically at 10–30 °C (optimum 26 °C) and at pH 6.5–8.5 (optimum 7.5), and has a doubling time of about 26 h. The 9.6 Mb genomic DNA of the type strain has a G + C content of 61%.
The type strain is Pla85_3_4T (DSM 103796T = LMG 29741T), which was isolated from the surface of wood incubated close to the discharge of a wastewater treatment plant in the Unterwarnow river in Rostock, Germany in September 2014. The genome (accession no. CP036433) and 16S rRNA sequence (GenBank accession no. MK559988) of strain Pla85_3_4T are available from GenBank.
References
Acehan D, Santarella-Mellwig R, Devos DP (2013) A bacterial tubulovesicular network. J Cell Sci 127:277–280
Bengtsson MM, Øvreås L (2010) Planctomycetes dominate biofilms on surfaces of the kelp Laminaria hyperborea. BMC Microbiol 10:261
Bengtsson MM, Sjøtun K, Lanzén A, Øvreås L (2012) Bacterial diversity in relation to secondary production and succession on surfaces of the kelp Laminaria hyperborea. ISME J 6:2188–2198
Blin K, Wolf T, Chevrette MG, Lu X, Schwalen CJ, Kautsar SA, Suarez Duran HG, de Los Santos EL, Kim HU, Nave M (2017) antiSMASH 4.0—improvements in chemistry prediction and gene cluster boundary identification. Nucl Acids Res 45:W36–W41
Boedeker C, Schuler M, Reintjes G, Jeske O, van Teeseling MC, Jogler M, Rast P, Borchert D, Devos DP, Kucklick M, Schaffer M, Kolter R, van Niftrik L, Engelmann S, Amann R, Rohde M, Engelhardt H, Jogler C (2017) Determining the bacterial cell biology of Planctomycetes. Nat Commun 8:14853
Boersma A, Kallscheuer N, Wiegand S, Rast R, Peeters S, Mesman R, Heuer A, Boedeker C, Jetten M, Rohde M, Jogler M, Jogler C (2019) Alienimonas californiensis gen. nov. sp. nov., a novel Planctomycete isolated from the kelp forest in Monterey Bay. Antonie van Leeuwenhoek. https://doi.org/10.1007/s10482-019-01367-4
Bondoso J, Harder J, Lage OM (2013) rpoB gene as a novel molecular marker to infer phylogeny in Planctomycetales. Antonie Van Leeuwenhoek 104:477–488
Bondoso J, Balague V, Gasol JM, Lage OM (2014) Community composition of the Planctomycetes associated with different macroalgae. FEMS Microbiol Ecol 88:445–456
Bondoso J, Albuquerque L, Nobre MF, Lobo-da-Cunha A, da Costa MS, Lage OM (2015) Roseimaritima ulvae gen. nov., sp. nov. and Rubripirellula obstinata gen. nov., sp. nov. two novel planctomycetes isolated from the epiphytic community of macroalgae. Syst Appl Microbiol 38:8–15
Bondoso J, Godoy-Vitorino F, Balague V, Gasol JM, Harder J, Lage OM (2017) Epiphytic Planctomycetes communities associated with three main groups of macroalgae. FEMS Microbiol Ecol 93:fiw255
Bordin N, González-Sánchez JC, Devos DP (2018) PVCbase: an integrated web resource for the PVC bacterial proteomes. Database 2018:bay042
Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552
Devos DP (2014) PVC bacteria: variation of, but not exception to, the Gram-negative cell plan. Trends Microbiol 22:14–20
Devos DP, Reynaud EG (2010) Evolution. Intermediate steps. Science 330:1187–1188
Devos DP, Jogler C, Fuerst JA (2013) The 1st EMBO workshop on PVC bacteria-Planctomycetes–Verrucomicrobia–Chlamydiae superphylum: exceptions to the bacterial definition? Antonie Van Leeuwenhoek 104:443–449
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res 32:1792–1797
