
Abstract
The calanoid copepod, Acartia tonsa Dana, 1849 is one of the most abundant and well-studied estuarian species with a worldwide distribution. In this research, we use the mitochondrial cytochrome oxidase subunit I gene to study the phylogeography of A. tonsa by analyzing sequences from specimens collected in the western Gulf of Mexico (GOM) along with all sequences from previous research. We reconstruct the phylogeny for the genus Acartia Dana, 1846 and highlight numerous potential misidentifications of Acartia species deposited in GenBank. The incorrect taxonomy assigned to some of these sequences results in apparently paraphyletic relationships. This study demonstrates that A. tonsa is a species complex with multiple, deeply diverging, lineages of varying geographic affinities. Multiple new lineages are found in the Texas GOM that is basal to northwestern Atlantic lineages with phylogenetic connectivity also observed between Brazil and the Texas GOM. Results show two major phylogeographic breaks in the North American continent, one at the border between the Gulf of Mexico and the Northwest Atlantic, and the other at about 35° N. One of the major clades in the A. tonsa species complex shows a clear pattern of divergence that follows the prevailing currents. Within this clade, older lineages are found in the western GOM while newer lineages are found in the eastern GOM and the southern coast of the northwest Atlantic, with the youngest lineages diversifying in the north. The results show that A. tonsa can be used as a model species for observing phylogeographical structuring of coastal plankton along the American continent.
Similar content being viewed by others

Introduction
Acartia tonsa Dana, 1849, is a calanoid copepod (Copepoda:Calanoida) in the family Acartiidae that is commonly found in coastal estuaries. Acartia tonsa is one of the most abundant and well-studied copepods in the world (Caudill and Bucklin 2004; Chen and Hare 2008, 2011; da Costa et al. 2011; da Costa et al. 2014; Zhou et al. 2016; Werbrouck et al. 2016; Minh et al. 2017; Øie et al. 2017; Rahlff et al. 2017; Franco et al. 2017; Jepsen et al. 2017; Pavlaki et al. 2017; Krause et al. 2017; Plough et al. 2018; Gomes et al. 2018; Sasaki and Dam 2019) with a global distribution along the coasts of the Indo-Pacific and Atlantic (Mauchline et al. 1998). It is often the dominant copepod species in estuaries playing a vital role in the trophic dynamics of these ecosystems as the main consumer of phytoplankton and the main food source for varied fish species (Mauchline et al. 1998). Previous research shows that A. tonsa has experienced geographical isolation resulting in the divergence of multiple, distinct cryptic lineages that in some cases live in sympatry (Chen and Hare 2011; Caudill and Bucklin 2004; and da Costa et al. 2014). Cryptic species, those that seem to be morphologically indistinguishable but genetically different, can pose challenges for taxonomists and biologists in terms of conservation, evolutionary theory, and biogeography (e.g., Avise et al. 1987; Bickford et al. 2007). Advances in molecular techniques and the ease with which we can identify species using molecular tools have greatly advanced the field of taxonomy. Phylogenetic and population analyses based on mitochondrial markers such as mtCOI, have been used to identify cryptic species in numerous marine organisms, including copepods (Lee 2000; Rocha-Olivares et al. 2001; Dawson and Jacobs 2001; Lee and Frost 2002; Caudill and Bucklin 2004; Holland et al. 2004; Chen and Hare 2008, 2011; Johnson et al. 2008; Dippenaar et al. 2010; Marrone et al. 2010; Blanco-Bercial et al. 2014; Cornils and Held 2014; Oyama et al. 2019; Govender et al. 2019; Han et al. 2019; Hupało et al. 2019; Figueroa et al. 2019; Chan et al. 2019). A comprehensive review of DNA barcoding of copepods based on mtCOI is presented by Blanco-Bercial et al. (2014), where they show that this marker was able to accurately identify to species level 1381 sequences for 195 species of copepods, showcasing the utility of this marker for taxonomic research.
The first evidence of deeply diverging genetic lineages in A. tonsa was presented by Caudill and Bucklin (2004), where they show four distinct lineages discovered from the coasts of the US Atlantic Ocean and the Gulf of Mexico (GOM) using mitochondrial 16S rRNA (mt16S) sequences. Chen and Hare (2008, 2011) identified three lineages (F, X, and S) of A. tonsa living in Chesapeake Bay and other Atlantic estuaries based on sequences of the mitochondrial cytochrome oxidase subunit I (mtCOI) gene. By applying a crustacean mtCOI molecular clock, Chen and Hare (2011) hypothesized that the northern Atlantic lineages of A. tonsa diversified pre-Pleistocene and the mid-Atlantic lineages diversified post-Pleistocene. As ice expanded southward, holoplankton species would have been pushed southward and some separated by falling sea levels and ice that caused geographical barriers. As the ice retreated and temperatures rose during warmer intervals of the Pleistocene, tropical species would have spread to higher latitudes (Chen and Hare 2011). Physiological and reproductive evidence that these deeply diverging lineages are different species is provided by a recent study by Plough et al. (2018) who performed pair crosses between individuals from these F and S lineages. They found a nearly complete lack of larvae production for between-lineage crosses, along with clear differences in size and chemical composition. They conclude that these lineages are likely reproductively isolated species.
Many other studies not only suggest that A. tonsa is likely a cryptic species complex, but also highlight several instances of misidentification and paraphyly (da Costa et al. 2011; Milligan et al. 2011; Blanco-Bercial et al. 2014; da Costa et al. 2014; Gomes et al. 2018). In Brazil, da Costa et al. (2014), identified three unique lineages of A. tonsa, also based on mtCOI sequences. Later, Gomes et al. (2018) demonstrate that two of the three lineages identified by da Costa et al. (2014) likely belong to a different species, Acartia lilljeborgi Giesbrecht, 1889. They provide morphological and genetic evidence to make this assertion. Milligan et al. (2011) show that based on the mitochondrial gene cytochrome B, Acartia hudsonica Pinhey, 1926 is paraphyletic because A. tonsa is included in an internal clade of A. hudsonica. In contrast, Blanco-Bercial et al. (2014) show that A. tonsa might be paraphyletic because A. hudsonica is included within internal clades of A. tonsa. This phylogenetic research presents a taxonomic conundrum: is Acartia tonsa a species complex with multiple monophyletic and cryptic lineages? Is Acartia tonsa paraphyletic, as suggested by the mixed genetic clades with A. hudsonica and A. lilljeborgi? How much of this taxonomic and phylogenetic uncertainty is due to species misidentification?
The processes and mechanisms underlying the diversification of A. tonsa lineages and the relationship of these lineages to (apparently) closely related species such as A. hudsonica and A. lilljeborgi cannot be inferred without additional data from lower latitudes such as the Gulf of Mexico that harbor distinct and potentially cryptic lineages from those identified by Chen and Hare (2008 and 2011), Caudill and Bucklin (2004) and da Costa et al. (2014). We hypothesize that the Gulf of Mexico was likely refuge for A. tonsa during glacial cycles and therefore contains older lineages that are basal to those found in the Northwest Atlantic. To test this hypothesis, we first resolve the phylogenetic status of A. tonsa, by reconstructing the phylogeny of the genus based on mtCOI sequences from all species of Acartia available in GenBank and from newly collected specimens from the Texas Gulf of Mexico. Then, we examine the genetic variation and phylogeographic structure of A. tonsa through phylogenetic and haplotype network analyses. Though additional markers, specially from nuclear regions, would undoubtedly help in elucidating the phylogeny and phylogeography of this species, we focus on mtCOI based on its utility as discussed above and because of the vast majority of available sequences from previous research have also used this marker. Focusing on this marker not only maximizes the number of sequences used in our analyses but also our taxonomic and geographic coverage.
Materials and methods
Sample collection
Acartia tonsa specimens were collected from July to November 2015 using a 300-μm simple plankton net either by hand tows from a fishing pier or by towing from a kayak (Fig. 1 and Supplemental Table 1); three 100-m tows were carried out at each of six sites to ensure at least ten A. tonsa specimens were collected. Specimens were immediately preserved in 95% ethanol in the field. Sorting and identification of A. tonsa individuals were accomplished using morphological analysis with the aid of dissecting and compound microscopes.

Distribution of specimens analyzed labeled by region: NW Atlantic (northern coast), NW Atlantic (southern coast), eastern Gulf of Mexico, western Gulf of Mexico, and Brazil (Europe and Pacific regions not shown). Arrows show major circulation patterns. Insert shows sampling sites in the Texas Gulf of Mexico, n denotes the number of specimens sequenced from each site
DNA extraction, PCR amplification, and sequencing
In the laboratory, A. tonsa individuals were rehydrated in molecular-grade water for 30 min, this ensures the removal of ethanol from the specimen. Genomic DNA was extracted by placing individuals in a 2-ml tube with 100 μl of Bio-Rad’s Instagene Matrix. The specimens were then placed in a thermomixer and incubated at 56 °C overnight. After incubation, samples were heated to 100 °C for 8 min. Samples were then centrifuged for 1 min at 10,000g. The supernatant containing the DNA was removed with a pipette and placed in a new 2-ml tube and were stored in a − 20 °C freezer until further use. Quantification of extracted DNA was carried out using Qubit fluorometer (Invitrogen) set to OD260. Samples containing at least 0.1 ng/μl of DNA underwent polymerase chain reaction (PCR) to amplify a ~ 658 base pair (bp) fragment of the mtCOI gene with consensus primers LCO1490 (5′-GGTCAACAAATCATAAAGATATTGG-3′) and HCO2198 (5′-TAAACTTCAGGGTGACCAAAAAATCA-3′) (Folmer et al. 1994). Polymerase chain reaction was carried out in a 25-μl reaction volume containing 7.55 μl PCR-grade water, Invitrogen’s 10X PCR Rxn Buffer (2.0 μl), 1.25 μl Invitrogen’s 50 mM MgCl2, 2.0 μl of 10 mM dNTP, 1.0 μl of 10 mM of each primer (LCO1490 and HCO2198), 0.2 μl Thermo Fisher’s Invitrogen Platinum TAQ DNA Polymerase, and 10.0 μl DNA. The following thermocycler conditions were employed: 94 °C for 2 min, followed by 40 cycles of 94 °C for 1 min, 46 °C for 1 min, 72 °C for 1.5 min, followed by a final elongation at 72 °C for one additional minute.
