
Abstract
Metal-mediated base pairs involving artificial nucleobases have emerged as a promising means for the site-specific functionalization of nucleic acids with metal ions. In this context, a GNA-appended (GNA: glycol nucleic acid) nucleoside analogue containing the artificial nucleobase 1H-imidazo[4,5-f][1,10]phenanthroline (P) has already been applied successfully in a variety of homo- and heteroleptic metal-mediated base pairs, mainly involving Ag(I) ions. Herein, we report a thorough investigation of the Hg(II)-binding properties of P when incorporated into antiparallel-stranded DNA duplexes. The artificial nucleobase P is able to form Hg(II)-mediated homoleptic base pairs of the type P–Hg(II)–P with a [2 + 2] coordination environment. In addition, the heteroleptic P–Hg(II)–T pair was investigated. The addition of a stoichiometric amount of Hg(II) to a duplex comprising either a P:P pair or a P:T pair stabilizes the DNA duplex by 4.3 °C and 14.5 °C, respectively. The P–Hg(II)–T base pair, hence, represents the most stabilizing non-organometallic Hg(II)-mediated base pair reported to date. The formation of the Hg(II)-mediated base pairs was investigated by means of temperature-dependent UV spectroscopy and CD spectroscopy.
Graphic abstract

Similar content being viewed by others
Introduction
Natural DNA is composed of four nucleobases and an anionic sugar-phosphate backbone. It forms a robust antiparallel-stranded double-helical structure based on self-assembly and molecular recognition. These unique properties render it an ideal building block in the context of supramolecular chemistry and DNA nanotechnology [1, 2]. The modular composition of DNA can be exploited to arrange metal ions within its helix in a predetermined fashion [3,4,5]. Historically, the interaction of transition metal ions with nucleic acids was first demonstrated using simple viscosimetry experiments of DNA solutions in the presence of various inorganic salts [6]. Subsequently, the discovery of the T–Hg(II)–T base pair pioneered the introduction of metal-based functionality into the nucleic acid scaffold (Fig. 1a) [7, 8]. The concept of applying nucleobases as ligands to locate metal ions inside a DNA duplex is nowadays referred to as metal-mediated base pairing. In such artificial base pairs, the natural hydrogen bonds between the complementary bases are formally replaced by metal–ligand coordinate bonds [9], leading to the desired site-specific functionalization. In fact, even duplexes consisting entirely of metal-mediated base pairs are feasible [10, 11]. Research on metal-mediated base pairing has not been restricted to natural nucleobases. It has rather been expanded by the introduction of artificial ligand-based nucleosides to bring in diversity in the form of site-specific functionalization [12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27], allowing the generation of different metal-induced DNA nanoarchitectures. A higher affinity of the artificial nucleosides towards a particular metal ion and the wide structural flexibility offered by them represent their largest advantages. Similarly, other nucleic acid topologies and nucleic acid analogues have been probed with respect to the site-specific incorporation of metal ions in analogy to metal-mediated base-pair formation [28,29,30,31]. Several promising applications have already been established in this area, including the construction of structures with a modified electrical response [32, 33], switchable devices [34], responsive devices [35, 36], regulated primer extension [37], the specific detection of canonical nucleobases [38, 39], the generation of DNA-templated metal nanoclusters [40], an expansion of genetic four-letter code [41], etc. Due to their accessibility from commercial resources, canonical nucleobases involved in metal-mediated base pairing have been most extensively studied, giving rise to C–Ag(I)–C and T–Hg(II)–T pairs [42]. Consequently, the first successful application of metal-mediated base pairing, namely a Hg(II)-sensor in water, was based on the well-established affinity of thymine towards Hg(II) [43]. Numerous structural studies resulted in a better understanding of the requirements for metal-mediated base-pair formation [9], indicating, for example, a structural flexibility particularly of base pairs involving monodentate ligands [44,45,46].

