
Abstract
The immune response to stroke is an exciting target for future stroke therapies. Stroke is a leading cause of morbidity and mortality worldwide, and clot removal (mechanical or pharmacological) to achieve tissue reperfusion is the only therapy currently approved for patient use. Due to a short therapeutic window and incomplete effectiveness, however, many patients are left with infarcted tissue that stimulates inflammation. Although this is critical to promote repair, it can also damage surrounding healthy brain tissue. In addition, acute immunodepression and subsequent infections are common and are associated with worse patient outcomes. Thus, the acute immune response is a major focus of researchers attempting to identify ways to amplify its benefits and suppress its negative effects to improve short-term recovery of patients. Here we review what is known about this powerful process. This includes the role of brain resident cells such as microglia, peripherally activated cells such as macrophages and neutrophils, and activated endothelium. The role of systemic immune activation and subsequent immunodepression in the days after stroke is also discussed, as is the chronic immune responses and its effects on cognitive function. The biphasic role of inflammation, as well as complex timelines of cell production, differentiation, and trafficking, suggests that the relationship between the acute and chronic phases of stroke recovery is complex. Gaining a more complete understanding of this intricate process by which inflammation is initiated, propagated, and terminated may potentially lead to therapeutics that can treat a larger population of stroke patients than what is currently available. The immune response plays a critical role in patient recovery in both the acute and chronic phases after stroke. In patients, the immune response can be beneficial by promoting repair and recovery, and also detrimental by propagating a pro-inflammatory microenvironment. Thus, it is critical to understand the mechanisms of immune activation following stroke in order to successfully design therapeutics.
Similar content being viewed by others


Introduction to Stroke
Stroke is a leading cause of death and disability worldwide. Ischemic stroke is a blockage of blood vessels that supply the brain, leading to a reduction or total loss of blood flow, while hemorrhagic stroke is caused by the rupture of intracerebral vessels [1]. Regardless of subtype, stroke causes rapid necrotic death of all cells within the core, which is the region of significant loss of oxygen and nutrients [2, 3]. Ultimately, the amount of brain tissue death is affected by the absolute area at risk, whether there is any residual tissue perfusion, the length of the occlusion, and the degree of reperfusion [4]. In some ischemic stroke patients, a “no-reflow” phenomenon is observed, where even after clot removal the tissue will not fully reperfuse, causing further damage [4]. In hemorrhagic stroke, hematomas expand and surrounding edema further compresses blood vessels resulting in more tissue damage [5]. Finally, both the size and location of the ischemic lesion are responsible for patient outcomes and observed symptoms.
Risk factors for stroke include aging, hypertension, obesity, atherosclerosis, type II diabetes mellitus, and peripheral infection. There are also gender-specific risks within different age groups [6,7,8]. Each of these risk factors is independently correlated with poor short-term stroke outcome in both clinical and experimental settings [9, 10]. All of these risk factors for stroke also activate the peripheral immune system; thus, there may be cross-talk between the brain and the systemic inflammatory milieu that contributes to worse outcomes [11].
Acute treatments for arterial ischemic stroke restore blood flow to hypo-perfused tissue through chemical or mechanical thrombolysis, sometimes with dramatic success [12, 13]. Although the treatment window for thrombectomy has recently been extended through the use of modern imaging techniques, many patients are still left untreated, and once ischemia has caused irreversible damage, therapies are limited [14,15,16]. In the past, only a minority of patients were eligible for intervention [17], and although this is improving, there are still many patients who are not eligible, and/or who have significant infarct burden even after successful revascularization. Because of the prominent role that the immune response plays in ischemic stroke pathology in both the subacute and chronic phases, it is a promising target for any patient with a completed infarction. The immune system can further injure damaged tissue, influence clearance of dead tissue, and either hinder or facilitate neuronal rewiring, all processes that are critical to recovery during the days and weeks after stroke.
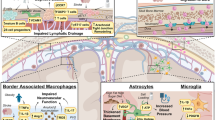
Various aspects of the immune response to stroke have been a focus for researchers, as summarized in Fig. 1. During the acute phase, the brain activates resident microglia and simultaneously recruits myeloid cells and neutrophils from the periphery. These events facilitate neuroinflammation and local repair after injury, and simultaneously activate the peripheral immune response. In the days after stroke, patients also become immunosuppressed, leading to high rates of infection. In addition, during this time, the active process of resolution is initiated to avoid excessive tissue damage. When inflammatory responses do not resolve properly, it may however result in chronic activation of the adaptive immune response, secondary neurodegeneration, and dementia in stroke survivors. Overall, understanding these different aspects of the immune response is essential for maximizing recovery of stroke patients. Finally, by modulating reperfusion-induced inflammation, these strategies have the potential to improve outcomes even after tPA therapy.

Summary of local and peripheral immune responses after stroke. Immediately following stroke, brain-resident immune cells such as microglia and astrocytes are activated to respond to injury. Subsequently, peripheral immune cells are activated and recruited to the brain to assist in the immune response. In the following days, peripheral immunodepression can occur, with a subsequent increased risk for systemic infections. The extent of these local and peripheral immune responses to stroke is variable, and this plays an important role in determining patient outcomes and overall functional recovery in the acute and chronic phases after stroke
Initiating Neuroinflammation After Stroke
Damage-Associated Molecular Patterns
Damage-associated molecular patterns (DAMPs), also known as alarmins, are released by necrotic cells and act as endogenous signals to stimulate the inflammatory response. Prior to secretion, these molecules also have physiological functions that are independent from their activities as rapid activators of the immune system. Immediately following arterial occlusion, intracellular ATP depletion and subsequent glutamate release to the extracellular space result in uncontrolled calcium-ion influx into cells [18]. Subsequently, dysregulated calcium signaling activates intracellular lipases and proteolytic enzymes, ultimately resulting in cell death [18]. Necrotic cells in the ischemic core release DAMPs into the extracellular circulation to assist in the activation of brain resident immune cells (microglia) through specific receptors on the extracellular surface. In addition, DAMPs recruit peripheral immune cells to the brain once they have been released into the bloodstream. Peripheral immune cells are activated outside the brain, and then traffic to the brain to assist in repair and recovery [19].
High-mobility group box 1 (HMGB1) and heat shock proteins (Hsps) are essential DAMPs for initiating the post-stroke inflammatory response via activating receptors on peripheral immune cells [20, 21]. Specifically, HMGB1 released from activated monocytes and macrophages or necrotic cells can bind to the receptor for advanced glycation end products (RAGEs) and toll-like receptors (TLRs) 2 and 4 once secreted into the bloodstream [21, 22]. Activation of these receptors results in upregulation and subsequent secretion of multiple cytokines that promote inflammation and cellular death after ischemia, including matrix metalloproteinase 9 (MMP-9), tumor necrosis factor-α (TNF-α), and interleukin (IL)-1β [20]. Activation of innate responses mediated by TLR-4 in monocytes, such as NF-κB activity and TNF synthesis, is associated with worse outcomes in stroke patients [23, 24]. Additionally, adequate clearance of DAMPs is required for proper resolution of inflammation following injury–deficiency in the clearance process results in severe inflammation and exacerbated neuronal injury in a murine model of ischemic stroke [25]. In contrast, astrocyte-produced HMGB1 may promote endothelial recovery [26]. HMGB1 release from activated astrocytes enhances neurite outgrowth, neuronal survival, and neurovascular recovery [26]. Taken together, these data suggest that HMGB1 likely plays a dual role in stroke pathophysiology and understanding the cell-specific role of this DAMP is critical to understanding the downstream signaling effects.