PCR products were visualized by electrophoresis on a 1% agarose TBE based gel followed by staining with 0.2 μg/mL ethidium bromide. Life Technology’s Invitrogen 1 KB Plus DNA ladder (1.0 μg/μl) was used with each gel to determine if a DNA band of the expected size (~ 700 bp) was amplified and to make sure other bands were not present. The PCR products with a single band of ~ 700 bp were purified using Sigma-Aldrich’s GenElute PCR clean-up kit. Purified PCR products were sequenced by Sanger sequencing technique by Eurofins MWG Operon LLC with the forward and reverse primers.
Phylogenetic analyses
For each specimen, the sequences for the forward and reverse strands were assembled with the software CLC Workbench 7.9.1 (CLC Bio, Aarhus, Denmark) using the settings: minimum aligned read length = 500 bp, alignment stringency = high, conflicts = ambiguity nucleotides, trim sequence ends, and trim using quality scores limit = 0.05 (note that assembler automatically generates a reverse complement sequence for the HCO2198 strand before assembly). Chromatograms were visually inspected for ambiguous bases between the two strands and conflicts were resolved manually. A cutoff was used where only bases with Phred scores of 20 or more were kept. A consensus sequence was generated from each assembly. The individual A. tonsa mtCOI sequences obtained from the Texas GOM, and sequences downloaded from GenBank (National Center for Biotechnology Information, NCBI, https://www.ncbi.nlm.nih.gov/genbank/) (see Supplemental Table 2) were aligned with MUSCLE v3.8 (Edgar 2004) using default parameters and visually inspected for consistency. This alignment was imported to the software dnaSP v5 (Librado and Rozas 2009) which was used to generate a haplotype list in NEXUS format with gaps and missing sites considered and invariable sites included.
To reconcile the taxonomic issues with A. tonsa noted from previous research (e.g., Blanco-Bercial et al. 2014; Gomes et al. 2018; Milligan et al. 2011), all A. tonsa haplotypes were aligned with additional sequences of mtCOI from all other species of Acartia available in GenBank with MUSCLE v3.8 (Supplemental alignment text file). This multi-species alignment was used in phylogenetic analyses using maximum-likelihood (ML) and Bayesian methods. Partitioned Maximum Likelihood analyses were done using Partition Finder v1.1.1 (Lanfear et al. 2014) and RAxML v8.0.0 (Stamatakis 2017). Blocks were defined by codon position and Partition Finder found two partitions: The first partition was for codon positions one and two and the second partition was for the third codon position. The best fit substitution model to use with RAxML was determined to be GTR + I + G. For the Bayesian analyses, MrModeltest 2.3 (Nylander 2004) determined the best fit substitution model to be the HKY + I + G. This model was implemented for the Bayesian analyses using Mr. Bayes 3.2 (Ronquist et al. 2012). The two chains were carried out for 1,000,000 generations, sampling every 500th generation. After inspecting the trace files generated by the Bayesian Markov Chain Monte Carlo (MCMC) runs, the initial 25% (2500) of sampled generations were omitted prior to building the consensus tree. Both phylogenies were rooted in Temora stylifera (GenBank accession number DQ889132) as an outgroup.
The phylogenetic reconstruction of the genus Acartia was inspected for possible paraphyly and misidentifications, which are noted in the “Results” and “Discussion.” The aim was to only retain sequences of Acartia tonsa and those of closely related species. The reduced data set included all sequences of Acartia hudsonica and Acartia lilljeborgi that formed clades with our haplotypes. One haplotype was removed, haplotype 7, because it is likely a distantly related species of Acartia, see “Results” and “Discussion”). This reduced data set was aligned and phylogenetic reconstruction was performed using the same methods as described above. Percent divergence between clades was calculated by summing the branch lengths of the Bayesian tree from the phylogenetic analysis. The branch lengths illustrate the model-adjusted differences between each branch.
A haplotype network analysis was performed using only haplotypes that were clearly A. tonsa, which resulted in the removal of haplotypes 1–6 and 10–24 as they are likely different species (see “Results” and “Discussion”). The haplotype network analyses were performed with PopArt v 1.7.2 (Leigh and Bryant 2015) with an epsilon of zero using a median-joining network that infers ancestral nodes by iteratively adding median sequence vectors (Leigh and Bryant 2015). By using inferred ancestors, the PopArt software deduces relationships between haplotypes and provides a straightforward visual representation of those relationships. Because the software PopArt ignores positions with missing data, the alignment was reduced to haplotypes that have at least 557 base pairs. Therefore, the following five haplotypes were removed from the network analyses: haplotypes 26 and 28 (522 bp), haplotype 30 (451 bp), haplotype 31 (473 bp), and haplotype 74 (291 bp). These haplotypes were not excluded from the phylogenetic analyses. A second haplotype network analysis was performed just using the haplotypes from the Texas GOM. It was necessary to take this approach because the data from GenBank did not contain the frequency distribution of haplotypes. Therefore, any network analyses using GenBank data are strictly based on the presence/absence of haplotypes. Our data from the Texas GOM has the specific number of individuals per haplotype so the size of the nodes in the network is weighted by the frequency of individuals occurring in each haplotype.
Results
Phylogeny of the genus Acartia
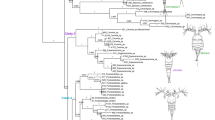
In total, 63 A. tonsa mtCOI sequences were obtained from the Texas GOM, and 321 sequences of A. tonsa were downloaded from GenBank totaling 384 sequences assigned to 195 haplotypes, 38 of which are present in the Texas GOM. The final alignment of 195 haplotypes was 557 bp long. An additional 215 sequences of mtCOI from all other species of Acartia in GenBank were added to this haplotype alignment for the phylogenetic reconstruction of the genus. Both ML and Bayesian methods resulted in similar trees with some minor differences as noted below (Fig. 2).

Phylogenetic reconstruction of the genus Acartia. The best ML tree is shown. Branch values indicate bootstrap support. Branches with bootstrap values less than 60 collapsed. Clades containing haplotypes from the present study shown in color, others are shown in black. In pink, blue, orange, and green are clades of uncertain taxonomy, which are resolved in subsequent analyses. In red are clades clearly belonging to Acartia tonsa. Tip labels for clades in color indicate species names, haplotype, source of sequences (GenBank or present study), and geographic source of specimen. Tip labels for clades in black only show species names and the number of sequences. All sequences belonging to clades shown in black were obtained from GenBank
The phylogenetic tree shows that there are 31 major clades with strong statistical support (Fig. 2). Relationships between these clades are unresolved, with low bootstrap and posterior probabilities (< 50 and < 0.5). Clades that contain haplotypes of A. tonsa are shown in color, while those that do not are shown in black (Fig. 2). Most of the major clades (23) contain single species: Acartia negligens Dana, 1849, Acartia danae Giesbrecht, 1889, Acartia steueri Smirnov, 1936, Acartia southwelli Sewell, 1914, Acartia tsuensis Ito, 1956, Acartia forticrusa Soh, Moon, Park, Bun & Maran, 2013, Acartia bispinosa Carl, 1907, Acartia fossae Gurney, 1927, Acartia discaudata Giesbrecht, 1881, Acartia margalefi Alcaraz, 1976, Acartia spinicauda Giesbrecht, 1889, Acartia erythraea Giesbrecht, 1889, Acartia pacifica Steuer, 1915, 2 clades of Acartia clausi Giesbrecht, 1889, 2 clades of Acartia bifilosa Giesbrecht, 1881 and 6 clades of A. tonsa (Haplotypes 8–9, 32–34, 10–19, 20–24, 35–73, and 114–193). The remaining 8 contain more than two species each, with unresolved internal branching, listed as they appear in clockwise order starting at the Temora stylifera Dana, 1849, root (Fig. 2): (1) 3 sequences of A. pacifica with 7 sequences of Acartia othsukai Ueda & Bucklin, 2006; (2) 1 sequence of A. erythrea with 2 sequences of Acartia amboinensis Carl, 1907; (3) 2 sequences of Acartia omori Bradford, 1976, with 7 sequences of A. clausi and 2 sequences of A. hudsonica; (4) 3 sequences of Acartia californiensis Trinast, 1976, with 1 sequence of Acartia longiremis Liljeborg, 1852; (5) 4 sequences of A. californiensis with 1 sequence of A. hudsonica and 2 sequences of A. tonsa (Haplotype 7); (6) 11 sequences of A. hudsonica with 4 sequences of A. tonsa (Haplotypes 2–5); (7) 1 sequence of A. hudsonica with 9 sequences of A. tonsa (Haplotypes 25–31); 8) 2 sequences of A. hudsonica with 184 sequences of A. tonsa (Haplotypes 74–113). Note that five of these 8 multispecies clades involve A. hudsonica.
Phylogeny of Acartia tonsa
The phylogenetic reconstruction of all haplotypes shows two major lineages (Fig. 3), one with Haplotype 6 as sister to A. hudsonica (in blue) and the other with A. lilljeborgi (in green) as sister to A. tonsa (in red). These two lineages are weakly supported, 0.88 pp and < 50 bs for the former and 0.92 pp and < 50 bs for the latter. The clades for A. hudsonica and A. lilljeborgi have high support (1 pp and 100 bs) while the clade for A. tonsa is weakly supported (88 pp and < 50 bs). Within the A. tonsa lineage, there are 5 well-supported clades (0.9–1 pp and 76–100 bs), but relationships between these 5 clades are weakly supported (0.57–0.88 pp and < 50–76 bs). We refer to the A. hudsonica clade as clade I, the A. lilljeborgi clade as clade II and the 5 well-supported A. tonsa clades as clades III–VII. Model-adjusted divergences between these clades, as calculated by summing the branch lengths of the Bayesian tree, range from 0.10 to 0.45 (Table 1).