Frank O, Michael V, Pauker O, Boedeker C, Jogler C, Rohde M, Petersen J (2014) Plasmid curing and the loss of grip—the 65-kb replicon of Phaeobacter inhibens DSM 17395 is required for biofilm formation, motility and the colonization of marine algae. Syst Appl Microbiol 38:120–127
Fuerst JA, Sagulenko E (2011) Beyond the bacterium: planctomycetes challenge our concepts of microbial structure and function. Nat Rev Microbiol 9:403–413
Fuerst JA, Webb RI (1991) Membrane-bounded nucleoid in the eubacterium Gemmata obscuriglobus. Proc Natl Acad Sci USA 88:8184–8188
Graça AP, Calisto R, Lage OM (2016) Planctomycetes as novel source of bioactive molecules. Front Microbiol 7:1241
Jeske O, Jogler M, Petersen J, Sikorski J, Jogler C (2013) From genome mining to phenotypic microarrays: planctomycetes as source for novel bioactive molecules. Antonie Van Leeuwenhoek 104:551–567
Jeske O, Schüler M, Schumann P, Schneider A, Boedeker C, Jogler M, Bollschweiler D, Rohde M, Mayer C, Engelhardt H, Spring S, Jogler C (2015) Planctomycetes do possess a peptidoglycan cell wall. Nat Commun 6:7116
Jeske O, Surup F, Ketteniß M, Rast P, Förster B, Jogler M, Wink J, Jogler C (2016) Developing techniques for the utilization of Planctomycetes as producers of bioactive molecules. Front Microbiol 7:1242
Jogler C (2014) The bacterial ‘mitochondrium’. Mol Microbiol 94:751–755
Jogler M, Jogler C (2013) Towards the development of genetic tools for Planctomycetes. In: Fuerst JA (ed) Planctomycetes: cell structure, origins and biology. Springer, Berlin, pp 141–164
Jogler C, Glöckner FO, Kolter R (2011) Characterization of Planctomyces limnophilus and development of genetic tools for its manipulation establish it as a model species for the phylum Planctomycetes. Appl Environ Microbiol 77:5826–5829
Jogler C, Waldmann J, Huang X, Jogler M, Glöckner FO, Mascher T, Kolter R (2012) Identification of proteins likely to be involved in morphogenesis, cell division, and signal transduction in Planctomycetes by comparative genomics. J Bacteriol 194:6419–6430
Kallscheuer N, Jogler M, Wiegand S, Peeters SH, Heuer A, Boedeker C, Jetten MS, Rohde M, Jogler C (2019a) Rubinisphaera italica sp. nov. isolated from a hydrothermal area in the Tyrrhenian Sea close to the volcanic island Panarea. Antonie van Leeuwenhoek. https://doi.org/10.1007/s10482-019-01329-w
Kallscheuer N, Jogler M, Wiegand S, Peeters SH, Heuer A, Boedeker C, Jetten MS, Rohde M, Jogler C (2019b) Three novel Rubripirellula species isolated from plastic particles submerged in the Baltic Sea and the estuary of the river Warnow in northern Germany. Antonie Van Leeuwenhoek. https://doi.org/10.1007/s10482-019-01368-3
Kallscheuer N, Wiegand S, Jogler M, Boedeker C, Peeters SH, Rast P, Heuer A, Jetten MSM, Rohde M, Jogler C (2019c) Rhodopirellula heiligendammensis sp. nov., Rhodopirellula pilleata sp. nov., and Rhodopirellula solitaria sp. nov. isolated from natural or artificial marine surfaces in Northern Germany and California, USA, and emended description of the genus Rhodopirellula. Antonie van Leeuwenhoek. https://doi.org/10.1007/s10482-019-01366-5
Kallscheuer N, Wiegand S, Peeters SH, Jogler M, Boedeker C, Heuer A, Rast P, Jetten MSM, Rohde M, Jogler C (2019d) Description of three bacterial strains belonging to the new genus Novipirellula gen. nov., reclassificiation of Rhodopirellula rosea and Rhodopirellula caenicola and readjustment of the genus threshold of the phylogenetic marker rpoB for Planctomycetaceae. Antonie van Leeuwenhoek. https://doi.org/10.1007/s10482-019-01374-5