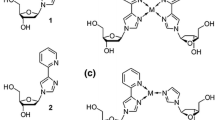
a Metal-mediated T–Hg(II)–T base pair, b artificial nucleoside analogue P ((S)-3-(1H-imidazo[4,5-f][1,10]phenanthrolin-1-yl)propane-1,2-diol), c model nucleobase 1 ((S)-3-(1H-imidazo[4,5-f][1,10]phenanthrolin-1-yl)-2-methoxypropan-1-ol)
We have previously reported the use of the artificial nucleoside analogue P in various contexts of metal-mediated base pairing [47,48,49,50], both in antiparallel- and parallel-stranded DNA duplexes (Fig. 1b). The ligand prefers to engage in a tetrahedral coordination geometry when forming [2 + 2] coordination complexes with Ag(I) [51], Cu(I) [52], and Zn(II) [53]. On the other hand, in the presence of a complementary monodentate nucleobase, it is also compatible with a [2 + 1] coordination pattern when a d10-configured metal ion is applied [47,48,49,50]. In this report, the homo- and heteroleptic base-pair formation of the nucleoside analogue P in the presence of Hg(II) is presented for antiparallel-stranded DNA duplexes. The initial goal was to generate a reversibly formed Hg(II)-mediated base pair that is more stable than the conventional T–Hg(II)–T base pair, so that a nanodevice with high-performance Hg(II)-sensing capacity becomes feasible.
Materials and methods
The (S)-configured imidazophenanthroline-containing GNA-based phosphoramidite and the model nucleobase 1 were synthesized according to the procedure published earlier [51]. DNA synthesis was performed in the DMT-off mode on a K&A Laborgeräte H8 DNA/RNA synthesizer. For the introduction of P into the oligonucleotide, an increased threefold coupling time was applied. After synthesis and purification, the desalted oligonucleotides were characterized by MALDI-TOF (matrix-assisted laser desorption-ionization time-of-flight) mass spectrometry (Table 1, Fig. S1, Supplementary material). MALDI-TOF mass spectra were recorded on Bruker Reflex IV or Bruker Autoflex Speed instruments using a 3-hydroxypicolinic acid/ammonium citrate matrix. During the quantification of the oligonucleotides, a molar extinction coefficient ε260 of 10.0 cm2 mmol−1 was used for P. UV measurements were performed on a CARY 100 Bio UV spectrometer using solutions containing 1 μM oligonucleotide duplex, 150 mM NaClO4, 2.5 mM Mg(ClO4)2, and 5 mM buffer (MOPS (pH 6.8), MES (pH 5.5) or borate (pH 9.0)). Prior to the assays, the samples were incubated with the added metal ions for 1 h at 5 °C. UV melting curves were recorded with a heating/cooling rate of 1 °C min−1 and a data interval of 1 °C. Absorbance was normalized according to Anorm = (A – Amin)/(Amax – Amin) at 260 nm. Melting temperatures were determined from a Gaussian fit of the maximum of the first derivative of the respective melting curve. The standard deviation of Tm represents the standard deviation of the Gaussian fit. CD spectra were recorded at 5 °C on a JASCO J-815 spectropolarimeter, smoothed, and a manual base line correction was applied.
Results and discussion
Characterization of the Hg(II)-binding behaviour of P
Model nucleobases have proven to be helpful in the determination of the metal-binding behaviour of the natural nucleosides [54]. In model nucleobases, the ribose moiety of the nucleoside is formally replaced by an alkyl group, eliminating a possible interference of the hydroxyl groups with metal binding. Similarly, model nucleobases have helped to elucidate the preferred metal-binding stoichiometry of artificial nucleosides and the geometry of the metal-mediated base pair [55,56,57,58]. In the context of the Zn(II)-binding capability of P, a suitably O-protected derivative 1 had been reported previously (Fig. 1c) [53]. This model nucleobase was now also titrated with Hg(II) in aqueous medium. As can be seen in the inset of Fig. 2, the UV spectrum of 1 shows two absorption maxima at 249 nm and 283 nm prior to the addition of Hg(II) ions. Their stepwise addition causes a significant shift of the absorption maximum at 283 nm alongside a disappearance of the absorption maximum at 249 nm. The spectral changes can be attributed to the formation of a new species into the solution. Isosbestic points at 223, 251, 261, 287, and 337 nm clearly indicate a direct equilibrium between the free ligand and its Hg(II) complex. The new absorption band at ~ 315 nm is most likely due to a metal-to-ligand charge transfer, based on the availability of a low-lying π* orbital of the aromatic phenanthroline moiety. When plotting the absorbance at 315 nm against the amounts of Hg(II), a levelling-off effect at 0.5 equivalents of Hg(II) can be observed (Fig. 2), suggesting the formation of a homoleptic 1:2 metal complex in aqueous medium (pH 7).