Phylogenetic reconstruction of Acartia tonsa species complex, including A. hudsonica and A. lilljeborgi, with A. dana as an outgroup. Tree based on mtCOI and inferred by Bayesian methods. Tree topology inferred by ML methods is similar. Branch values show posterior probabilities and bootstrap statistics. * represents bootstrap values < 0.50. Tip labels indicate species names and haplotypes. Internal branching of seven major clades (I–VII) collapsed. In blue, clade I representing A. hudsonica, in green clade II representing A. lilljeborgi. In red clades III–VII representing A. tonsa species complex. In parenthesis lineages as defined by Chen and Hare (2011)
Clade I (shown in blue, Fig. 3), contains Haplotypes 2–5 from the NW Atlantic and 11 sequences of Acartia hudsonica, also from the NW Atlantic. Clade II (shown in green, Fig. 3), has one branch that contains Haplotypes 10–19 of A. lilljeborgi from Brazil (these are deposited as A. tonsa in GenBank, but see correction by Gomes et al. 2018 and discussion in present study) that is sister to a branch that contains Haplotypes 1 and 20–24 from Port Mansfield, Texas GOM. Clade III (Figs. 3 and 4), contains lineage S as defined by Chen and Hare (2008). Clade III has 16 haplotypes from the Texas GOM (Bahia Grande, Port Aransas, and Galveston Bay) and 23 haplotypes from the NW Atlantic (Fig. 4). The basal lineage of clade III is a single haplotype from the NW Atlantic. The relationships within clade III are largely unresolved but the median-joining network analysis shows three clear groups (Fig. 4). The first group has 17 haplotypes from the NW Atlantic and was previously defined as the S Clade (Chen and Hare 2008). The second group contains one haplotype from the NW Atlantic, three haplotypes from the Eastern GOM, and 12 haplotypes from Texas, with haplotype 55 sharing individuals from the Texas GOM and the eastern GOM. The third group has three haplotypes from the Texas GOM; haplotype 40 has two individuals, one from the Texas GOM and one from the NW Atlantic.

Phylogeographic analyses of Acartia tonsa species complex clade III (S). Colors represent the geographic source of specimens (see legend). a Phylogenetic tree based on mtCOI and inferred by Bayesian methods. Tree topology inferred by ML methods is similar. Branch values show posterior probabilities and bootstrap statistics. Branches with posterior probabilities < 0.50 collapsed. * represents bootstrap values < 0.50; b median-joining network based on the presence or absence of haplotypes by geographic region. The size of the nodes represents the number of regions where each haplotype is present. The small, black nodes represent an inferred ancestral haplotype. Notches on each branch represent the number of mutations between haplotypes
Clade III is basal to the remaining A. tonsa, which includes two branches, with two clades each. Clade IV sister to clade V and clade VI sisters to clade VII (Fig. 3). Clade IV (Fig. 5) has two lineages; one lineage contains clade L3 as defined by da Costa et al. (2014) with three haplotypes from the Brazilian coast and the other lineage has Haplotypes 8 and 9 from Baffin Bay, Texas GOM. Sister to clade IV, clade V (Fig. 5) consists of seven Haplotypes, 25–31, from Canada and New Jersey, along with a single sequence of A. hudsonica. The haplotype network supports the phylogenetic reconstruction (Fig. 5). The final branch in the phylogeny contains clades VI and VII. Clade VI corresponds to the X clade defined by Chen and Hare (2011), which includes 40 haplotypes (74–113) from the North-East Atlantic Coast and Europe. Two sequences of A. hudsonica are also found within this clade. No specimens from the Texas GOM belong to this clade, therefore our analyses have nothing new to contribute in regard to this clade beyond the work by Chen and Hare (2011). Sister to this clade is clade VII (Fig. 6) which contains the F clade as defined by Chen and Hare (2008 and 2011). Seven major lineages emerged within this clade; F1 through F5 were described by Chen and Hare (2008 and 2011) whereas lineages F6 and F7 are newly identified basal lineages in this group. Samples from the Texas GOM belong to the three basal lineages (F5, F6, and F7). The basal lineage, F7 splits into six haplotypes from the Texas GOM including; five from Port Mansfield, two from Bahia Grande, six from the Baffin Bay, three from Port Aransas, and two haplotypes from the Florida GOM (Fig. 6). The second branch splits into two main lineages; one contains F5 and F6 and the other contains F1 through F4. The newly defined clade F6 comprises haplotypes from the Texas GOM including ten haplotypes from Port Lavaca and two from Galveston Bay (Fig. 6). Clade F5 includes three haplotypes from the Texas GOM with three individuals from Bahia Grande and one individual from Port Lavaca (Fig. 6). Clade F5 also consists of four haplotypes from the South Florida east Atlantic coast, and one haplotype from Georgia and the North Florida East Coast. The next major split contains two well-supported clades; clade F4 and clades F1 through F3. Clade F4 branches into five haplotypes from South Florida East Coast, seven haplotypes from Georgia and the North Florida East Coast, and one haplotype from the North-East Atlantic Coast (Fig. 6). The last major lineage comprising clades F1 through F3 is well supported, but the relationships between clades F1-F3 are not well defined. All 44 haplotypes from clades F1 to F3 are from the NW Atlantic (Fig. 6). The median-joining network analysis shows comparable results.

Phylogeographic analyses of Acartia tonsa species complex clades IV and V. Colors represent the geographic source of specimens (see legend). a Phylogenetic tree based on mtCOI and inferred by Bayesian methods. Tree topology inferred by ML methods is similar. Branch values show posterior probabilities and bootstrap statistics. Branches with posterior probabilities < 0.50 collapsed. * represents bootstrap values < 0.50; b median-joining network based on the presence or absence of haplotypes by geographic region. The size of the nodes represents the number of regions where each haplotype is present. The small, black nodes represent an inferred ancestral haplotype. Notches on each branch represent the number of mutations between haplotypes

Phylogeographic analyses of Acartia tonsa species complex clade VII. Colors represent the geographic source of specimens (see legend). a Phylogenetic tree based on mtCOI and inferred by Bayesian methods. Tree topology inferred by ML methods is similar. Branch values show posterior probabilities and bootstrap statistics. Branches with posterior probabilities < 0.50 collapsed. * represents bootstrap values < 0.50; b median-joining network based on presence or absence of haplotypes by geographic region. The size of the nodes represents the number of regions where each haplotype is present. The small, black nodes represent an inferred ancestral haplotype. Notches on each branch represent the number of mutations between haplotypes
Texas GOM haplotype network analyses
The median-joining network with the samples sequenced from the Texas GOM shows seven distinct groups (Fig. 7). For this network analysis, the size of the nodes is representative of the haplotype frequencies. Bahia Grande has representatives from clade III (S) and clade VII (F5 and F7). It is the only sampling location with individuals belonging to the sub-group F5 within clade VII. Baffin Bay is the only Texas site sampled with individuals belonging to clade IV and has representatives from clade VII (F7). Port Mansfield samples revealed the only individuals belonging to clade II which are recognized as Acartia lilljeborgi, and it has individuals from clade VII (F6 and F7). Port Lavaca only contained individuals from clade VII (F6 and F7). Port Aransas has one individual belonging to haplotype 6, the phylogeny showed this haplotype as sister to A. hudsonica. Port Aransas also has individuals from clade III (S) and clade VII (F7). Galveston Bay has individuals within clade III (S) and clade V (F6). The most diverse is clade VII with representatives from all six sampling sites contained within three of its subgroups (F5, F6, and F7).

Median-joining network analysis of the haplotypes of Acartia tonsa species complex found in the Texas GOM based on mtCOI. The color of each node represents the geographic source of the specimen (see legend). Seven major groups are indicated by black circles labeled with corresponding clades from the phylogenetic analyses. The size of the node reflects the haplotype frequency. Black nodes represent an inferred ancestor. Notches on each branch represent the number of mutations between haplotypes. Note that according to our phylogenetic analyses, clade II corresponds to A. lilljeborgi and haplotype 6 is a sister clade to clade I which corresponds to A. hudsonica
Discussion
Genus Acartia: Phylogeny, taxonomy, and misidentification
The phylogenetic tree for the genus Acartia based on mtCOI shows 30 major clades (Fig. 2). The relationship between these branches is not supported, demonstrating that mtCOI cannot resolve phylogenetic relationships for this genus, but it does support the monophyly of many species. Of the 24 species in this phylogeny, 13 are monophyletic: A. negligens, A. dana, A. steueri, A. southwelli, A. tsuensis, A. forticrusa, A. bispinosa, A. fossae, A. discaudata, A. margalefi, A. spinicauda, A. levequei, and A. jilleti. The first 11 comprise 12 major clades while the last 2 have a single specimen each and form independent branches (Fig. 2). In addition to these, two clades appear to be paraphyletic but, upon further analyses (see discussion below of Acartia tonsa species complex), it is clear that the paraphyly is due to misidentifications. One clade belongs to A. hudsonica (shown in blue, Fig. 2) and the other to A. lilljeborgi (shown in green, Fig. 2). There are 4 clades that split two species into two clades each, 2 independent clades of A. clausi, and 2 independent clades of A. bifilosa. The clade of A. clausi with 18 sequences is all from samples in the Mediterranean and Black Sea (Fig. 2). The clade of A. clausi with 19 sequences is all from the North Sea (NE Atlantic, Fig. 2). It is clearly apparent that these are two clades of A. clausi with different geographic affinities. A similar scenario occurs with A. bifilosa, where the clade with 7 sequences are all samples from the North Sea (NE Atlantic) while the one with 9 sequences is all from China in the Western Pacific (Fig. 2).