Kallscheuer N, Wiegand S, Heuer A, Rensink S, Boersma AS, Jogler M, Boedeker C, Peeters SH, Rast P, Jetten MS, Rhode M, Jogler C (2020) Blastopirellula retiformator sp. nov. isolated from the shallow-sea hydrothermal vent system close to Panarea Island. Antonie van Leeuwenhoek. https://doi.org/10.1007/s10482-019-01377-2
Kohn T, Heuer A, Jogler M, Vollmers J, Boedeker C, Bunk B, Rast P, Borchert D, Glöckner I, Freese HM, Klenk HP, Overmann J, Kaster AK, Wiegand S, Rohde M, Jogler C (2016) Fuerstia marisgermanicae gen. nov., sp. nov., an unusual member of the phylum Planctomycetes from the German Wadden Sea. Front Microbiol 7:2079
Kohn T, Wiegand S, Boedeker C, Rast P, Heuer A, Jetten M, Schüler M, Becker S, Rohde C, Müller R-W (2019) Planctopirus ephydatiae, a novel Planctomycete isolated from a freshwater sponge. Syst Appl Microbiol 43:126022
König E, Schlesner H, Hirsch P (1984) Cell wall studies on budding bacteria of the Planctomyces/Pasteuria group and on a Prosthecomicrobium sp. Arch Microbiol 138:200–205
Konstantinidis KT, Tiedje JM (2005) Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci USA 102:2567–2572
Lachnit T, Fischer M, Kunzel S, Baines JF, Harder T (2013) Compounds associated with algal surfaces mediate epiphytic colonization of the marine macroalga Fucus vesiculosus. FEMS Microbiol Ecol 84:411–420
Lage OM, Bondoso J (2014) Planctomycetes and macroalgae, a striking association. Front Microbiol 5:267
Lage OM, Albuquerque L, Lobo-da Cunha A, da Costa MS (2017) Mariniblastus fucicola gen. nov., sp. nov. a novel planctomycete associated with macroalgae. Int J Syst Evol Micribol 67:1571–1576
Lechner M, Findeiss S, Müller L, Marz M, Stadler P, Prohaska S (2011) Proteinortho: detection of (co)orthologs in large-scale analysis. BMC Bioinform 12:124
Lee HW, Roh SW, Shin NR, Lee J, Whon TW, Jung MJ, Yun JH, Kim MS, Hyun DW, Kim D, Bae JW (2013) Blastopirellula cremea sp. nov., isolated from a dead ark clam. Int J Syst Evol Microbiol 63:2314–2319
Lee I, Ouk Kim Y, Park SC, Chun J (2016) OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol 66:1100–1103
Lindsay MR, Webb RI, Fuerst JA (1997) Pirellulosomes: a new type of membrane-bounded cell compartment in planctomycete bacteria of the genus Pirellula. Microbiology 143:739–748
Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucl Acids Res 42:D490–D495
Lonhienne TG, Sagulenko E, Webb RI, Lee KC, Franke J, Devos DP, Nouwens A, Carroll BJ, Fuerst JA (2010) Endocytosis-like protein uptake in the bacterium Gemmata obscuriglobus. Proc Natl Acad Sci USA 107:12883–12888
Luo C, Rodriguez RL, Konstantinidis KT (2014) MyTaxa: an advanced taxonomic classifier for genomic and metagenomic sequences. Nucl Acids Res 42:e73
Neumann S, Wessels HJ, Rijpstra WI, Sinninghe Damste JS, Kartal B, Jetten MS, van Niftrik L (2014) Isolation and characterization of a prokaryotic cell organelle from the anammox bacterium Kuenenia stuttgartiensis. Mol Microbiol 94:794–802
Oberbeckmann S, Kreikemeyer B, Labrenz M (2018) Environmental factors support the formation of specific bacterial assemblages on microplastics. Front Microbiol 8:2709
Overmann J, Abt B, Sikorski J (2017) Present and future of culturing bacteria. Annu Rev Microbiol 71:711–730