UV absorbance of an aqueous solution of compound 1 at 315 nm in the presence of increasing amounts of Hg(II) at pH 7. Inset: UV/Vis spectrum of compound 1 in the presence of various amounts of Hg(II). Arrows indicate the direction of the changes
Investigation of the Hg(II)-binding properties of the DNA duplexes
The metal-binding ability of P inside antiparallel-stranded DNA duplexes was investigated in the context of both homo- and hetero-base pairs by introducing P as a central nucleoside surrogate into two short oligonucleotide sequences (Table 1). We selected these previously investigated sequences to allow a better comparison with other metal-mediated base pairs [47, 48], in particular because the sequence context is known to play an important role in the net stabilization of metal-mediated base pairs [49]. In this study, duplex I comprises one P:P homo base pair, whereas duplex II contains one central P:T hetero base pair. In addition, duplex III bearing natural base pairs only was investigated as a reference. The propensity towards the formation of a Hg(II)-mediated base pair was probed by an analysis of the thermal duplex denaturation (as derived by UV spectroscopy) and by a CD-spectroscopic analysis indicating the impact of the metal complex formation on the duplex conformation.
UV-dependent thermal denaturation analyses
Homo base pair
In the absence of any Hg(II), the melting temperature of duplex I amounts to 36.7(2) °C (Fig. 3). For comparison, duplex III lacking the artificial P:P pair melts at 43.6(4) °C (Fig. S2b, Supplementary material). This difference in melting temperature Tm can most likely be attributed to the distortion around the P:P pair upon the incorporation of the bulky imidazophenanthroline moiety with an acyclic backbone into the duplex. When solutions containing duplex I are incubated with Hg(II) prior to the thermal denaturation analysis, a significant increase of the melting temperature Tm is observed. In the presence of one Hg(II) per duplex, a transition of the sigmoidal melting profile towards higher Tm can be observed, leading to a Tm of 41.0(4) °C. Addition of excess Hg(II) ions confers a minor additional thermal stabilization only. In combination with the established stoichiometry of the model nucleobase complex [Hg(1)2]2+, the incorporation of one Hg(II) into the P:P pair can be safely concluded, leading to a P–Hg(II)–P base pair. The generation of this metal-mediated base pair causes a duplex stabilization of 4.3(4) °C.

Denaturation of duplex I as determined UV-spectroscopically [solid black line: 0 equiv of Hg(II); broken red line: 1 equiv of Hg(II); dotted blue line: 2 equiv of Hg(II)]. The inset shows the melting temperature Tm depending on the amount of Hg(II). Experimental conditions: 1 μM duplex, 150 mM NaClO4, 2.5 mM Mg(ClO4)2, and 5 mM MOPS (pH 6.8)
The stoichiometry of the P–Hg(II)–P base pair within duplex I is further confirmed by a plot of the UV absorbance of the duplex at 254 nm vs. the added equivalents of Hg(II) (Fig. 4). Again, a drastic change in absorbance is observed up to the addition of one Hg(II) per duplex, whereas the addition of excess Hg(II) has a less pronounced effect and can most likely be attributed to non-specific binding.

Change of the UV absorbance of duplex I at 254 nm upon the stepwise addition of Hg(II), clearly confirming the binding of one Hg(II) per duplex (and hence per P:P pair). Inset: UV spectrum of duplex I in the presence of various amounts of Hg(II). The arrow indicates the direction of the change. Experimental conditions: 1 μM duplex, 150 mM NaClO4, 2.5 mM Mg(ClO4)2, and 5 mM MOPS (pH 6.8)
Hetero base pair
Duplex II comprises essentially the same oligonucleotide sequence as duplex I, differing only in one nucleoside. In duplex II, one of the artificial nucleoside analogues P is formally replaced by a thymine residue. As the thymine moiety must be deprotonated at its N3 position to enable a coordination of the Hg(II) ion, the Hg(II)-binding studies were performed at different pH values.
The UV-based thermal denaturation studies of duplex II show a sigmoidal duplex melting with different melting temperatures depending on the pH of the medium (Fig. S3, Supplementary material). In the absence of Hg(II), the duplex becomes less stable with increasing pH. This trend had been observed previously for the same duplex under slightly different experimental conditions [i.e., in the absence of Mg(ClO4)2]. It can be explained by the fact that P becomes protonated under acidic conditions to form a stabilizing PH+–T base pair involving two hydrogen bonds (Fig. S5b, Supplementary material). Under neutral conditions, the formation of one bifurcated hydrogen bond is assumed, whereas deprotonated thymine cannot engage in hydrogen bonding with P at all.