Another species with multiple clades is Acartia californiensis split into three independent clades (Fig. 2). One clade has 21 sequences from Southern California and Baja California. The other two clades have a mix of A. californiensis with other species. One has 3 sequences of A. californiensis with one A. longiremis and 6 unidentified species. There is no taxonomic or geographic commonality in this clade. The A. californiensis are all from Oregon, but the unidentified species are from the NW Altantic, while the single sequence of A. longiremis is from the North Sea (NE Atlantic). Therefore, the taxonomy of this clade remains uncertain. However, the third clade contains 4 sequences of A. californiensis from San Francisco Bay, along with one sequence of A. hudsonica (KC287254), also from San Francisco Bay, and two sequences of A. tonsa (KC287386 and KC287387) from San Diego (sole members of haplotype 7 in the present study). In the ML tree, this clade is sister to A. jilleti (from Australia), while in the Bayesian tree (not shown) this clade is sister to A. levequei (from the Galapagos Islands). According to our phylogeny, these sequences of dubious taxonomy are not closely related to the other clades containing A. tonsa. They are likely a unique species from the Eastern Pacific. The commonality within this clade is geographic, all sequences are from bays in southern California. It seems that the single A. hudsonica sequence and the two A. tonsa sequences are a result of misidentifications and that this clade belongs to a closely related or subspecies of A. californiensis (Table 2).
Three additional clades have multiple species, which appear to be a result of incorrect species attribution to sequences obtained from community samples. One of these involves 7 sequences of A. clausi (EU599493-EU599499) and 2 sequences of A. hudsonica (HM045294) from unpublished work on molecular identification of copepod species from Chinese coastal waters (Fig. 2). These sequences form a strongly supported clade with two sequences of A. omori from Korean waters (AY145426 and JX98226) the latter, according to its GenBank record, is based on unpublished morphological analyses. Given that the 7 sequences of A. clausi and the 2 sequences of A. hudsonica are a result of molecular identification alone, it is very likely that they were assigned an incorrect taxonomy and should be attributed to species of A. omori (Table 2). Another major clade with the same issue includes 3 sequences of A. pacifica (EU599509, DQ665251 and DQ665252) embedded with 7 sequences of A. othsukai and 1 sequence of an unidentified Acartia (Fig. 2). The sequences derived from A. ohtsukai include a morphological analysis, while those assigned to A. pacifica are strictly from molecular identification. This is another case of sequence attribution to the incorrect species (Table 2). The third clade with the same taxonomic issues involves one sequence of A. erythrea (HM045353) from molecular identification, forming a clade with two sequences of A. amboinensis. The GenBank records for the A. amboinensis sequences state that taxonomic work was included with the molecular work, therefore, it is likely that the sequence attributed to A. erythrea belongs to A. amboinensis (Table 2).
The remaining 6 major clades (4 in red and 2 in orange, Fig. 2) contain haplotype lineages of A. tonsa. There are 3 sequences of A. hudsonica (KC287251-KC287253) distributed in two of these clades, clade V and clade VI. These three sequences of A. hudsonica are mixed with almost 400 sequences of A. tonsa. It would be a poor argument to say that the presence of A. hudsonica makes these two A. tonsa clades paraphyletic. Clade V contains a single sequence, downloaded from GenBank, attributed to A. hudsonica (KC287253). This sequence was generated from a specimen collected in the same region in the NW Atlantic as the specimens of A. tonsa that make up the rest of the clade V (Haplotypes 25–31). Clade VI contains two sequences attributed to A. hudsonica (KC287251 and KC287252), both downloaded from GenBank, obtained from specimens collected in the NW Atlantic. These two sequences are embedded with over 200 sequences of A. tonsa, primarily from the NW Atlantic (some are from Europe). The most likely scenario is that these three sequences are incorrectly attributed to A. hudsonica, when in fact they are from A. tonsa (Table 2).
We have discussed how Acartia hudsonica is a problematic taxon not just for A. tonsa, but for clades of several other species. The clades where these few sequences of A. hudsonica appear within do not have a taxonomic pattern, the sequences are interspersed with random taxa. The only commonality between these A. hudsonica sequences and the sequences for the other species that they form a clade with is the region where the specimens were collected. This taxonomic randomness but geographic coherence suggests that they are not a case of paraphyly but a case of misidentification (Table 2). Additional work with other genetic markers that includes more taxa and specimens from additional regions would help elucidate the phylogenetic relationships of this genus. Nevertheless, mtCOI does prove useful in recovering the monophyly of most Acartia species and it proves to be a good marker for determining potential species misidentifications. Based on our phylogenetic analyses, we urge the authors of the GenBank sequences with dubious taxonomic placement to check their data; and in the absence of clear morphological observations, to either change the species attribution in their GenBank record to an unidentified Acartia (e.g., Acartia sp.) or to follow the taxonomic recommendations based on our phylogenetic reconstruction (Table 2).
Acartia tonsa species complex
Genetic variation of mtCOI between species has been shown useful to distinguish closely related species from one another (e.g., Bucklin et al. 2003, 2016; Blanco-Bercial et al. 2014). The mtCOI sequence divergence between species of calanoid copepods ranges from 9 to 25% (Bucklin et al. 1996, 1999, 2001, 2003; Hill et al. 2001; Blanco-Bercial et al. 2014; Hirai et al. 2015). Bucklin et al. (2003) sequenced mtCOI for 34 calanoid copepod species and found that mtCOI sequence divergence between genera ranged from 12 to 25%. Congeneric species that are very similar or morphologically indistinguishable (cryptic species) can display significant mitochondrial genes sequence divergence (Hill et al. 2001). For example, mtCOI sequence divergence ranged between 8 and 12% among 18 calanoid copepod species of six genera (Bucklin et al. 1999). Similarly, Hirai et al. (2015) found 18 divergent lineages of Pleuromamma abdominalis that they hypothesized to be 18 distinct species based on percent divergences ranging from 4.3–28.6%. The phylogenetic reconstruction of our haplotype data along with sequences of A. hudsonica and A. lilljeborgi show divergences between major clades that are consistent with the upper range of the divergences found for interspecific calanoid copepods across different genera (Fig. 3 and Table 1). Clade I is comprised of sequences from specimens collected in the NW Atlantic that have been attributed to both, A. hudsonica and A. tonsa. We believe that these A. tonsa specimens were misidentified and that clade I corresponds to A. hudsonica (Figs. 2 and 3). There are several lines of evidence that support our claim. Clade I is strongly supported and deeply diverging, with model-corrected per site divergence from clades II-VII ranging between 0.30 and 0.45 (Table 1 and Fig. 3). Of the 17 sequences of A. hudsonica deposited in GenBank, 11 of them form part of this clade. The other 6 are interspersed across 4 different clades with different species as discussed in the previous section (Fig. 2). Additionally, the 4 sequences of A. tonsa within this clade come from specimens collected near the same locality as the A. hudsonica specimens. The most likely scenario is not a case of paraphyly, but a case of misidentification (Table 2). Sister to clade I is a single specimen of Acartia collected in Texas that corresponds to Haplotype 6 (Fig. 3). Given the phylogenetic position of Haplotype 6 and the degree of genetic differentiation, it is highly unlikely that this specimen belongs to A. tonsa. Haplotype 6 is most likely a sibling species (or subspecies) of A. hudsonica. Further specimen collection along with morphological analyses are needed to reach a definite conclusion.
Clade II, which belongs to A. lilljeborgi, ranges in divergence from other clades from 0.16 to 0.27 (Fig. 3). This is higher than the average divergence for calanoid copepods as discussed above. Within this clade are two lineages, one from Brazil and one from Texas (Fig. 3). The assertion that this clade belongs to A. lilljeborgi is largely due to research by Gomes et al. (2018). In their research, Gomes et al. (2018) suggest that the sequences of the Brazilian lineage in clade II that were deposited in GenBank as A. tonsa by da Costa et al. (2014) are in fact, Acartia lilljeborgi. Unfortunately, Gomes et al. (2018) do not provide accession numbers for their mtCOI sequences, and a search for A. lilljeborgi in GenBank returns zero results; but their findings can be analyzed with respect to our work based on the cladding of their sequences with those of da Costa et al.’s (2014). Gomes et al. (2018) carefully identified specimens of Acartia collected from the Brazilian coast as either A. lilljeborgi or A. tonsa. The phylogeny presented is based solely on sequences from Brazilian specimens including those generated by da Costa et al. (2014), and it shows two separate, monophyletic clades, one including their specimens of A. lilljeborgi with da Costa et al.’s (2014) lineages I and II and the other including their specimens of A. tonsa with da Costa et al.’s (2014) lineage III. Their main conclusion is that da Costa et al. (2014) misidentified their specimens of lineages I and II as A. tonsa when in fact they are A. lilljeborgi. Their argument is quite convincing and since our clade II consists of da Costa et al.’s (2014) lineages I and II identified by Gomes et al. (2018) as A. lilljeborgi in a sister relationship with a clade of seven haplotypes from Texas (Haplotypes 1 and 20–24, Figs. 2 and 3), it suggests that we have also misidentified specimens belonging to these haplotypes as being A. tonsa and therefore, must conclude that the strongly supported and monophyletic clade II is indeed A. lilljeborgi.
Acartia lilljeborgi is a sister to the A. tonsa species complex that includes clades III–VII (Fig. 3). Clades III–VII are strongly supported and deeply diverging. The model-corrected divergence ranges from 0.10 to 0.24. Because divergences among clades III–VII are within or above the range described for congeneric divergences in this study and others (e.g., Bucklin et al. 1999, 2001, 2003; Hill et al. 2001; Hirai et al. 2015), each clade should be considered a distinct species based on the evolutionary species concept; where species can be identified based on their level of genetic differences (Hill et al. 2001). A recent study by Plough et al. (2018) supports this conclusion. Their study focused on A. tonsa specimens from Chesapeake Bay belonging to Chen and Hare’s (2008) S and F lineage (corresponding to clades III and VII in our study). Their research shows reproductive isolation between these lineages by an extremely reduced hatching rate from between-lineage crosses, leading to an almost complete lack of nauplii production. They also report significant morphological and chemical differences, with members of the F clade being 13–24% shorter and having 70% less carbon than those belonging to the S clade. Future work using specimens from other lineages for crossbreeding, morphological, and biochemical analyses is likely to provide further evidence to elevate these various clades to species status.