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW (2015) CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055
Peeters SH, van Niftrik L (2018) Trending topics and open questions in anaerobic ammonium oxidation. Curr Opin Chem Biol 49:45–52
Peeters SH, Wiegand S, Kallscheuer N, Jogler M, Heuer A, Jetten MSM, Rast P, Boedeker C, Rohde M, Jogler C (2019) Three marine strains constitute the novel genus and species Crateriforma conspicua in the phylum Planctomycetes. Antonie Van Leeuwenhoek. https://doi.org/10.1007/s10482-019-01375-4
Pilhofer M, Rappl K, Eckl C, Bauer AP, Ludwig W, Schleifer KH, Petroni G (2008) Characterization and evolution of cell division and cell wall synthesis genes in the bacterial phyla Verrucomicrobia, Lentisphaerae, Chlamydiae, and Planctomycetes and phylogenetic comparison with rRNA genes. J Bacteriol 190:3192–3202
Pruesse E, Peplies J, Glockner FO (2012) SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28:1823–1829
Qin QL, Xie BB, Zhang XY, Chen XL, Zhou BC, Zhou J, Oren A, Zhang YZ (2014) A proposed genus boundary for the prokaryotes based on genomic insights. J Bacteriol 196:2210–2215
Rast P, Glockner I, Boedeker C, Jeske O, Wiegand S, Reinhardt R, Schumann P, Rohde M, Spring S, Glockner FO, Jogler C, Jogler M (2017) Three novel species with peptidoglycan cell walls form the new genus Lacunisphaera gen. nov. in the family Opitutaceae of the verrucomicrobial subdivision 4. Front Microbiol 8:202
Rensink S, Wiegand S, Kallscheuer N, Rast P, Peeters SH, Heuer A, Boedeker C, Jetten MS, Rohde M, Jogler M, Jogler C (2020) Description of the novel planctomycetal genus Bremerella, containing Bremerella volcania sp. nov., isolated from an active volcanic site, and reclassification of Blastopirellula cremea as Bremerella cremea comb. nov. Antonie van Leeuwenhoek. https://doi.org/10.1007/s10482-019-01378-1
Rivas-Marín E, Devos DP (2018) The paradigms they are a-Changin’: past, present and future of PVC bacteria research. Antonie Van Leeuwenhoek 111:785–799
Rivas-Marin E, Canosa I, Devos DP (2016a) Evolutionary cell biology of division mode in the bacterial Planctomycetes–Verrucomicrobia–Chlamydiae superphylum. Front Microbiol 7:1964
Rivas-Marin E, Canosa I, Santero E, Devos DP (2016b) Development of genetic tools for the manipulation of the planctomycetes. Front Microbiol 7:914
Rodriguez-R LM, Konstantinidis KT (2016) The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Preprints 4:e1900v1
Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, Eliceiri KW (2017) Image J2: imageJ for the next generation of scientific image data. BMC Bioinform 18:529
Santarella-Mellwig R, Pruggnaller S, Roos N, Mattaj IW, Devos DP (2013) Three-dimensional reconstruction of bacteria with a complex endomembrane system. PLoS Biol 11:e1001565
Schlesner H, Hirsch P (1984) Assignment of ATCC 27377 to Pirella gen. nov. as Pirella staleyi comb. nov. Int J Syst Evol Microbiol 34:492–495
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Strous M, Fuerst JA, Kramer EH, Logemann S, Muyzer G, van de Pas-Schoonen KT, Webb R, Kuenen JG, Jetten MS (1999) Missing lithotroph identified as new planctomycete. Nature 400:446–449
Tlaskal V, Zrustová P, Vrška T, Baldrian P (2017) Bacteria associated with decomposing dead wood in a natural temperate forest. FEMS Microbiol Ecol 93:fix157