Several Hsps have also been implicated in initiating the innate immune response after ischemic stroke [20]. After secretion, Hsp70 binds TLRs on the surface of macrophages and dendritic cells in the periphery and microglia in the brain. Via TLR signaling, Hsp70 activates NF-κB in addition to other pro-inflammatory pathways [27]. It can also directly polarize CD4+ T cells to a pro-inflammatory Th1 state through interaction with extracellular receptors [28]. In contrast, overexpressing Hsp70 in mice with stroke improves long-term outcomes and knockout of Hsp70 worsens them [28, 29]. Hsp27 and Hsp32 have also been implicated in stroke. Extracellular Hsp27 binds various receptors on peripheral immune cells such as TLRs on the surface of activated macrophages, and activates the pro-inflammatory NF-κB pathway [30]. In contrast, Hsp32, or heme oxygenase-1 (HO-1), increases ischemic brain damage via formation of reactive oxygen species [31]. Interestingly, treatment of wild-type microglia with deferoxamine (a hypoxia mimetic) decreases neuronal damage, but this effect is abolished in HO-1(−/−) microglia, indicating cell-specific roles for this DAMP [32].
Due to their prominent role in the acute immune response to stroke, DAMPs have been proposed as promising therapeutic targets. However, it is often difficult to distinguish between the beneficial and injurious roles of inflammation in stroke recovery, as evidenced by the dual roles of many of these proteins, and their abilities to target multiple pathways to modulate the immune response. These studies highlight the need for further work in distinguishing cell-type specific roles and downstream signaling pathways activated by DAMPs to selectively target the harmful aspects of the immune response.
Blood–Brain Barrier
Once peripheral immune cells have been activated by DAMPs, they must access the brain to assist in repair. In the healthy CNS, the blood–brain barrier (BBB) regulates the movement of nutrients and blood-borne molecules into the brain, and metabolic waste out of the brain, both maintaining a healthy environment and protecting the brain from peripheral toxins [33, 34]. Under physiological conditions, the BBB also excludes peripheral cells, but during pathological conditions such as stroke, traumatic brain injury, or multiple sclerosis, this function changes to attract immune cells [35].
Initially, circulating lymphocytes contact inflamed vessels via P-selectin glycoprotein ligand-1 (PSGL-1), and interact with endothelial cells via E-selectin and P-selectin [36]. This interaction tethers peripheral cells to the endothelial surface. Subsequently, integrins such as lymphocyte function-associated antigen 1 (LFA-1) and very late antigen-4 (VLA-4) on lymphocytes bind to ICAM-1 and VCAM-1 on endothelial cells, respectively, causing firm lymphocyte adherence to the endothelium and subsequent lymphocyte crawling [37]. Finally, PECAM-1 (CD31) and CD99 homophilic interactions facilitate structural changes to tight junctions and endothelial cell structure, and subsequently promote both paracellular and transcellular diapedesis [38]. Several animal studies indicate that trafficking of leukocytes into the brain contributes to neuroinflammation-mediated neuronal cell death [39, 40].
Independent of cellular infiltration, the structural integrity of the BBB is altered by MMP-2 activation within 6 h of reperfusion, resulting in increased permeability [41]. MMPs degrade basal lamina proteins such as fibronectin, laminin, and heparan sulfate, which contributes to the vessel damage associated with BBB breakdown and subsequent dysfunction [41,42,43]. Additional degradation and remodeling of tight junctions, and further increases in BBB permeability, occur within 48 h after reperfusion [41, 42, 44]. While the initial BBB opening is transient, the later increase is the result of severe damage to blood vessels by expression and activation of MMP-9 and MMP-3 [41, 45].
Innate Inflammation After Stroke
Initial Acute Brain Inflammation
In the acute phase of stroke, peripheral immune cells infiltrating the brain parenchyma contribute to the inflammatory environment [33]. Signaling via the chemokine receptor CCR2 recruits both monocytes and neutrophils from the periphery to the site of injury [46]. Neutrophils are the primary cell type adhered to endothelial cells in brain vessels following the initial myeloid cell response, and they are found in significant numbers in the brain parenchyma by 3 days post-stroke in animal models [39, 47].
Accumulating evidence from both human and rodent models suggests that post-stroke accumulation of neutrophils in the brain parenchyma worsens stroke outcomes [39, 48, 49]. Neutrophils obstruct capillaries in the microvascular bed following reperfusion, suggesting that they play a causative role in the “no-reflow” phenomenon observed in humans [4]. Pharmacological ablation of neutrophils reduces infarct volumes and improves neurological outcomes, further supporting a pathogenic role of neutrophil accumulation and activation after stroke [50, 51]. Likewise, genetic deficiencies in adhesion molecules such as ICAM-1 and P-selectin reduce neutrophil trafficking into brain parenchyma, resulting in smaller infarct volumes 48 h after stroke [52, 53]. Neutrophil-mediated damage may be mediated through their high proteolytic enzyme content, their capacity to generate ROS, and ability to activate the complement system [54, 55].
Myeloid cells are also among the first immune cells to respond to injury [47, 56, 57]. Both brain resident microglia and monocyte-derived macrophages (MDMs) that are derived from peripheral monocyte ingress recognize DAMPs and other stress signals and become activated to migrate to the site of injury. Initially, microglia in the stroke penumbra are activated by cytokines and various DAMPs from the necrotic debris of local dead or dying cells. Microglia are also recruited from the surrounding brain regions by neuronal-derived chemokines such as fractalkine (CX3CL1) [58,59,60,61].
Studies investigating the beneficial or detrimental effects of microglia on the brain have been contradictory. This may be due to the different stroke models utilized and the outcomes assessed. Also, tools to distinguish MDMs from microglia in the injured brain have historically been limited [62, 63]. Newly developed chimeric models and microglia-specific markers such as Tmem119 are helping to identify the individual roles of each cellular subtype, and will hopefully lend clarity [62].
Overall, microglia likely have an early pro-inflammatory role, while MDMs primarily serve a clearance and immune resolving function [64]. However, there is more to learn about the heterogeneity within each cell population [65]. Promoting early inflammation may be critical, as when microglia are ablated before ischemic stroke it causes exaggerated neurological deficits and increased infarct volume [66], perhaps because they, via cross-talk with astrocytes, are critical for termination of inflammation by neutrophil engulfment [67].