Upon the addition of one Hg(II) per duplex, a considerable increase in the melting temperature is observed for all investigated pH values (see Fig. 5 for pH 6.8 and Fig. S3, Supplementary material, for all pH values under investigation). Again, excess Hg(II) leads to hardly any additional change in Tm. These data are supportive of the formation of a mononuclear complex in the designated binding site of the duplex. To confirm the site-specific incorporation of Hg(II) into the P:T pair, additional titration experiments were performed using duplex III, lacking the artificial pair. Here, the addition of Hg(II) does not affect the melting temperature within the standard deviation (Fig. S2, Supplementary material), with ΔTm essentially being 0 °C. This further corroborates the selective formation of a P–Hg(II)–T base pair in duplex II. Interestingly, the stability of duplex II bearing the P–Hg(II)–T pair strongly depends on the pH, with the Tm at pH 6.8 being significantly higher than under acidic or alkaline conditions (Table 2). The maximum increase ΔTm of 14.5(6) °C is observed under these near-neutral conditions. Hence, the stability of duplex II bearing a P–Hg(II)–T pair follows the trend known for duplexes with canonical nucleobases only, in which both alkaline and acidic conditions lead to a decreased stability of the Watson–Crick base pairs. It is interesting to note that the thermal stabilization of 14.5(6) °C is more than twice as large as that observed previously for the same base pair in a parallel-stranded duplex [ΔTm = 11 °C for a duplex comprising two P–Hg(II)–T pairs] [47]. At first, this discrepancy appears surprising, considering that fact that the base pairs should only differ in the relative orientation of their glycosidic bonds (cisoid vs. transoid). However, this may very well be due to the different length of the oligonucleotides under investigation, as previous publications had indicated that the thermal stabilization induced by metal-mediated base pair formation typically is larger for shorter oligonucleotides [59], as is the case here.

Denaturation of duplex II as determined UV-spectroscopically at pH 6.8 [solid black line: 0 equiv of Hg(II); broken red line: 1 equiv of Hg(II); dotted blue line: 2 equiv of Hg(II)]. The inset shows the melting temperature Tm depending on the amount of Hg(II). Experimental conditions: 1 μM duplex, 150 mM NaClO4, 2.5 mM Mg(ClO4)2, and 5 mM MOPS (pH 6.8)
Previous computational studies on the mechanism of formation of a T–Hg(II)–T base pair suggested that Hg(II) acts as a Lewis acid [60]. Hence, at least one of the aqua ligands in its hydration shell becomes deprotonated. The resulting hydroxido ligand facilitates the removal of the thymine N3 proton, yielding an in situ deprotonated thymine which can then be easily mercurated. Hence, alkaline conditions are not required for the formation of a T–Hg(II)–T base pair. It is highly likely that a similar mechanism also takes place during the formation of the P–Hg(II)–T base pair in duplex II. Hence, an elevated pH value may not necessarily be required for the formation of the P–Hg(II)–T pair. In fact, alkaline conditions destabilize the duplex, probably due to the usual destabilization of the regular Watson–Crick pairs. The reduced stability of duplex II at pH 5.5 compared to pH 6.8 can likely be explained by the generally lower stability of DNA duplexes under acidic conditions, too. These explanations are in good agreement with the melting temperatures determined for duplex III without any artificial nucleosides.
CD-spectroscopic analyses
CD spectroscopy was employed to elucidate the impact of the formation of the Hg(II)-mediated base pair on the secondary structure of the DNA duplexes.
Homo base pair
The CD spectrum of the Hg(II)-free duplex I shows an intense negative Cotton effect at ca. 249 nm and two positive Cotton effects at 258 nm and 275 nm, accompanied by a broad negative Cotton effect at ca. 292 nm (Fig. 6, black spectrum). This CD spectral pattern varies significantly from that of reference duplex III bearing canonical base pairs only (Fig. 6, grey spectrum). The deviation from the usual B-DNA conformation must be due to the incorporation of the bulky imidazophenanthroline nucleoside analogue. Upon the addition of one equivalent of Hg(II) to the solution, the spectrum of duplex I (Fig. 6, red spectrum) shows a significantly more negative molar ellipticity around 300 nm along with a red shift of the characteristic Cotton effect previously found at ca. 292 nm (Δλ = + 8 nm). Such an effect had previously been seen for the Ag(I)-, Cu(I)-, and Zn(II)-mediated homo base pairs of P and had been assigned to the enantio-specific formation of a chiral tetrahedrally distorted metal complex [51,52,53]. In addition, the molar ellipticity of the two positive Cotton effects increases significantly. When performing this experiment with reference duplex III, no considerable changes in the CD spectra are detected (Fig. S4, Supplementary material). These observations further confirm the anticipated site-specific incorporation of the Hg(II) ion into duplex to yield a P–Hg(II)–P base pair. Due to the strong fingerprint of the chiral metal complex in the CD spectrum, a detailed conclusion regarding a possible change of the DNA duplex conformation is not possible.