Making such distinctions are essential before undertaking any population-specific study on A. tonsa. For example, a recent study by Sasaki and Dam (2019) investigated patterns of thermal tolerance and phenotypic plasticity in A. tonsa. They collected specimens of A. tonsa from the Northwest Atlantic (north of 40° N) and Florida (south of 30° N) including coasts in the Atlantic and the Gulf of Mexico. They included a haplotype analysis based on mtCOI, (unfortunately their sequences have not been made available and therefore could not be directly compared to our findings). They show the presence of four major clades. This is in agreement with our observations where the four corresponding clades would presumably be clades III (S), V, VI (X), and VII (F), note that clade IV in our study is only found in Texas and Brazil, not sampled by Sasaki and Dam (2019). In their study, they analyze their data under the assumption that these clades are the same species and treat their samples from each location as a population. They show how the various mtCOI clades of Acartia are widespread with some sites sharing haplotypes and how thermal tolerance decreases at higher latitudes, while phenotypic plasticity increases. One of their main conclusions is that the large plasticity in A. tonsa results in the wide geographic distribution of this species in varying environmental conditions and that this, combined with high levels of genetic diversity increases migration success and reduces vulnerability to stressors such as climate change (Sasaki and Dam 2019). Evidence from the present study and previous research indicates that these lineages are not the same species (e.g., Caudill and Bucklin 2004; Chen and Hare 2008, 2011; Plough et al. 2018) and each has its own evolutionary history and likely has been influenced by different speciation processes. This means that instead of having a widespread geographic distribution of genotypes of the same species, there is an overlapping distribution of different species.
In the case of Sasaki and Dam (2019) if each of the four major lineages is analyzed independently as four distinct species, geographic structuring becomes readily apparent and estimated gene flow between the various locations would be reduced. For example, Sasaki and Dam (2019) show that the specimens from lower latitudes largely belong to lineages A and B which are infrequent in northern sites (absent in the case of lineage A in the 3 northernmost sites), with one and zero haplotypes shared between northern and southern sites respectively. Specimens from lineage D and C are mostly found in northern sites, they are only present in low numbers in one and two of the five southern sites respectively. This generates similar patterns as found in our present study (see discussion below on phylogeography of A. tonsa species complex) and that by Chen and Hare (2011) where analyses were divided by lineage resulting in high geographic genetic structuring. Assuming that the lineages in Sasaki and Dam (2019) represent unique species, it could be argued that A and B are species that evolved in a tropical climate and are therefore more tolerant to higher temperatures, while C and D are species that evolved in a temperate climate and therefore less tolerant to higher temperatures. If the lineages presented by Sasaki and Dam (2019) are analyzed as unique species, independent of each other, then the results would lead to a different interpretation. It is unlikely that the thermal tolerance spans the same range in each lineage as it does for all lineages combined. The more likely scenario is that each lineage has its own unique response. This is congruent with the results reported by Sasaki and Dam (2019), where they show a trend of increasing thermal tolerance with decreasing latitude, but the interpretation would differ, instead of having one species with wide-ranging environmental tolerance, there are multiple species with overlapping ranges that have varying and in some cases overlapping, environmental tolerances. Some may be more susceptible to climate change than others and the loss of one lineage means the loss of a species that cannot be replaced by the remaining lineages.
Therefore, future studies should treat A. tonsa as a species complex. Detailed phenotypic analyses of all the lineages are necessary to identify any morphological characteristics that may differ between the various clades. Performing crossbreeding experiments and studying biochemical properties and behavioral characteristics of these clades, similar to the work by Plough et al. (2018) will help to further elucidate their speciation and taxonomic status. Phylogenetic analyses using other markers, including nuclear regions, would also aid in these efforts.
Phylogeography of Acartia tonsa species complex
The phylogeographic patterns of clades III–VII of Acartia tonsa’s species complex differ and each lineage has a unique evolutionary history. The diversification within clade III (Fig. 4) which corresponds to Chen and Hare’s (2011) lineage S remains largely unresolved due to low posterior probabilities and bootstrap support, with most haplotypes from the NW Atlantic collapsing into single branches (Fig. 4). The haplotype network analyses do show some geographic structuring with two main groups, one in the NW Atlantic and another in the GOM, both with numerous haplotypes radiating from one central haplotype (Fig. 4). But any assertions on diversification patterns are obscured. Chen and Hare (2011) proposed that the lack of resolution in their S lineage was likely due to higher migration rates because of higher salinity tolerances of members of this lineage. More sampling and additional genetic markers may resolve these relationships and elucidate patterns of divergence within this clade.
The next clade in the phylogeny, clade IV has two lineages, one from Brazil and the other from the GOM (Fig. 5). The Brazilian lineage is based on sequences by da Costa et al. (2014) that were confirmed as belonging to A. tonsa by the comparative morphological study of Gomes et al. (2018). There are no representatives of this clade in the NW Atlantic. This suggests that this clade represents a strictly tropical member of the A. tonsa species complex. Its sister clade, clade V (Fig. 5), only contains haplotypes from one region, the NW Atlantic suggesting that this is a temperate member of the A. tonsa species complex. While clade V shows a restricted distribution, clade IV demonstrates a close phylogenetic relationship between two geographic regions, the Gulf of Mexico and Brazil. Zooplankton connectivity between the western GOM and Brazil is expected due to ocean circulation (Fig. 1), where the North Brazil Current flows northwest along the Brazilian coast, entering the Caribbean Sea in the southeast and becoming the main branch of the Caribbean Current (Chérubin and Richardson 2007). The Caribbean Current flows northwest, arriving at the Yucatan Channel and entering the Gulf of Mexico on the eastern side, where it becomes the Loop Current flowing north and contributing to eddies that travel to the western GOM (Alvera-Azcárate et al. 2009). This circulation pattern also helps explain the geographic distribution of the sister clades of A. lilljeborgi as discussed earlier, one from Brazil and one from the GOM. There are several examples of marine organisms that demonstrate close phylogenetic relationships between these two regions. For example, the night shark (Carcharhinus signatus) from the Western Atlantic has two major maternal lineages based on the mitochondrial control region; one lineage is prevalent in Brazil, while the other lineage is prevalent in the USA, including the Gulf of Mexico (Domingues et al. 2019). A phylogenetic analysis of Atlantic oyster species, based on mtCOI and mitochondrial 16 s ribosomal RNA revealed that the North American species of the American oyster Crassotrea virginica is sister to the north Brazilian species Crassotrea rhizophorae (Lazoski et al. 2011). Some species show even closer phylogenetic relationships between Brazil and the Gulf of Mexico. The swimming crabs Portunus vossi and Portunus spinimanus show no genetic differences between specimens collected in the GOM and those collected in Brazil, based on mitochondrial 16S rRNA (Mantelatto et al. 2007). These examples demonstrate that genetic connectivity between Brazil and the GOM can be found among marine organisms of wide-ranging motility and life history, from sessile to planktonic to nektonic.
The next diverging clade, Clade VI is composed of haplotypes exclusively from the NW Atlantic and Europe (Fig. 3). This clade corresponds to Chen and Hare’s (2011) lineage X. Chen and Hare (2011) suggest that the restricted distribution of lineage X (clade VI) to higher latitudes, missing from well-sampled, mid-Atlantic estuaries, points to a recent invasion and likely not an endemic to North America. They also suggest that the connection with Europe is from a secondary invasion and that the ultimate source of this group will remain unknown until more regions are sampled. The only contribution that we can add to their discussion of this lineage is that it is absent from the Texas GOM.
The clearest diversification pattern is observed within clade VII (Fig. 6). The diversification pattern of clade VII is the only one that provides support to our hypothesis that southern lineages are basal to NW Atlantic lineages. The observed pattern suggests a range of expansion and species divergence that follows the prevailing circulation pattern in the GOM and northwestern Atlantic (Fig. 1). Genetic structuring of copepods due to ocean circulation has been observed from regional to global scales (e.g., Goetze 2005; Blanco-Bercial and Bucklin 2016; Questel et al. 2016). Goetze (2005) shows how two species of copepods, Eucalanus spinifer, and Eucalanaus hyalinus have distinct populations across hemispheres due to retention within subtropical gyres and also demonstrates how in the southern hemisphere, E. hyalinus populations have greater connectivity across ocean basins due to transport by the Antarctic circumpolar current. In the North Pacific, a study by Questel et al. (2016) demonstrates how circulation patterns can explain the observed gene flow within populations of four species of the copepod Pseudocalanus. Their gene flow model demonstrates how in P. newmani, there is primarily a northward flow to the Pacific Arctic Region that reflects the dominant northward flow of water masses from the North Pacific, through the Bering Strait, into the Arctic. They also note how bidirectional gene flow in some species, such as P. acuspes and P. minutus may be maintained by seasonal changes in circulation.
The divergence and speciation pattern observed in clade VII of the A. tonsa species complex can also be explained by ocean circulation. In the northwestern GOM, circulation across the shelf varies spatially and seasonally (e.g., Xue et al. 2013). The southern region is on the Tamaulipas-Veracruz (TAVE) shelf. During spring and summer, circulation is to the north and during fall and winter circulation reverses to the south (Xue et al. 2013). In the northernmost region over the Louisiana-Texas (LATEX) shelf, circulation is dominated by currents flowing south during fall, winter, and spring, mostly associated with coastal-trapped waves (Rivas 2017). During summer the currents reverse and flow north, a consequence of the onset of an atmospheric high-pressure system over the eastern Gulf which drives an intensified wind stream over the western Gulf (Carrasco-Díaz et al. 2015). At the meeting point of the LATEX and TAVE shelfs and their associated circulation patterns, there is a confluence of currents in the spring; one current coming from the south and the other coming from the north. These spatially and seasonally varying currents provide a mechanism for multidirectional dispersal within the Texas coast with export to the eastern side of the GOM in the summer and to the southern GOM in the fall and winter. In the eastern GOM, circulation is dominated by the Loop Current, which is formed by the Caribbean Current, entering the GOM through the Yucatan channel (Oey et al. 2013). The Loop Current begins its flow northeast but changes direction as it approaches the continent and diverts southeast, around Florida, exiting through the straits of Florida and on to the northwestern Atlantic were it becomes the Gulf Stream, flowing north along the North American continent (Oey et al. 2013).