Subacute Innate Neuroinflammation
Myeloid cells can also transition from a naïve state to an anti-inflammatory phenotype in response to interleukin-4 (IL-4) release from damaged neurons via interferon regulatory factor (IRF)-4 signaling [68, 69]. IL-4 treatment of cultured microglia facilitated both trophic factor expression and peroxisome proliferator–activated receptor (PPAR)-γ-dependent phagocytosis, which are hallmark characteristics of anti-inflammatory myeloid cells [68, 70]. Anti-inflammatory microglia may also reduce activation of immune cells via release of the anti-inflammatory cytokines IL-10 or transforming growth factor (TGF)-β [71, 72]. There may also be other, as yet undiscovered, factors that induce myeloid cell polarization to an overall anti-inflammatory phenotype, and may aid in repair following injury [68, 73].
There are also factors that polarize peripheral cells to pro-inflammatory phenotypes, which are likely detrimental in the context of stroke. Monocytes are recruited from the periphery by monocyte chemoattractant protein-1 (MCP-1), also known as CCL2 [46]. Within days after the initial injury, neurons release factors including ATP and Fas ligand, which promote pro-inflammatory MDM phenotypes and activate pro-apoptotic pathways [1, 74]. These MDMs release inflammatory mediators such as TNF-α and IL-1β, various MMPs such as MMP-9 and MMP-3, and reactive oxygen species (ROS), all of which are implicated in ischemic damage [75, 76]. Clinical studies suggest that blocking the IL-1β receptor may improve long-term outcomes after stroke, suggesting these IL-1β secreting macrophages are detrimental to patient recovery [77,78,79]. However, the fact that clinical outcomes improved in a trial conducted prior to widespread use of thrombolysis, but were neutral in a later trial with alteplase as the standard of care with similar patient demographics, suggests a potential detrimental interaction between tPA and IL-1β pathways [78, 79]. This was suggested in the results of the SCIL-STROKE trial with IL-RA where the effect of IL1-RA on lowering IL-6 levels seemed to be beneficial but was counteracted by a potentially harmful effect. This was not proven to be due to tPA; however, 70% of the subjects were treated with alteplase [79].
There is still controversy, however, about when and which myeloid cells are pro- vs anti-inflammatory and when and why they transition from one phenotype to another. One study reported transition of both microglia and MDMs towards a pro-inflammatory state 4 days post-stroke [80]. However, using more advanced tools, Wattananit et al. suggest that this phenotypic shift may be limited to resident microglia [63]. By 2 weeks post-stroke, it appears that MDMs are primarily anti-inflammatory, and this is associated with improved long-term functional recovery. Likewise, when monocyte recruitment to the brain was inhibited, no functional recovery was observed, and there was decreased expression of anti-inflammatory genes such as TGFβ, CD163, and Ym1 [63]. Additionally, age-related changes to IRF activity may change the polarization state of microglia, therefore altering the inflammatory milieu both prior to and post-injury [69].
In summary, therapies targeted towards innate immune cells can have a significant beneficial effect in animal models; however, accumulating evidence suggests that, in order to achieve clinical success, they must be able to promote resolution and an anti-inflammatory phenotype rather than block inflammation completely. There remain controversies and knowledge gaps as to when and how these cells change function, and also about which functions will ultimately be beneficial.
The Effects of Stroke on the Acute Peripheral Response
Immunodepression After Stroke
Infection, predominantly pneumonia and UTIs, complicate stroke recovery in up to 30% of cases [81]. Pneumonia is the most common and fatal post-stroke complication, and correlates with poor stroke outcomes and possibly death (Table 1; [81, 102, 103]). It is clear that pathophysiological immunodepression, rather than pre-existing infection or aspiration of oral microbiota, is a major contributing factor [104]. For example, the severity of post-stroke infection is directly correlated with stroke size, which itself is correlated to the severity of leukocytopenia [105]. This is evident in murine stroke models, where one thousand-fold fewer bacteria are needed to cause pneumonia after stroke [106]. In addition, infection-induced inflammation exacerbates ischemic damage and acute neurological deficits in an IL-1- and neutrophil-dependent manner [9, 107].
Curiously, clinical studies with prophylactic antibiotic treatment to combat this phenomenon have yielded mixed results (Table 2). In a rodent study, treatment with ceftiofur (a β-lactam antibiotic), enrofloxacin (a fluoroquinolone antibiotic), or a vehicle control yielded no change in functional outcomes at 24 h or 1 week post-stroke [113]. Interestingly, enrofloxacin-treated rats had worse motor performance at 1 month post-stroke, suggesting this effect may be independent of the antibiotic mechanism and due to off-target effects of this antibiotic class [113]. In stroke patients, treatment with some antibiotics has reduced infarct size or infection prevalence; however, these outcomes were not associated with better overall recovery [114, 115]. In contrast, prophylactic administration of the broad spectrum mezlocillin along with the beta-lactamase inhibitor sulbactam decreased both the incidence and severity of fevers within 3–4 days of stroke, and improved long-term outcome [116]. Finally, minocycline treatment significantly improved clinical outcomes in patients, although this effect was not mediated through a decreased infection rate and actually may be due to an anti-inflammatory effect on microglia [117]. Overall, post-stroke infection is a significant risk for poor patient outcomes, and better understanding the mechanisms of immunodepression would greatly benefit patient recovery.
Leukocytopenia, or a reduction in white blood cell numbers, has been well documented in stroke patients [118, 119]. Studies have also detected a decrease in lymphocyte proliferation in response to mitogens and decreased ex vivo secretion of pro-inflammatory cytokines, suggesting that lymphocyte function is also impaired by stroke [105, 120, 121]. Consistent with this idea, T cell secretion of TNF, as well as other Th1-associated cytokines such as IL-2, IL-12, and interferon (IFN)-γ may be decreased in patients in the days after stroke [122, 123]. This could be beneficial in the absence of infection as pro-inflammatory responses in peripheral organs can exacerbate brain damage and result in worse neurological outcomes in stroke patients [124]. Thus, understanding the relationship between immune activation and suppression in peripheral organs following stroke is essential to successfully treating patients.
In contrast, other studies show that despite decreased lymphocyte numbers, their function remains intact following stroke [122, 125]. Many studies show a dramatic increase in plasma and cerebrospinal fluid (CSF) levels of TNF-α and other pro-inflammatory cytokines, which correlates with stroke severity [125,126,127,128]. Thus, it is still unclear whether lymphocytes respond normally to typical infectious pathogens, such as those responsible for pneumonia or UTIs. It is clear, however, that lymphocytopenia correlates with the occurrence of post-stroke infections, suggesting a role for this phenomenon in the development of infections, and subsequent poor patient outcomes [105, 119].