CD spectrum of duplex I in the absence (black solid line) and presence of one equiv of Hg(II) (red broken line). For comparison, the CD spectrum of reference duplex III is shown (grey dotted line), too. Experimental conditions: 1 μM duplex, 150 mM NaClO4, 2.5 mM Mg(ClO4)2, and 5 mM MOPS (pH 6.8)
Hetero base pair
A comparison of the CD spectra of duplex II under acidic, near-neutral, and alkaline conditions prior to the addition of Hg(II) shows slight differences (Fig. S5a, Supplementary material). A negative Cotton effect at ca. 295 nm is observed at pH 5.5 and 6.8, whereas no such peak is observed at pH 9.0. In contrast, a negative Cotton effect at ca. 245 nm is found under all experimental conditions, but this effect is more prominent at pH 6.8 and 9.0. It is difficult to assign these spectroscopic differences to particular changes in the DNA duplex structure. However, as previously discussed in the context of the formation of related Ag(I)-mediated base pairs [48], different hydrogen-bonding patterns between P and T are feasible, depending on the protonation state of the nucleobases and, hence, on the pH value (Fig. S5b, Supplementary material). It is, therefore, likely that P:T pairs of different structure are formed at the different pH values, giving rise to the different CD spectra of duplex II.
Interestingly, the CD spectra of duplex II are essentially identical in the presence of one equivalent of Hg(II) at pH 6.8 and 9.0 (Fig. 7), indicating that the same duplex conformation is adopted under these conditions upon the formation of the P–Hg(II)–T base pair. Even at pH 5.5, the same overall shape of the CD spectrum is observed, albeit with slight differences in the intensities of the Cotton effects. These differences follow the same trends as observed in the absence of Hg(II) (Fig. S5a, Supplementary material). Nonetheless, clear changes take place upon the formation of the P–Hg(II)–T base pair under all experimental conditions, as evidenced by a more negative molar ellipticity at ca. 245 nm and a less intense positive Cotton effect between 260 and 280 nm (Fig. S6, Supplementary material).

CD spectrum of duplex II in the presence of one equiv. of Hg(II) at pH 5.5 (solid back line), pH 6.8 (red broken line), and pH 9.0 (dotted grey line). Experimental conditions: 1 μM duplex, 150 mM NaClO4, 2.5 mM Mg(ClO4)2, and 5 mM buffer [MOPS (pH 6.8), MES (pH 5.5), or borate (pH 9.0)]
Conclusions
The present study confirms that a P:P homo base pair within an antiparallel-stranded DNA double helix is capable of binding Hg(II), forming a P–Hg(II)–P base pair. This Hg(II)-mediated base pair significantly increases the thermal stability of the duplex. Based on the CD data, the enantio-specific formation of a chiral Hg(II) complex can be concluded. Even though the T–Hg(II)–T pair is a paradigm of a metal-mediated base pair, examples for other Hg(II)-mediated base pair are scarce. The only other instances involve pyridine [61] or organometallic-mercurated ligands [62,63,64,65]. Hence, Hg(II)-mediated base pairs formed from an artificial mispair are a rarity. The P–Hg(II)–P pair represents the first example of a Hg(II)-mediated base pair comprising a bidentate ligand and thereby increases the pool of artificial nucleobase capable of forming Hg(II)-mediated base pairs. Moreover, an antiparallel-stranded DNA duplex comprising a central P:T pair is capable of forming a highly stabilizing Hg(II)-mediated base pair. The thermal stabilization of the duplex depends on the acidity of the medium and is most pronounced at pH 6.8. Under these conditions, the P–Hg(II)–T pair turns out to be the most stabilizing non-organometallic Hg(II)-mediated base pair reported to date, with an increase in the melting temperature ΔTm of + 14.5(6) °C. Hence, it fulfils the initial objective of creating a metal-mediated base pair that is more stabilizing than the T–Hg(II)–T base pair (ΔTm up to 10 °C, depending on the DNA sequence) [66].