This geographic progression of the dominant circulation from the western GOM to the eastern GOM and finally north, up the North American coast, is matched by the phylogenetic diversification of Clade VII (Fig. 6), the basal lineage is found exclusively in the GOM and it is predominantly from the western GOM. The second diverging lineage has one clade that is exclusive to the western GOM and the other is present in both western and eastern GOM and on the southern coast of the NW Atlantic. The third diverging branch has basal branches with a representative from the eastern GOM, and southern coast of the NW Atlantic while the last set of diverging branches are exclusively found in the northern coast of the northwest Atlantic. The northward diversification pattern along the North American coast was already recognized by Chen and Hare (2011). Our data shows that the origin of these lineages lies within the Gulf of Mexico (Fig. 6). Chen and Hare (2008) describe the F clade as having an affinity for lower salinities (0.3–12 psu); but, our study shows that lineages that are basal to the F clade can survive in hypersaline waters in the Texas GOM (Supplementary Table 1) suggesting that the F clade originated in high salinity waters of the GOM and that the divergence and affinity to lower saline waters is a more recent one that only occurs in estuaries of the northeast Atlantic coast.
The pattern of phylogeographical structuring in A. tonsa’s species complex clade VII (Fig. 6) has been observed in many coastal organisms, including ctenophores (Bayha et al. 2015), amphipods (Kelly et al. 2006), barnacles (Govindarajan et al. 2015), squid (Herke and Foltz 2002) and others summarized in Soltis et al., (2006). This common pattern can be attributed to the southerly displacement and isolation of populations during the last glacial cycle in the Pleistocene epoch (Jacobson et al. 1987; Soltis et al. 2006) (Jacobson et al. 1987; Soltis et al. 2006). Bayha et al. (2015) looked at the worldwide phylogeographic patterns of the ctenophore M. leidyi using cytochrome oxidase b (cytb) and six micronuclear satellites. Both showed a phylogeographic separation at the Labrador current suggesting that endemic lineages of M. leidyi from North America’s Atlantic coast are kept separate by the collision of the warm Gulf Current flowing north and the cooler Labrador Current flowing south. Basal lineages of M. leidyi were found in the Florida GOM while newer lineages were identified in Cape Hatteras, North Carolina. This phylogenetic break at Cape Hatteras is likely due to organism’s inability to swim against the currents or their intolerance to thermal gradients (Bayha et al. 2015); and it is found in many other species including fish (Avise et al. 1987), crustaceans (Kelly et al. 2006; Chen and Hare 2011), and mollusks (Baker et al. 2008). Kelly et al. (2006) showed a divergence between southern and northern lineages of the amphipod Gammarus tigrinus in the mid-Atlantic supported with the congruence of COI and ITSI sequencing. Govindarajan et al. (2015) found a similar pattern in the wide-spread barnacle, Chthamalus fragilis. Based on COI, they revealed a distinct pattern of speciation between southern and northern sites ranging from Tampa Bay, Florida to Cape Cod, Massachusetts. Chen and Hare (2011) pointed to this break in the Cape Hatteras region by highlighting how F1–F3 lineages (in clade VII) are only present near the Chesapeake Bay while the F4 lineage (in clade VII) is found south of Cape Hatteras in the Carolinas and eastern GOM (Alabama). As in other species, this oceanographic barrier could be the explanation as to why newer lineages of A. tonsa are only found in the NW Atlantic and why no representatives from those newer lineages are present in the Texas GOM.
Another major phylogenetic break occurs where the eastern GOM meets the northwestern Atlantic. Chen and Hare (2011) show how the F5 lineage (in clade VII) is primarily found in the eastern GOM. To this lineage, our study adds a sister lineage, clade VII’s F6, only present in the western GOM (Fig. 6). Furthermore, the newly discovered basal lineage, F7, is primarily found in the western GOM with only two representatives from the eastern GOM. This phylogenetic break between the GOM and the northwest Atlantic is also present in clade III, which has two main mitochondrial groups composed of haplotypes radiating from two central haplotypes, one with all members from the NW Atlantic, and the other with all but one member from the GOM (Fig. 4). Finally, clade V, strictly form the NW Atlantic is sister to clade IV from the GOM and Brazil (Fig. 5). Avise (1992) highlighted the importance of this phylogenetic break by demonstrating a similar pattern in a wide range of marine taxa, including the horseshoe crab (Limulus polyphemus), American oyster (Crassostrea virginica), seaside sparrow (Ammodramus maritimus), diamondback terrapin (Malaclemys terrapin), toadfish (Opsanus tau and Opsanus beta), and black sea bass (Centropristis striata). Soltis et al. (2006) expand on this list to include an additional 23 taxa with a phylogenetic break between the GOM and NW Atlantic, which includes 11 species of fish, 6 species of mollusks, and 6 species of crustaceans. For some species, this phylogenetic break occurs exactly at the southern tip of Florida, while for others, it occurs on the northeast coast of Florida with the Gulf Stream responsible for the transport of GOM haplotypes into the southern coasts of the NW Atlantic (Avise 2000). This export of haplotypes from the GOM by the Gulf Stream would explain the presence of single haplotypes on the southern coast of the NW Atlantic from clades of the A. tonsa species complex that are otherwise exclusive to the GOM.
In summary, this study emphasizes the utility of mtCOI in discerning species of Acartia and it highlights numerous misidentifications of Acartia species deposited in GenBank. The incorrect taxonomy assigned in some of these sequences results in apparently paraphyletic relationships and therefore the taxonomic attribution of these sequences needs to be corrected. This study also demonstrates that A. tonsa is a species complex and presents new lineages found within the Texas GOM. Many of these are basal to NW Atlantic lineages. The geographic distribution of clades highlights two major phylogeographic breaks, one in the Atlantic coast of North America at about 35° N, near Cape Hatteras; and the other in Florida, at the border of the Gulf of Mexico and the NW Atlantic. One of the major clades in this Acartia tonsa species complex (clade VII) shows a clear pattern of divergence and speciation that follows the prevailing currents. Older lineages are found in the western Gulf of Mexico. Diversification follows the path of the Loop Current in the GOM going east. As the Loop Current joins the Gulf Stream and flows north along the North American continent, diversification also follows this same path with the youngest lineages found in the northern coast of the NW Atlantic. Phylogenetic connectivity is also observed between Brazil and the Gulf of Mexico. Ocean circulation provides a clear path of dispersal that can explain the observed sister clades between Brazil and the Gulf of Mexico, with the North Brazil current flowing up the coast of Brazil and into the Caribbean, becoming the Caribbean Current which then enters the Gulf of Mexico as the Loop Current. The results show that A. tonsa can be used as a model species for observing phylogeographical structuring of coastal plankton along the American continent. Further sampling in South America, the Caribbean, Mexico and along Pacific coastal estuaries and the use of additional genetic markers that include nuclear regions will provide more resolution for understanding phylogeographical patterns of the A. tonsa species complex.