Stroke-induced activation of the autonomic nervous system (ANS) may mediate immunodepression [129, 130]. The ANS is comprised of the parasympathetic nervous system (PSNS) and the sympathetic nervous system (SNS), and both affect immune responses in the periphery [102]. Ischemic injury immediately activates the SNS, leading to splenic contraction and shrinkage [131]. The PSNS antagonizes the pathways activated by the SNS, and is suppressed following stroke [129, 132, 133]. Splenectomy prior to MCAO significantly reduces infarct size, as well as the number of neutrophils and activated microglia in the brain [134, 135]. Likewise, irradiation of the spleen following stroke significantly reduces infarct size in rats [136]. Thus, in the acute phase after ischemic injury, splenic contractions contribute to immune cell efflux and mobilization in the blood stream, which contributes to inflammatory injury in the brain.
In the days after stroke, the supply of spleen-derived immune cells becomes exhausted and spleen size is dramatically reduced, contributing to immunodepression [137]. The β2-adrenergic receptor (ADRβ2), the primary adrenoreceptor on both innate and adaptive immune cells, suppresses immune function upon activation by the SNS [138]. For example, treatment with the ADRβ2 antagonist propranolol reduced bacterial infections and subsequent mortality, supporting a role for adrenergic signaling in post-stroke immunodepression [102, 106]. Moreover, adrenergic signaling mediates a decrease in proliferation of spleen-derived immune cells after stroke [139, 140]. Treatment of rats with carvedilol (a pan-adrenergic receptor antagonist), prazosin (an α1-receptor antagonist), or propranolol independently prevented the reduction in spleen size, but only carvedilol significantly reduced infarct volume [131]. Together, these results suggest that the splenic response to stroke is mediated through the activation of both α and β adrenergic receptors, which results in an impaired antibacterial immune response following stroke [102].
Additionally, the brain can modulate the gastrointestinal system through SNS hyperactivation following stroke [129, 141]. In various murine stroke models, this results in endothelial cell necrosis, mucosal dissociation, and inflammatory cell infiltration [129, 142]. The loss of mucosal integrity and immune cell dysfunction allows bacterial translocation and increases the risk for systemic infections [143]. SNS activation and increased risk of infection has also been observed in clinical studies [103, 144]. Another mechanism of gut-brain cross-talk is mediated via the microbiome. Microbes in the gut synthesize neurotransmitters, short-chain fatty acids, and bile acids that enter the blood stream and signal to neurons, astrocytes, and microglia in the CNS [145]. The makeup of microbes in the gut not only influences stroke outcomes, but is also itself altered after stroke [146,147,148]. Many of the signaling mechanisms between the microbiome are not yet understood, and these studies are not yet translated to human populations. Both are active areas of current research.
Peripheral Immune Activation by Stroke
Although post-stroke immunosuppression has been confirmed in both clinical and animal models, brain signaling to the periphery following stroke can also activate the peripheral immune system. Following CNS innate immune activation, secreted chemokines and cytokines not only assist in local immune activation but also signal back through the blood stream to recruit peripheral cells, primarily from the spleen, to the site of injury [149, 150]. This cross-talk between organ systems occurs throughout the body, and it can be detrimental, as severe neurological stress resulting from stroke, traumatic brain injury, or infection can cause multiple organ failure [124]. In humans, there is a long-lasting peripheral immune activation, and the degree of activation is associated with later cognitive decline [151].
In addition to the SNS, the brain and spleen communicate via activation of the hypothalamic–pituitary–adrenal (HPA) axis. Both induce release of catecholamines and steroids that trigger immune cell efflux from the spleen and trafficking to the brain [152]. Cytokines such as IL-1, IL-6, IL-10, and TNF-α are released by both brain resident and peripheral immune cells following stroke, and mediate immune activation [103, 153,154,155]. For example, IL-1 increases after stroke, and signals to the hypothalamus to release corticotropin-releasing hormone (CRH) into the blood [156, 157]. Subsequently, CRH stimulates secretion of adrenocorticotropic hormone (ACTH) from the anterior pituitary gland, which circulates to the adrenal glands and induces secretion of glucocorticoids by the adrenal cortex [158,159,160]. Interestingly, the adrenal gland may also activate peripheral immune responses through humoral mechanisms or catecholamines [103].
Adaptive Immune Cell Roles in Neuroinflammation After Stroke
T Lymphocytes
Adaptive immune cells such as B and T lymphocytes also traffic from the blood into the brain after stroke. T cells are heterogeneous and include subsets with various functions—regulatory (Treg), pro-inflammatory (Th1 and Th17), and anti-inflammatory (Th2). Upon entering the brain, their roles are determined by the specific populations of cells that become activated. Although it has been suggested that infiltration of T cells into the brain begins 3–4 days post-stroke, following monocyte and neutrophil infiltration [161], Tregs accumulate within 24 h in rodent stroke models [61, 162, 163].
T cells are a key mediator of acute stroke pathology, and ablating these cells after stroke may be neuroprotective [164]. In Rag1(−/−) mice, a reduction in infarct volume and an improvement in the neurological deficit of wild-type mice occurred 24 h post-injury. This protection was reversed in Rag1(−/−) mice reconstituted with wild-type splenocytes [162]. Inhibition of lymphocyte trafficking results in fewer T cells in the CNS following stroke, and reduced pro-inflammatory cytokine production [162].
T cells can be subdivided into different populations based on surface markers and cytokine expression, which determines their impact following stroke. For example, CD4+CD25+FoxP3+ T cells proliferate and accumulate in the ischemic brain up to 30 days post injury and promote an anti-inflammatory environment [165]. Additionally, adoptive transfer of Tregs reduces inflammatory responses both intrinsic and extrinsic to the CNS [166]. Moreover, Tregs provide neurovascular protection against stroke by inhibiting peripheral neutrophil-derived MMP-9 production [166]. Thus, Tregs may be promising candidates for cell-based therapies targeting post-stroke inflammatory dysregulation and neurovascular dysfunction. In contrast, CD8+ T cells are first observed in the brain at 3 h post-stroke and maintain their numbers in the following days. CD8+ effector T cells are likely detrimental in the acute phase of stroke due to pro-inflammatory properties [165]. Thus, a primary role of Tregs is likely to provide neuroprotection by suppressing these effector cells [165].
Interestingly, Treg-mediated neuroprotection might depend primarily on their ability to produce the anti-inflammatory cytokine IL-10. Treg transfer from IL-10-deficient mice into mice with stroke had a minimal effect on recovery, whereas IL-10-producing Tregs reduced stroke damage by antagonizing the pro-inflammatory effects of IFN-γ and TNF-α [167, 168]. Several studies in various disease states have demonstrated that antigen-specificity is necessary for Tregs to produce IL-10, which may further clarify why this cell population is only beneficial in certain microenvironments [2, 169]. In contrast, Treg depletion immediately after stroke may improve stroke outcomes up to 1 week later, as Tregs are adherent to cerebral blood vessels in the hours after stroke, and may contribute to microvascular dysfunction and the “no-reflow” phenomenon [170]. These discrepancies may be due to the different stroke models utilized resulting in different stroke sizes, and different methods of Treg depletion [167, 170]. Ultimately, it is apparent that the immune response is specific to the stroke model utilized and the extent and severity of ischemia accomplished in a given model [171].