References
Bandy TJ, Brewer A, Burns JR, Marth G, Nguyen T, Stulz E (2011) Chem Soc Rev 40:138–148
Chidchob P, Sleiman HF (2018) Curr Opin Chem Biol 46:63–70
Naskar S, Guha R, Müller J (2020) Angew Chem Int Ed 59:1397–1406
Takezawa Y, Müller J, Shionoya M (2017) Chem Lett 46:622–633
Jash B, Müller J (2017) Chem Eur J 23:17166–17178
Yamane T, Davidson N (1961) J Am Chem Soc 83:2599–2607
Katz S (1963) Biochim Biophys Acta 68:240–253
Miyake Y, Togashi H, Tashiro M, Yamaguchi H, Oda S, Kudo M, Tanaka Y, Kondo Y, Sawa R, Fujimoto T, Machinami T, Ono A (2006) J Am Chem Soc 128:2172–2173
Müller J (2019) Coord Chem Rev 393:37–47
Ono A, Kanazawa H, Ito H, Goto M, Nakamura K, Saneyoshi H, Kondo J (2019) Angew Chem Int Ed 58:16835–16838
Kondo J, Tada Y, Dairaku T, Hattori Y, Saneyoshi H, Ono A, Tanaka Y (2017) Nat Chem 9:956–960
Su M, Tomás-Gamasa M, Carell T (2015) Chem Sci 6:632–638
Müller SL, Zhou X, Leonard P, Korzhenko O, Daniliuc C, Seela F (2019) Chem Eur J 25:3077–3090
Zhao H, Leonard P, Guo X, Yang H, Seela F (2017) Chem Eur J 23:5529–5540
Méndez-Arriaga JM, Maldonado CR, Dobado JA, Galindo MA (2018) Chem Eur J 24:4583–4589
Santamaría-Díaz N, Méndez-Arriaga JM, Salas JM, Galindo MA (2016) Angew Chem Int Ed 55:6170–6174
Kim E-K, Switzer C (2014) Org Lett 16:4059–4061
Takezawa Y, Nishiyama K, Mashima T, Katahira M, Shionoya M (2015) Chem Eur J 21:14713–14716
Kobayashi T, Takezawa Y, Sakamoto A, Shionoya M (2016) Chem Commun 52:3762–3765
Schmidt OP, Benz AS, Mata G, Luedtke NW (2018) Nucleic Acids Res 46:6470–6479
Schmidt OP, Mata G, Luedtke NW (2016) J Am Chem Soc 138:14733–14739
Sandmann N, Defayay D, Hepp A, Müller J (2019) J Inorg Biochem 191:85–93
Sandmann N, Bachmann J, Hepp A, Doltsinis NL, Müller J (2019) Dalton Trans 48:10505–10515
Röthlisberger P, Levi-Acobas F, Sarac I, Marlière P, Herdewijn P, Hollenstein M (2019) J Inorg Biochem 191:154–163
Mandal S, Hebenbrock M, Müller J (2017) Chem Eur J 23:5962–5965
Tan X, Litau S, Zhang X, Müller J (2015) Langmuir 31:11305–11310
Räisälä H, Lönnberg T (2019) Chem Eur J 25:4751–4756
Müller J (2017) Beilstein J Org Chem 13:2671–2681
Schlegel MK, Zhang L, Pagano N, Meggers E (2009) Org Biomol Chem 7:476–482
Jayarathna DR, Stout HD, Achim C (2018) Inorg Chem 57:6865–6872
Engelhard DM, Nowack J, Clever GH (2017) Angew Chem Int Ed 56:11640–11644
Liu S, Clever GH, Takezawa Y, Kaneko M, Tanaka K, Guo X, Shionoya M (2011) Angew Chem Int Ed 50:8886–8890
Léon JC, She Z, Kamal A, Shamsi MH, Müller J, Kraatz H-B (2017) Angew Chem Int Ed 56:6098–6102
Takezawa Y, Yoneda S, Duprey J-LHA, Nakama T, Shionoya M (2016) Chem Sci 7:3006–3010
Takezawa Y, Nakama T, Shionoya M (2019) J Am Chem Soc 141:19342–19350
Schönrath I, Tsvetkov VB, Zatsepin TS, Aralov AV, Müller J (2019) J Biol Inorg Chem 24:693–702
Funai T, Nakamura J, Miyazaki Y, Kiriu R, Nakagawa O, Wada S-i, Ono A, Urata H (2014) Angew. Chem Int Ed 53:6624–6627