van Teeseling MC, Mesman RJ, Kuru E, Espaillat A, Cava F, Brun YV, VanNieuwenhze MS, Kartal B, van Niftrik L (2015) Anammox Planctomycetes have a peptidoglycan cell wall. Nat Commun 6:6878
Vollmers J, Frentrup M, Rast P, Jogler C, Kaster AK (2017) Untangling genomes of novel planctomycetal and verrucomicrobial species from monterey bay kelp forest metagenomes by refined binning. Front Microbiol 8:472
Wagner M, Horn M (2006) The Planctomycetes, Verrucomicrobia, Chlamydiae and sister phyla comprise a superphylum with biotechnological and medical relevance. Curr Opin Biotechnol 17:241–249
Wiegand S, Jogler M, Jogler C (2018) On the maverick Planctomycetes. FEMS Microbiol Rev 42:739–760
Wiegand S, Jogler M, Boedeker C, Pinto D, Vollmers J, Rivas-Marín E, Kohn T, Peeters SH, Heuer A, Rast P, Oberbeckmann S, Bunk B, Jeske O, Meyerdierks A, Storesund JE, Kallscheuer N, Lücker S, Lage OM, Pohl T, Merkel BJ, Hornburger P, Müller R-W, Brümmer F, Labrenz M, Spormann AM, Op den Camp H, Overmann J, Amann R, Jetten MSM, Mascher T, Medema MH, Devos DP, Kaster A-K, Øvreås L, Rohde M, Galperin MY, Jogler C (2020) Cultivation and functional characterization of 79 planctomycetes uncovers their unique biology. Nat Microbiol 5:126–140
Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, Schleifer K-H, Whitman WB, Euzéby J, Amann R, Rosselló-Móra R (2014) Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol 12:635
Zaicnikova MV, Berestovskaya YY, Akimov VN, Kostrikina NA, Vasilieva LV (2011) Singulispaera mucilagenosa sp. nov., a novel acid-tolerant representative of the order Planctomycetales. Microbiol 80:101–107
Acknowledgements
Open Access funding provided by Projekt DEAL. Part of this research was funded by the Deutsche Forschungsgemeinschaft Grants KA 4967/1-1 and JO 893/4-1, Grant ALWOP.308 of the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO), SIAM (Soehngen Institute for Anaerobic Microbiology) Grant No. 024002002 and the Radboud Excellence fellowship. We thank Ina Schleicher for skillful technical assistance. Brian Tindall and Regine Fähnrich from the DSMZ as well as the BCCM/LMG Bacteria collection we thank for support during strain deposition. We are grateful to our collaborators Sonja Oberbeckmann and Matthias Labrenz (IOW Warnemünde, Germany) for sampling support.
Author information
Authors and Affiliations
Contributions
SHP and SW wrote the manuscript, analysed the data and prepared the figures, SW performed the genomic and phylogenetic analysis, AH and MJ isolated the strains and performed the initial cultivation and strain deposition, SHP and CB performed the light microscopic analysis, NK and MSMJ contributed to text preparation and revised the manuscript, MR performed the electron microscopic analysis. CJ supervised AH and the study. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical statement
This article does not contain any studies with animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peeters, S.H., Wiegand, S., Kallscheuer, N. et al. Lignipirellula cremea gen. nov., sp. nov., a planctomycete isolated from wood particles in a brackish river estuary. Antonie van Leeuwenhoek 113, 1863–1875 (2020). https://doi.org/10.1007/s10482-020-01407-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-020-01407-4