Inhibiting specific T cell subtypes associated with detrimental cytokine signaling reduces infarct size [172,173,174]. Adoptive transfer of Th1- and Th17-type T cells into stroke mice increases infarct volume suggesting that both IFN-γ- and IL-17-mediated responses impact stroke damage [1, 164, 174]. Supporting this, post-mortem sections of human stroke tissue identified IL-17 localization in CD4+ T cells and astrocytes [166]. Th1 cells are major producers of IFN-γ, and potentiate damage by increasing monocyte expression of IFN-inducible protein 10 (IP-10), or CXCL10, which is itself a chemoattractant for activated T cells [162, 175]. Similarly, mice lacking CCL5, which is critical for recruiting and activating T lymphocytes, had smaller infarct volumes compared to wild-type mice, supporting a detrimental role of T cells following stroke [176]. Furthermore, CD4+ T cell–induced IL-21 has been observed in post-mortem human stroke tissue [173]. Inhibition of IL-21 before transient MCAO in mice is neuroprotective, supporting a detrimental role [173]. Overall, these findings highlight the importance of identifying and targeting specific T cell subpopulations in order to develop appropriate therapeutic targets for stroke.
B Lymphocytes After Stroke
B cells also infiltrate the brain in the days after ischemic injury. B cell deficiency worsens histological damage and functional outcomes after transient cerebral ischemia [177]. B cell–deficient μMT mice had larger infarct volumes, higher mortality, more severe functional deficits, and increased numbers of activated T cells, macrophages, microglia, and neutrophils compared to wild-type mice, and this effect was reversed following adoptive transfer of B cells from wild-type mice [177]. In contrast, other studies have not observed any differences in stroke size or severity in μMT mice [178, 179]. Interestingly, when IL-10-deficient B cells were transferred into μMT mice, this effect was not observed, supporting a role for IL-10-producing B cells in stroke recovery [177]. Although regulatory B cells represent only ~ 0.5% of CD19+ B cells, they are an important subset that secrete high levels of IL-10 [180]. There is evidence that early regulatory B cell responses to stroke are beneficial. Regulatory B cells also limit inflammation in animal models of MS via the production of IL-10 [181]. Together, these data suggest that the role of B lymphocytes in cerebral ischemia may be protective rather than pathogenic. It is notable, however, that this effect was observed only 48 h after MCAO, before B cells are typically observed in the brain. In a different study, B cell–deficient mice failed to show improvement against ischemic injury, although this may be because the analysis was performed only 24 h post-injury [162]. It may be that early effects exerted by B cells take place outside the brain, perhaps in lymphatic tissue. CNS antigens can indeed be detected in the cervical lymph nodes of stroke patients within 3 days of stroke in concentrations proportional to the extent of brain damage [182].
Overall, it is clear that all of these inflammatory components (DAMPs, myeloid cells, and lymphocytes) are inter-related, and altering the activity of one population will influence the others. While T cell responses following ischemic stroke can directly affect stroke recovery, they also influence B cell activation. For B cell activation and auto-antibody responses to occur in response to stroke, CNS antigens released from dead and damaged cells must bind the receptor of autoreactive cells that have escaped negative selection. These CNS antigens may reach secondary lymphoid tissue directly, or within antigen-presenting cells that have phagocytosed dead tissue. An impaired BBB may also facilitate direct antigen exit to circulation. Additionally, autoreactive Th cells that have also escaped negative selection must be present to provide a reciprocal secondary signal, and to fully activate the B cell [183]. Furthermore, DAMP-mediated activation of TLRs can increase the expression of co-stimulatory molecules on B cells, to further facilitate activation and autoimmunity [184].
Resolution Versus Chronic Neuroinflammation After Stroke
Resolution of Neuroinflammation
Proper resolution of inflammation and subsequent repair require both debris clearance and pro-inflammatory signaling [185]. Phagocytosis of cellular debris by myeloid cells results in the production of cytokines such as TGF-β and IL-10 [186, 187]. While these cytokines do have pro-inflammatory properties, in the context of inflammatory resolution they suppress Th1 responses and promote Treg development, thereby promoting repair and survival rather than cell death and damage [186, 187]. Similarly, the same cells (astrocytes, microglia, macrophages) that initially secrete pro-inflammatory cytokines transition phenotypically to become critical suppliers of growth factors and other molecules required for neurogenesis, angiogenesis, and neuronal sprouting, among other repair-oriented processes [188]. For example, astrocyte production of vascular endothelial growth factor (VEGF) is essential for angiogenesis, and microglial production of insulin-like growth factor-1 is required for neuronal sprouting following ischemia [188].
Resolving inflammation is an active process, rather than simply a decline in inflammatory signaling. In the brain, one cue for resolution of the inflammatory response is phagocytic removal of apoptotic neutrophils [189]. Interestingly, as in neutrophil activation, it appears macrophages require both integrin-ligand interactions and TNF-α in order to properly induce apoptosis [189]. Thus, proper inflammatory resolution relies on each of these signals, and consequent removal of cytotoxic cells. Unfortunately, however, the initiation of inflammatory resolution is otherwise not well described after stroke.
There may be similar mechanisms to those important for immune resolution in other conditions, such as wound healing. Following tissue damage, glucocorticoid production increases, which can induce macrophages to transition from a pro-inflammatory to an anti-inflammatory phenotype [190, 191]. Moreover, IL-4 secretion by Th2 cells provides an additional signal for macrophages to transition to a phenotype that suppresses inflammation and favors wound healing [192]. The role of these molecules in triggering this transition following ischemia still needs more attention.
Resolving inflammation after stroke is crucial. When resolution does not properly occur, a deleterious chronic condition develops. This happens more frequently after brain ischemia than ischemia in other organs [193], and several mechanisms may be involved. Inflammation may be exacerbated by a failure of neutrophils to undergo apoptosis, or by a failure of macrophages to phagocytose and clear the apoptotic cells or extracellular myelin and cholesterol in a timely manner [188, 193]. Additionally, premature dampening of T cell effector functions may allow inflammatory stimuli to persist, and result in non-resolving inflammation [188]. A chronic condition may then develop and result in further tissue damage.
Post-stroke Dementia and B Lymphocytes
Chronic neuroinflammation is associated with cognitive decline and dementia after stroke. Dementia is a significant problem in stroke survivors [194]. Furthermore, stroke not only has the potential to immediately affect cognition due to direct tissue damage in the acute phase, it also worsens the trajectory of cognitive decline [195]. Much work has been done to understand acute recovery from the initial infarct, but this later chronic stage is less well studied. While initial B cell activation likely occurs in the periphery, the ischemic lesion may become an additional site of B cell activation and auto-antibody production in the weeks after stroke if the immune response does not resolve properly.