Jash B, Müller J (2017) Eur J Inorg Chem 3857–3861
Taherpour S, Golubev O, Lönnberg T (2016) Inorg Chim Acta 452:43–49
Léon JC, González-Abradelo G, Strassert CA, Müller J (2018) Chem Eur J 24:8320–8324
Kaul C, Müller M, Wagner M, Schneider S, Carell T (2011) Nat Chem 3:794–800
Tanaka Y, Kondo J, Sychrovský V, Šebera J, Dairaku T, Saneyoshi H, Urata H, Torigoe H, Ono A (2015) Chem Commun 51:17343–17360
Ono A, Togashi H (2004) Angew Chem Int Ed 43:4300–4302
Yamaguchi H, Šebera J, Kondo J, Oda S, Komuro T, Kawamura T, Dairaku T, Kondo Y, Okamoto I, Ono A, Burda JV, Kojima C, Sychrovský V, Tanaka Y (2014) Nucleic Acids Res 42:4094–4099
Liu H, Cai C, Haruehanroengra P, Yao Q, Chen Y, Yang C, Luo Q, Wu B, Li J, Ma J, Sheng J, Gan J (2017) Nucleic Acids Res 45:2910–2918
Schmidt OP, Jurt S, Johannsen S, Karimi A, Sigel RKO, Luedtke NW (2019) Nat Commun 10:4818
Jash B, Müller J (2018) Chem Eur J 24:10636–10640
Jash B, Scharf P, Sandmann N, Fonseca Guerra C, Megger DA, Müller J (2017) Chem Sci 8:1337–1343
Scharf P, Jash B, Kuriappan JA, Waller MP, Müller J (2016) Chem Eur J 22:295–301
Naskar S, Müller J (2019) Chem Eur J 25:16214–16218
Jash B, Neugebauer J, Müller J (2016) Inorg Chim Acta 452:181–187
Jash B, Müller J (2018) Angew Chem Int Ed 57:9524–9527
Jash B, Müller J (2018) J Inorg Biochem 186:301–306
Schöllhorn H, Raudaschl-Sieber G, Müller G, Thewalt U, Lippert B (1985) J Am Chem Soc 107:5932–5937
Mandal S, Wang C, Prajapati RK, Kösters J, Verma S, Chi L, Müller J (2016) Inorg Chem 55:7041–7050
Sinha I, Hepp A, Kösters J, Müller J (2015) J Inorg Biochem 153:355–360
Seubert K, Böhme D, Kösters J, Shen W-Z, Freisinger E, Müller J (2012) Z Anorg Allg Chem 638:1761–1767
Clever GH, Söltl Y, Burks H, Spahl W, Carell T (2006) Chem Eur J 12:8708–8718
Schweizer K, Kösters J, Müller J (2015) J Biol Inorg Chem 20:895–903
Šebera J, Burda J, Straka M, Ono A, Kojima C, Tanaka Y, Sychrovský V (2013) Chem Eur J 19:9884–9894
Tanaka K, Clever GH, Takezawa Y, Yamada Y, Kaul C, Shionoya M, Carell T (2006) Nat Nanotechnol 1:190–194
Ukale DU, Lönnberg T (2018) Angew Chem Int Ed 57:16171–16175
Ukale D, Shinde VS, Lönnberg T (2016) Chem Eur J 22:7917–7923
Aro-Heinilä A, Lönnberg T, Virta P (2019) Bioconjugate Chem 30:2183–2190
Ukale DU, Tähtinen P, Lönnberg T (2020) Chem Eur J 26:2164–2168
Takezawa Y, Shionoya M, Müller J (2017) In: Atwood JL (ed) Comprehensive supramolecular chemistry II, vol 4. Elsevier, Oxford, pp 259–293
Acknowledgements
Open Access funding provided by Projekt DEAL. Funding by the Deutsche Forschungsgemeinschaft (SFB 858) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jash, B., Müller, J. Stable Hg(II)-mediated base pairs with a phenanthroline-derived nucleobase surrogate in antiparallel-stranded DNA. J Biol Inorg Chem 25, 647–654 (2020). https://doi.org/10.1007/s00775-020-01788-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-020-01788-x