References
Alcaraz M (1976) Description of Acartia margalefi, a new species of pelagic copepod, and its relationship with A. clausi. (Descripción de Acartia margalefi, una especie nueva de Copépodo pelagico, y su relación con A. clausi. Investigación Pesquera, Barcelona
Alvera-Azcárate A, Barth A, Weisberg RH (2009) The Surface Circulation of the Caribbean Sea and the Gulf of Mexico as Inferred from Satellite Altimetry. J Phys Oceanogr 39(3):640–57. https://doi.org/10.1175/2008JPO3765.1
Avise JC (2000) Phylogeography: the history and formation of species. Harvard University Press
Avise JC, Reeb CA, Saunders NC (1987) Geographic population structure and species differences in mitochondrial DNA of mouthbrooding marine catfishes (Ariidae) and demersal spawning toadfishes (Batrachoididae). Evolution 41:991–1002. https://doi.org/10.1111/j.1558-5646.1987.tb05872.x
Avise JC (1992) Molecular population structure and the biogeographic history of a regional fauna: A case history with lessons for conservation biology. Oikos 63(1):62–76. https://doi.org/10.2307/3545516
Baker P, Austin JD, Bowen BW, Baker SM (2008) Range-wide population structure and history of the northern quahog (Merceneria merceneria) inferred from mitochondrial DNA sequence data. ICES J Mar Sci 65:155–163. https://doi.org/10.1093/icesjms/fsn007
Bayha KM, Chang MH, Mariani CL et al (2015) Worldwide phylogeography of the invasive ctenophore Mnemiopsis leidyi (Ctenophora) based on nuclear and mitochondrial DNA data. Biol Invasions 17:827–850. https://doi.org/10.1007/s10530-014-0770-6
Bickford D, Lohman DJ, Sodhi NS et al (2007) Cryptic species as a window on diversity and conservation. Trends Ecol Evol 22:148–155. https://doi.org/10.1016/j.tree.2006.11.004
Blanco-Bercial L, Bucklin A (2016) New view of population genetics of zooplankton: RAD-seq analysis reveals population structure of the North Atlantic planktonic copepod Centropages typicus. Mol Ecol 25:1566–1580. https://doi.org/10.1111/mec.13581
Blanco-Bercial L, Cornils A, Copley N, Bucklin A (2014) DNA barcoding of marine copepods: assessment of analytical approaches to species identification. PLoS Curr
Bradford JM (1976) Partial revision of the Acartia subgenus Acartiura (Copepoda: Calanoida: Acartiidae). New Zealand Journal of Marine and Freshwater Research 10:159–202
Bucklin A, LaJeunesse TC, Curry E et al (1996) Molecular diversity of the copepod, Nannocalanus minor: genetic evidence of species and population structure in the North Atlantic Ocean. J Mar Res 54:285–310
Bucklin A, Guarnieri M, Hill RS et al (1999) Taxonomic and systematic assessment of planktonic copepods using mitochondrial COI sequence variation and competitive, species-specific PCR. In: Zehr JP, Voytek MA (eds) Molecular ecology of aquatic communities. Springer Netherlands, Dordrecht, pp 239–254
Bucklin A, Guarnieri M, McGillicuddy DJ, Sean Hill R (2001) Spring evolution of Pseudocalanus spp. abundance on Georges Bank based on molecular discrimination of P. moultoni and P. newmani. Deep-Sea Res II 48:589–608. https://doi.org/10.1016/S0967-0645(00)00128-4
Bucklin A, Frost BW, Bradford-Grieve J et al (2003) Molecular systematic and phylogenetic assessment of 34 calanoid copepod species of the Calanidae and Clausocalanidae. Mar Biol 142:333–343
Bucklin A, Lindeque PK, Rodriguez-Ezpeleta N et al (2016) Metabarcoding of marine zooplankton: prospects, progress and pitfalls. J Plankton Res 38:393–400
Carrasco-Díaz M, Rivas D, Orozco-Contreras M, Sánchez-Montante O (2015) An assessment of wind power potential along the coast of Tamaulipas, northeastern Mexico. Renew Energy 78:295–305. https://doi.org/10.1016/j.renene.2015.01.007
Carl J (1907) Copépodes d’Amboine. Revue Suisse de Zoologie:15:7–18
Caudill CC, Bucklin A (2004) Molecular phylogeography and evolutionary history of the estuarine copepod, Acartia tonsa, on the Northwest Atlantic coast. Hydrobiologia 511:91–102. https://doi.org/10.1023/B:HYDR.0000014032.05680.9d
Chan IZW, Chang JJM, Huang D, Todd PA (2019) Colour pattern measurements successfully differentiate two cryptic Onchidiidae Rafinesque, 1815 species. Mar Biodivers 49:1743–1750. https://doi.org/10.1007/s12526-019-00940-4
Chen G, Hare MP (2008) Cryptic ecological diversification of a planktonic estuarine copepod, Acartia tonsa. Mol Ecol 17:1451–1468. https://doi.org/10.1111/j.1365-294X.2007.03657.x
Chen G, Hare MP (2011) Cryptic diversity and comparative phylogeography of the estuarine copepod Acartia tonsa on the US Atlantic coast. Mol Ecol 20:2425–2441. https://doi.org/10.1111/j.1365-294X.2011.05079.x
Chérubin LM, Richardson PL (2007) Caribbean current variability and the influence of the Amazon and Orinoco freshwater plumes. Deep-Sea Res I 54:1451–1473. https://doi.org/10.1016/j.dsr.2007.04.021
Cornils A, Held C (2014) Evidence of cryptic and pseudocryptic speciation in the Paracalanus parvus species complex (Crustacea, Copepoda, Calanoida). Front Zool 11:19. https://doi.org/10.1186/1742-9994-11-19
da Costa KG, Vallinoto M, da Costa RM (2011) Molecular identification of a new cryptic species of Acartia tonsa (Copepoda, Acartiidae) from the northern coast of Brazil, based on mitochondrial COI gene sequences. J Coast Res:359–363
da Costa KG, Rodrigues Filho LFS, Costa RM et al (2014) Genetic variability of Acartia tonsa (Crustacea: Copepoda) on the Brazilian coast. J Plankton Res 36:1419–1422. https://doi.org/10.1093/plankt/fbu080
Dana JD (1846) Notice of some genera of Cyclopacea. Ann Mag Nat Hist 18:181–185
Dana JD (1849) United States Exploring Expedition: During the Years 1838, 1839, 1840, 1841, 1842 Under the Command of Charles Wilkes. Atlas zoophytes. C. Sherman
Dawson MN, Jacobs DK (2001) Molecular evidence for cryptic species of Aurelia aurita (Cnidaria, Scyphozoa). Biol Bull 200:92–96. https://doi.org/10.2307/1543089
Dippenaar SM, Mathibela RB, Bloomer P (2010) Cytochrome oxidase I sequences reveal possible cryptic diversity in the cosmopolitan symbiotic copepod Nesippus orientalis Heller, 1868 (Pandaridae: Siphonostomatoida) on elasmobranch hosts from the KwaZulu-Natal coast of South Africa. Exp Parasitol 125:42–50. https://doi.org/10.1016/j.exppara.2009.08.017
Domingues RR, Bruels CC, Gadig OBF et al (2019) Genetic connectivity and phylogeography of the night shark (Carcharhinus signatus) in the western Atlantic Ocean: implications for conservation management. Aquat Conserv Mar Freshw Ecosyst 29:102–114. https://doi.org/10.1002/aqc.2961
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Figueroa DF, McClure A, Figueroa NJ, Hicks DW (2019) Hiding in plain sight: invasive coral Tubastraea tagusensis (Scleractinia:Hexacorallia) in the Gulf of Mexico. Coral Reefs 38:395–403. https://doi.org/10.1007/s00338-019-01807-7
Folmer O, Black M, Hoen W et al (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metozoan invertebrates. Mol Mar Biol Biotechnol 3:294–299
Franco SC, Augustin CB, Geffen AJ, Dinis MT (2017) Growth, egg production and hatching success of Acartia tonsa cultured at high densities. Aquaculture 468:569–578. https://doi.org/10.1016/j.aquaculture.2016.10.044
Giesbrecht W (1881) Vorläufige Mitteilung aus einer Arbeit über die freilebenden Copepoden des Kieler Hafens. Zool Anz 4(83):254–258
Giesbrecht W (1889) Elenco dei Copepodi pelagici raccolti dal Tenente di vascello Gaetano Chierchia durante il viaggio della R. Corvetta ‘Vettor Pisani’negli anni 1882–1885 e dal Tenente di vascello Francesco Orsini nel Mar Rosso, nel 1884. Rendiconti delle Sedute della R. Accademia dei Lincei (4) 7 sem. Atti della Accademia Nazionale dei Lincei, Classe di Scienze Fisiche Matematiche e Naturali Rendiconti 7 (4) sem 1 474–481
Goetze E (2005) Global population genetic structure and biogeography of the oceanic copepods Eucalanus hyalinus and E. spinifer. Evolution 59:2378–2398. https://doi.org/10.1554/05-077.1
Gomes CM, da Costa KG, Ferreira GS et al (2018) Hidden diversity in cryptic Brazilian lineages of Acartia (Copepoda, Calanoida, Acartiidae). J Sea Res 141:21–25. https://doi.org/10.1016/j.seares.2018.08.005
Govender A, Groeneveld J, Singh S, Willows-Munro S (2019) The design and testing of mini-barcode markers in marine lobsters. PLoS ONE 14:e0210492. https://doi.org/10.1371/journal.pone.0210492
Govindarajan AF, Bukša F, Bockrath K et al (2015) Phylogeographic structure and northward range expansion in the barnacle Chthamalus fragilis. PeerJ 3:e926. https://doi.org/10.7717/peerj.926
Gurney R (1927) Report on the Crustacea Copepoda (littoral and semiparasitic). Zoological Results of the Cambridge Expedition to the Suez Canal, 1924, no. 35. Trans Zool Soc 22:451–577
Han S-Y, Kim J-K, Tashiro F et al (2019) Relative importance of ocean currents and fronts in population structures of marine fish: a lesson from the cryptic lineages of the Hippocampus mohnikei complex. Mar Biodivers 49:263–275. https://doi.org/10.1007/s12526-017-0792-2
Herke S, Foltz D (2002) Phylogeography of two squid (Loligo pealei and L. plei) in the Gulf of Mexico and northwestern Atlantic Ocean. Mar Biol 140:103–115. https://doi.org/10.1007/s002270100680
Hill R, Allen L, Bucklin A (2001) Multiplexed species-specific PCR protocol to discriminate four N. Atlantic Calanus species, with an mtCOI gene tree for ten Calanus species. Mar Biol 139:279–287. https://doi.org/10.1007/s002270100548
Hirai J, Tsuda A, Goetze E (2015) Extensive genetic diversity and endemism across the global range of the oceanic copepod Pleuromamma abdominalis. Prog Oceanogr 138:77–90. https://doi.org/10.1016/j.pocean.2015.09.002
Holland BS, Dawson MN, Crow GL, Hofmann DK (2004) Global phylogeography of Cassiopea (Scyphozoa: Rhizostomeae): molecular evidence for cryptic species and multiple invasions of the Hawaiian islands. Mar Biol 145:1119–1128. https://doi.org/10.1007/s00227-004-1409-4
Ito T (1956) Three new copepods from brackish-water lakes of Japan. Pac Sci 10(4):468–473