In models of permanent distal MCAO, adult wild-type mice with a cortical stroke are cognitively normal at 1 week post-stroke [178, 196]. They then develop a cognitive deficit 8 weeks later [178, 196]. Importantly, the fact that mice are cognitively normal 1 week after stroke suggests that cognitive decline is not a direct effect of damage from the stroke itself. When this same stroke model is utilized in μMT mice which lack mature B cells, or in wild-type mice treated with anti-CD20 to abolish B cells, the cognitive deficit does not develop [178]. Together, these data suggest that B cells are critical to the development of a delayed cognitive deficit following stroke. Interestingly, there are increased numbers of B cells in the brain in approximately two thirds of human stroke survivors who develop dementia after stroke [178].
In both animal models and in humans, antibodies specific for CNS self-antigens can develop following stroke [197,198,199], although negative studies have also been recently published [200]. The specific B cell isotype is determined by cytokines produced by Th cells [201]. Auto-antibodies of the IgG, IgM, and IgA isotype have all been detected at an increased frequency in the CSF of stroke patients, suggesting an important role for T cell signaling in this pathogenesis [202, 203].
In the months and years following the initial injury, the neuroimmune response may trigger a progressive neurodegenerative process driven by dysregulated adaptive immune responses, and this may be a factor in vascular cognitive impairment. Post-stroke cognitive impairment is indeed very common, impacting 30–50% of stroke survivors [194, 204, 205], but a tight link to autoreactive antibodies against brain proteins, found in both the periphery and the CNS in patients, has not been established. Still, there are a few small studies that strongly suggest a connection.
For example, one study analyzed serum antibodies from a cohort of stroke patients who were subjected to cognitive testing at multiple time points up to 365 days after stroke [206]. It found that 17% of patients had a decline in cognitive ability in the year after stroke, while 83% were stable or improved [207]. Cognitive decline was predicted by antibody titers to anti-myelin basic protein (MBP) but not anti-proteolipid protein (PLP) [207]. Additional clinical studies have identified auto-antibodies against neurofilament, glial fibrillary acidic protein, S100B, MBP, and PLP in plasma and CSF of stroke patients [208,209,210]. The presence of auto-antibodies in the CNS could be harmful via multiple mechanisms, including antibody-mediated cellular toxicity, fixation of complement, inhibition of signal transduction, or direct induction of apoptosis.
Finally, it is unlikely that B lymphocytes act alone in the pathogenesis of immune mediated post-stroke cognitive decline. Innate immune cells, T cells, and even neuronal responses likely also play a role and are present late after stroke. Larger studies with more detailed cognitive testing and longer follow-up periods will be required to determine whether serum and/or CSF auto-antibodies are predictive and causative in post-stroke cognitive decline and dementia.
A working model is that a pro-inflammatory state in the periphery acts as an adjuvant to brain proteins when they are released into the bloodstream post-stroke. To test this, we used single-cell mass cytometry to characterize the peripheral immune response to stroke in the year following stroke, and subsequently correlated this response with cognitive changes [19]. Using Elastic Net regression modelling, the systemic immune response was categorized into three distinct phases: acute, intermediate, and late. The acute phase (2 days post-stroke) was characterized by increased signal transducer and activator of transcription-3 (STAT3) signaling in innate immune cells. The intermediate phase (5 days post-stroke) was characterized by increased CREB signaling in adaptive immune cells. The late phase (90 days post-stroke) was characterized by elevated neutrophil numbers, and IgM+ B cells [19]. When aligned with changes in cognitive function between days 90 and 365 post-stroke, the acute inflammatory phase correlated with post-stroke cognitive trajectories, supporting the idea that the early immune response may influence later adaptive immune response and subsequently alter post-stroke cognition [19]. Further studies involving longer term cognitive testing and immune analyses will be critical in understanding these relationships and translating them into therapeutics.
Immunomodulation as a Potential Stroke Therapy
A variety of strategies have been utilized to target the immune response to stroke, such as antioxidant therapies, statins, and inhibition of leukocyte trafficking (Table 3). These studies in murine models of stroke have yielded conflicting results, as well as limited translational success in human patients in the acute phase. The apparently contradictory results between mouse and human studies are likely due to multiple factors: the heterogeneity in stroke pathogenesis in different murine models, the different time points assessed in each both animal and human studies, the different models of stroke utilized in the laboratory, the multitude of immune cells involved, and the variety of factors that immune cells can produce. Additionally, most trials were done before it was possible to choose patients based on the volume of stroke core (dead tissue) and tissue at risk (the brain region not functioning due to low perfusion that is potentially salvageable). With modern imaging techniques to select patients, it is possible that some previously tried therapies might be more successful in select subgroups of stroke patients.
Antioxidant treatment in stroke hinges on the idea that free radical production, which occurs during tissue re-perfusion, causes severe tissue damage and subsequent cell death [227]. In rodent models, immediate treatment with ROS-scavenging molecules reduces infarct volume and improves acute recovery within 3 days of stroke [227]. While many of these treatments seem to be safe and well-tolerated in human patients, trials demonstrated at best only minor, short-term improvement but were given later after stroke [223, 226, 228]. In contrast, studies of the lipid peroxidation inhibitor tirilazad resulted in increased mortality and worse outcomes in patients [225]. The reason for this lack of translation is perplexing. Further work is necessary to improve the similarities between animal models and human disease and understand the differences.
Additionally, inhibition of lymphocyte trafficking has received a lot of attention due to promising results in rodent stroke models. Fasudil, a Rho kinase inhibitor, prevented neutrophil and monocyte infiltration into the brain, increased regional cerebral blood flow, and upregulated eNOS activity in endothelial cells in rodent studies [218, 229]. Animal studies have demonstrated efficacy of Fasudil in reducing pulmonary arterial pressure in models of pulmonary hypertension, but effects on systemic blood pressure were not evaluated in clinical trials targeting stroke recovery [230, 231]. While a clinical trial showed minor functional improvement in patients, the short-term study (a 30-day assessment) makes it hard to assess the overall patient benefit, as it is clear that the acute immune response plays a critical role in determining long-term outcomes [218].
In a clinical trial of UK-279,276 (a recombinant glycoprotein that inhibits leukocyte infiltration), no effect on infarct size or overall recovery was observed 90 days after stroke [217]. This is in contrast to rodent studies which observed a reduction in neutrophil accumulation and infarct volume in rats 7 days after stroke [232]. However, these studies did not assess functional outcomes in rodent models. Therefore, if future murine studies are to become translatable to human trials, they should focus on longer term outcomes and functional recovery which are the main endpoints assessed in clinical trials, rather than just a reduction in infarct size.
Antibody-mediated blockade of adhesion molecules on endothelial cells has been utilized to reduce lymphocyte trafficking into rodent brains after stroke. ICAM-1-deficient mice have reduced infarct volume, increased survival, and reduced neurological deficits following transient MCAO [52]. However, a clinical trial assessing the efficacy of a murine ICAM-1 antibody, enlimomab, revealed worsened neurological performance, an increased number of stroke-related deaths, and more adverse occurrences such as infections and fever [212, 233]. These effects are thought to be mediated by an immune response to the murine antibody treatment, however, rather than an effect of blocking ICAM-1.
Likewise, studies of VCAM-1 have yielded conflicting results. Reducing VCAM-1 expression diminished T cell infiltration into the CNS and decreased infarct volume in one rodent study [168]. However, other studies using anti-VCAM-1 antibodies revealed no change in outcomes compared to IgG-treated animals [168, 234]. Unfortunately, none of these studies examine the effects of adhesion molecule inhibition on endothelial cell activation and signaling, as lack of target engagement could explain some of these disparate results. While anti-VCAM1 has not yet been tested in humans, there have been two phase 2 clinical trials of Natalizumab (anti-VLA-4, the ligand for VCAM-1 on lymphocytes). The first 2a trial, ACTION, failed to reduce stroke size but demonstrated safety and a trend towards better 30- and 90-day outcomes [216]. ACTION II was a phase 2b trial aimed at replicating these later favorable outcomes and is not yet published but was reported to be negative [235]. In this case, animal studies utilized shorter drug administration times and yielded mixed results with different animal stroke models [168, 236,237,238,239].
Another immunomodulatory agent, the sphingosine-1-phosphate inhibitor Fingolimod, inhibits leukocyte trafficking and is approved for use in multiple sclerosis patients [214]. Both rodent and human studies showed reductions in infarct volumes and neurological recovery; however, the patient trials have been small and were open label [213,214,215, 240].
In addition to the specific reasons above, there are systematic issues that may be critical to improve in order to develop successful immunomodulatory therapies. Preclinical stroke researchers primarily utilize younger, healthy male mice and give drugs at the time of or 3 h after ischemia, while human trials often choose later time points (Table 3). Animal studies should include both sexes, permanent and transient stroke models, and later dosing time points, and should also consider inter-species differences in immune responses and the effects of anesthetic use [241]. In addition, comorbid conditions are understudied in animals. Common comorbidities in stroke patients include age, obesity, diabetes, and hypertension, and each is independently associated with increased peripheral inflammation, which results in worse outcomes for stroke patients [242,243,244] and likely would also affect the patient response to immune modulating drugs. Research incorporating these different considerations will hopefully lead to improved translation between preclinical animal and clinical human studies and facilitate therapeutic development.
Future Research Priorities
The vast majority of clinical studies of immunomodulation after stroke have focused on the acute phase, assessing outcomes up to 90 days (Table 3). While this timing matches the timing of assessments in many pre-clinical stroke models, the lack of clinical translation highlights the need for longer term, chronic follow-ups in both the clinic and the laboratory so that we can better understand outcomes following stroke. Additionally, many clinical studies begin treatment within 6 h of stroke onset. While this timing may be appropriate for therapies targeting the innate immune response, it may be detrimental in its inhibition of proper inflammatory resolution. Further work is needed to understand the complexities of immune activation and how it transitions to successful inflammatory resolution after stroke, as well as targeted therapies to promote anti-inflammatory immune cell phenotypes. Additionally, for therapies targeting adaptive immune cells, later treatment in the days/weeks after stroke, when these cells begin accumulating in the brain, may be more appropriate. Finally, pre-clinical models that better mimic the human disease by incorporating both permanent and transient ischemia together, using both sexes and humanized immune responses, and incorporate pro-inflammatory comorbidities such as age, obesity, and diabetes will facilitate clinical translation. Further human studies are also necessary to optimize these changes and lead to a subsequent redesign of human trials that will be better suited to beneficially modulate inflammation after stroke.
Abbreviations
- ACTH:
-
adrenocorticotropic hormone
- ADRβ2:
-
β2-adrenergic receptor
- AIS:
-
acute ischemic stroke
- ANS:
-
autonomic nervous system
- ATP:
-
adenosine triphosphate
- BBB:
-
blood–brain barrier
- BI:
-
Barthel Index
- CHF:
-
congestive heart failure
- CNS:
-
central nervous system
- CRH:
-
corticotropin releasing hormone
- CSF:
-
cerebrospinal fluid
- CX3CL1:
-
fractalkine
- DAMP:
-
damage-associated molecular pattern
- GFAP:
-
glial fibrillary acidic protein
- GPCR:
-
G-protein coupled receptor
- HMGB1:
-
high-mobility group box 1
- HO-1:
-
heme oxygenase-1
- HPA:
-
hypothalamic-pituitary-adrenal
- Hsp:
-
heat shock protein
- ICAM-1:
-
intracellular adhesion molecule-1
- IFN:
-
interferon
- IL:
-
interleukin
- IP-10:
-
interferon-inducible protein-10
- IV:
-
intravenous
- LFA-1:
-
lymphocyte function-associated antigen 1
- LPS:
-
lipopolysaccharide
- MBP:
-
myelin basic protein
- MCAO:
-
middle cerebral artery occlusion
- MCP-1:
-
monocyte chemoattractant protein-1
- MICU:
-
medical intensive care unit
- MMP:
-
matrix metalloproteinase
- MoCA:
-
Montreal Cognitive Assessment
- MRI:
-
magnetic resonance image
- mRS:
-
modified Rankin Scale
- NICU:
-
neurological intensive care unit
- NIHSS:
-
National Institute of Health Stroke Scale
- PLA:
-
platelet-leukocyte aggregate
- PLP:
-
proteolipid protein
- PPAR:
-
peroxisome proliferator–activated receptor
- PSGL-1:
-
P-selectin glycoprotein ligand-1
- PSNS:
-
parasympathetic nervous system
- PSP:
-
post-stroke pneumonia
- RAGE:
-
receptor for advanced glycation end products
- ROS:
-
reactive oxygen species
- SAI:
-
stroke-associated infection
- siRNA:
-
small interfering ribonucleic acid
- SNS:
-
sympathetic nervous system
- STAT3:
-
signal transducer and activator of transcription-3
- SU:
-
stroke unit
- TGF:
-
transforming growth factor
- Th:
-
T helper cell
- TIA:
-
transient ischemic attack
- TLR:
-
toll-like receptor
- TNF:
-
tumor necrosis factor
- tPA:
-
tissue plasminogen activator
- Treg:
-
T regulatory cell
- UTI:
-
urinary tract infection
- VCAM-1:
-
vascular cell adhesion molecule-1
- VEGF:
-
vascular endothelial growth factor
- VLA-4:
-
very late antigen-4
References
Rayasam A, Hsu M, Hernández G, et al. Contrasting roles of immune cells in tissue injury and repair in stroke: The dark and bright side of immunity in the brain. Neurochem Int 2017;107:104–116.