Hupało K, Teixeira MAL, Rewicz T et al (2019) Persistence of phylogeographic footprints helps to understand cryptic diversity detected in two marine amphipods widespread in the Mediterranean basin. Mol Phylogenetics Evol 132:53–66. https://doi.org/10.1016/j.ympev.2018.11.013
Jacobson GL, Webb T, Grimm EC (1987) Patterns and rates of vegetation change during the deglaciation of eastern North America. In: Ruddiman WF, Wright HE (eds) North America and adjacent oceans during the last deglaciation, vol 80301. Geological Society of America, Boulder, pp 277–288
Jepsen PM, Bjørbæk NS, Rayner TA et al (2017) Recommended feeding regime and light climate in live feed cultures of the calanoid copepod Acartia tonsa Dana. Aquacult Int 25:635–654. https://doi.org/10.1007/s10499-016-0063-4
Johnson SB, Warén A, Vrijenhoek RC (2008) DNA barcoding of Lepetodrilus limpets reveals cryptic species. J Shellfish Res 27:43–51. https://doi.org/10.2983/0730-8000(2008)27[43:DBOLLR]2.0.CO;2
Kelly DW, MacIsaac HJ, Heath DD (2006) Vicariance and dispersal effects on phylogeographic structure and speciation in a widespread estuarine invertebrate. Evolution 60:257–267. https://doi.org/10.1111/j.0014-3820.2006.tb01104.x
Krause KE, Dinh KV, Nielsen TG (2017) Increased tolerance to oil exposure by the cosmopolitan marine copepod Acartia tonsa. Sci Total Environ 607–608:87–94. https://doi.org/10.1016/j.scitotenv.2017.06.139
Lanfear R, Calcott B, Kainer D et al (2014) Selecting optimal partitioning schemes for phylogenomic datasets. BMC Evol Biol 14:82. https://doi.org/10.1186/1471-2148-14-82
Lazoski C, Gusmão J, Boudry P, Solé-Cava A (2011) Phylogeny and phylogeography of Atlantic oyster species: evolutionary history, limited genetic connectivity and isolation by distance. Mar Ecol Prog Ser 426:197–212. https://doi.org/10.3354/meps09035
Lee CE (2000) Global phylogeography of a cryptic copepod species complex and reproductive isolation between genetically proximate “populations.”. Evolution 54:2014–2027. https://doi.org/10.1111/j.0014-3820.2000.tb01245.x
Lee CE, Frost BW (2002) Morphological stasis in the Eurytemora affinis species complex (Copepoda: Temoridae). Hydrobiologia 480:111–128. https://doi.org/10.1023/A:1021293203512
Leigh JW, Bryant D (2015) popart : full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116. https://doi.org/10.1111/2041-210X.12410
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Liljeborg W (1852) Hafs-Crustaceer vid Kullaberg. Öfversigt af Kongliga Vetenskaps-Akademiens Förhandlingar 9:1–13
Mantelatto FL, Robles R, Felder DL (2007) Molecular phylogeny of the western Atlantic species of the genus Portunus (Crustacea, Brachyura, Portunidae). Zool J Linnean Soc 150:211–220. https://doi.org/10.1111/j.1096-3642.2007.00298.x
Marrone F, Brutto SL, Arculeo M (2010) Molecular evidence for the presence of cryptic evolutionary lineages in the freshwater copepod genus Hemidiaptomus G.O. Sars, 1903 (Calanoida, Diaptomidae). Hydrobiologia 644:115–125. https://doi.org/10.1007/s10750-010-0101-6
Mauchline J, Blaxter JHS, Southward AJ and Tyler PA 1998 The biology of calanoid copepods—introduction. Advances in marine biology [33rd Ed]. Elsevier Academic Press. USA
Milligan PJ, Stahl EA, Schizas NV, Turner JT (2011) Phylogeography of the copepod Acartia hudsonica in estuaries of the northeastern United States. Hydrobiologia 666:155–165. https://doi.org/10.1007/s10750-010-0097-y
Minh TTV, Hansen BW, Kiørboe T (2017) The constraints of high density production of the calanoid copepod Acartia tonsa Dana. J Plankton Res 39:1028–1039. https://doi.org/10.1093/plankt/fbx056
Nylander JAA (2004) MrModeltest v2. Program distributed by the author
Oey L-Y, Ezer T, Lee H-C (2013) Loop current, rings and related circulation in the Gulf of Mexico: a review of numerical models and future challenges. Geophys Monograph Am Geophys Union 161:31–56
Øie G, Galloway T, Sørøy M et al (2017) Effect of cultivated copepods (Acartia tonsa) in first-feeding of Atlantic cod (Gadus morhua) and ballan wrasse (Labrus bergylta) larvae. Aquac Nutr 23:3–17. https://doi.org/10.1111/anu.12352
Oyama H, Itoi S, Ueda H et al (2019) Genetic difference between African and Japanese scombropid populations based on cytochrome c oxidase subunit I gene sequences. Mitochondrial DNA B 4:1016–1020. https://doi.org/10.1080/23802359.2019.1584056
Pavlaki MD, Morgado RG, van Gestel CAM et al (2017) Influence of environmental conditions on the toxicokinetics of cadmium in the marine copepod Acartia tonsa. Ecotox Environ Safe 145:142–149. https://doi.org/10.1016/j.ecoenv.2017.07.008
Pinhey KF (1926) No. 6: Entomostraca of the Belle Isle Strait Expedition, 1923, with notes on other planktonic species: Part I. Contrib Can Biol Fish 3:179–233
Plough LV, Fitzgerald C, Plummer A, Pierson JJ (2018) Reproductive isolation and morphological divergence between cryptic lineages of the copepod Acartia tonsa in Chesapeake Bay. Mar Ecol Prog Ser 597:99–113. https://doi.org/10.3354/meps12569
Questel JM, Blanco-Bercial L, Hopcroft RR, Bucklin A (2016) Phylogeography and connectivity of the Pseudocalanus (Copepoda: Calanoida) species complex in the eastern North Pacific and the Pacific Arctic region. J Plankton Res 38:610–623. https://doi.org/10.1093/plankt/fbw025
Rahlff J, Peters J, Moyano M et al (2017) Short-term molecular and physiological responses to heat stress in neritic copepods Acartia tonsa and Eurytemora affinis. Comp Biochem Physiol A Mol Integr Physiol 203:348–358. https://doi.org/10.1016/j.cbpa.2016.11.001
Rivas D (2017) Wind-driven coastal-trapped waves off southern Tamaulipas and northern Veracruz, western Gulf of Mexico, during winter 2012–2013. Est Coast Shelf Sci 185:1–10. https://doi.org/10.1016/j.ecss.2016.12.002
Rocha-Olivares A, Fleeger JW, Foltz DW (2001) Decoupling of molecular and morphological evolution in deep lineages of a meiobenthic harpacticoid copepod. Mol Biol Evol 18:1088–1102. https://doi.org/10.1093/oxfordjournals.molbev.a003880
Ronquist F, Teslenko M, Van der M et al (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542
Sasaki MC, Dam HG (2019) Integrating patterns of thermal tolerance and phenotypic plasticity with population genetics to improve understanding of vulnerability to warming in a widespread copepod. Glob Change Biol 25:4147–4164. https://doi.org/10.1101/619775
Sewell R (1914) Notes on the surface Copepoda of the Gulf of Mannar. Spolia Zeylanica 191:pls–17
Smirnov SS (1936) Beschreibung einer neuen Acartia-Art aus dem Japanischen Meer nebst einigen Bemerkungen über die Untergattung Euacartia Steuer. Zool Anz 114:87–92
Soltis DE, Morris AB, McLachlan JS et al (2006) Comparative phylogeography of unglaciated eastern North America. Mol Ecol 15:4261–4293. https://doi.org/10.1111/j.1365-294X.2006.03061.x
Soh HY, Yong Moon S, Ok Park E, Venmathi Maran BA (2013) A New Species of Acartia subgenus Euacartia (Copepoda: Calanoida: Acartiidae) from korean estuaries based on morphological and molecular evidence. J Crustacean Biol 33:718–729
Stamatakis A (2017) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Steuer A (1915) 3. Revision der Gattung Acartia Dana. (Copepoden der Valdivia Expedition.). Zool Anz, 45(9):392–7
Trinast EM (1976) A preliminary note on Acartia californiensis, a new calanoid copepod from Newport Bay, California. Crustaceana 54–58
Ueda H, Bucklin AC (2006) Acartia (Odontacartia) ohtsukai, a new brackish-water calanoid copepod from Ariake Bay, Japan, with a redescription of the closely related A. pacifica from the Seto Inland Sea. Hydrobiologia 560:77–91
Werbrouck E, Tiselius P, Van Gansbeke D et al (2016) Temperature impact on the trophic transfer of fatty acids in the congeneric copepods Acartia tonsa and Acartia clausi. J Sea Res 112:41–48. https://doi.org/10.1016/j.seares.2016.03.001
Xue Z, He R, Fennel K et al (2013) Modeling ocean circulation and biogeochemical variability in the Gulf of Mexico. Biogeosciences 10:7219–7234. https://doi.org/10.5194/bg-10-7219-2013
Zhou C, Vitiello V, Casals E et al (2016) Toxicity of nickel in the marine calanoid copepod Acartia tonsa: nickel chloride versus nanoparticles. Aquat Toxicol 170:1–12. https://doi.org/10.1016/j.aquatox.2015.11.003
Acknowledgements
We would like to thank the anonymous reviewers whose suggestions helped improve and clarify this manuscript.
Funding
The research reported in this publication was supported by startup funds from the University of Texas Rio Grande Valley to Diego Figueroa and by the Gulf Research Program of the National Academies of Sciences, Engineering, and Medicine under the Grant Agreement number 2000007266 to Diego Figueroa. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Gulf Research Program or the National Academies of Sciences, Engineering, and Medicine.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed by the authors.
Sampling and field studies
All necessary permits for sampling and observational field studies were obtained by the authors from the competent authorities and are mentioned in the acknowledgments, if applicable.
Data availability
All sequences generated by this study have been deposited in GenBank – Accession numbers MK309279-MK309316 and MK310021-MK319927.
Author contribution statement
DFF conceived and designed research. NJF collected the specimens, generated the genetic data, and analyzed the data. DFF and DH supervised research and analyses. NJF and DFF wrote the manuscript. DFF, NJF, and DH contributed to the interpretation of data. All authors contributed critically to the drafts and gave final approval for publication.
Additional information
Communicated by R. R. Hopcroft
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Figueroa, N.J., Figueroa, D.F. & Hicks, D. Phylogeography of Acartia tonsa Dana, 1849 (Calanoida: Copepoda) and phylogenetic reconstruction of the genus Acartia Dana, 1846. Mar. Biodivers. 50, 23 (2020). https://doi.org/10.1007/s12526-020-01043-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12526-020-01043-1