Chamorro A, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R. The immunology of acute stroke. Nat Rev Neurol. 2012;8(7):401–410. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22664787
Lee S. Pathophysiology of Ischemic Stroke. 2017;(1).
del Zoppo GJ, Schmid-Schönbein GW, Mori E, Copeland BR, Chang CM. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke. 1991;22(10):1276–1283. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1926239
Gliem M, Mausberg AK, Lee JI, et al. Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann Neurol. 2012;71(6):743–752. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=22718543
Bravo-Alegria J, McCullough LD, Liu F. Sex differences in stroke across the lifespan: The role of T lymphocytes. Neurochem Int 2017;107:127–137. https://doi.org/10.1016/j.neuint.2017.01.009
Macrez R, Ali C, Toutirais O, et al. Stroke and the immune system: From pathophysiology to new therapeutic strategies. Lancet Neurol 2011;10(5):471–480. https://doi.org/10.1016/S1474-4422(11)70066-7
Gorelick PB, Scuteri A, Black SE, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke. 2011;42(9):2672–2713. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=21778438
McColl BW, Rothwell NJ, Allan SM. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1- and neutrophil-dependent mechanisms. J Neurosci. 2007;27(16):4403–4412. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17442825
Terao S, Yilmaz G, Stokes KY, Ishikawa M, Kawase T, Granger DN. Inflammatory and injury responses to ischemic stroke in obese mice. Stroke 2008;39(3):943–950. Available from: https://www.ahajournals.org/doi/10.1161/STROKEAHA.107.494542
O’Donnell MJ, Xavier D, Liu L, et al. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): a case-control study. Lancet 2010;376(9735):112–123. Available from: https://www.sciencedirect.com/science/article/pii/S0140673610608343
Saver JL, Albers GW, Dunn B, Johnston KC, Fisher M. Stroke Therapy Academic Industry Roundtable (STAIR) recommendations for extended window acute stroke therapy trials. Stroke 2009;40(7):2594–2600. Available from: https://www.ahajournals.org/doi/10.1161/STROKEAHA.109.552554
Papadopoulos S, Chandler W, Salamat M, Tool E, Sackellares J. Recombinant human tissue-type plasminogen activator therapy in acute thromboembolic stroke. J Neurosurg 1987;67:394–398. Available from: https://thejns.org/view/journals/j-neurosurg/67/3/article-p394.xml
Nogueira RG, Jadhav AP, Haussen DC, et al. Thrombectomy 6 to 24 Hours after Stroke with a Mismatch between Deficit and Infarct. N Engl J Med 2018;378(1):11–21. Available from: http://www.nejm.org/doi/10.1056/NEJMoa1706442
Albers GW, Marks MP, Kemp S, et al. Thrombectomy for stroke at 6 to 16 hours with selection by perfusion imaging HHS public access. N Engl J Med 2018;378(8):708–718. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6590673/pdf/nihms-947728.pdf
Campbell BC V, Ma H, Ringleb PA, et al. Extending thrombolysis to 4·5-9 h and wake-up stroke using perfusion imaging: a systematic review and meta-analysis of individual patient data. Lancet (London, England). 2019;394(10193):139–147. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31128925
Adeoye O, Hornung R, Khatri P, Kleindorfer D. Recombinant tissue-type plasminogen activator use for ischemic stroke in the United States: a doubling of treatment rates over the course of 5 years. Stroke. 2011;42(7):1952–1955. Available from: https://www.ahajournals.org/doi/10.1161/STROKEAHA.110.612358
Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17(7):796–808. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=21738161
Tsai AS, Berry K, Beneyto MM, et al. A year-long immune profile of the systemic response in acute stroke survivors. Brain. 2019;142(4):978–991. Available from: https://academic.oup.com/brain/article/142/4/978/5373058
Gülke E, Gelderblom M, Magnus T. Danger signals in stroke and their role on microglia activation after ischemia. Ther Adv Neurol Disord 2018;11:1–14.
Yang H, Lundbäck P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME, et al. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol Med. 2012 18(1):250–259. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22105604
Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418(6894):191–195. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12110890
Murray KN, Buggey HF, Denes A, Allan SM. Systemic immune activation shapes stroke outcome. Mol Cell Neurosci 2013;53:14–25. https://doi.org/10.1016/j.mcn.2012.09.004
Ke X, Fei F, Chen Y, et al. Hypoxia upregulates CD147 through a combined effect of HIF-1 α and Sp1 to promote glycolysis and tumor progression in epithelial solid tumors. Carcinogenesis 2012;33(8):1598–1607.
Shichita T, Ito M, Morita R, et al. MAFB prevents excess inflammation after ischemic stroke by accelerating clearance of damage signals through MSR1. Nat Med 2017;23(6):723–732. https://doi.org/10.1038/nm.4312
Hayakawa K, Qiu J, Lo EH. Biphasic actions of HMGB1 signaling in inflammation and recovery after stroke. 2010; Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3705575/pdf/nihms484418.pdf.
Asea A, Rehli M, Kabingu E, et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277(17):15028–15034. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11836257
Kim JY, Yenari MA. The immune modulating properties of the heat shock proteins after brain injury. Anat Cell Biol 2013.
Zheng Z, Kim JY, Ma H, Lee JE, Yenari MA. Anti-inflammatory effects of the 70 kDa heat shock protein in experimental stroke. J Cereb Blood Flow Metab 2008.
Batulan Z, Pulakazhi Venu VK, Li Y, et al. Extracellular Release and Signaling by Heat Shock Protein 27: Role in Modifying Vascular Inflammation. Front Immunol 2016;7:285. Available from: http://journal.frontiersin.org/Article/10.3389/fimmu.2016.00285/abstract
Sharp FR, Zhan X, Liu D-Z. Heat shock proteins in the brain: role of Hsp70, Hsp 27, and HO-1 (Hsp32) and their therapeutic potential. Transl Stroke Res. 2013;4(6):685–692. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24323422
LeBlanc RH, Chen R, Selim MH, Hanafy KA. Heme oxygenase-1-mediated neuroprotection in subarachnoid hemorrhage via intracerebroventricular deferoxamine. J Neuroinflammation 2016;13(1):244. Available from: http://jneuroinflammation.biomedcentral.com/articles/10.1186/s12974-016-0709-1
Lopes Pinheiro MA, Kooij G, Mizee MR, et al. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim Biophys Acta Mol basis Dis 2016;1862(3):461–471. https://doi.org/10.1016/j.bbadis.2015.10.018
Salmeron KE, Edwards DN, Fraser JF, Bix GJ. Edema and BBB Breakdown in Stroke [Internet]. Brain Edema: From Molecular Mechanisms to Clinical Practice. Elsevier Inc.; 2017. 219–233 p. https://doi.org/10.1016/B978-0-12-803196-4.00012-6.
Hawkins BT, Egleton RD. Pathophysiology of the Blood-Brain Barrier: Animal Models and Methods. Curr Top Dev Biol 2007;80(07):277–309.
Rodrigues SF, Granger DN. Blood cells and endothelial barrier function. Tissue Barriers. 2015;3(1–2):e978720. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25838983
Shulman Z, Shinder V, Klein E, et al. Lymphocyte crawling and transendothelial migration require chemokine triggering of high-affinity LFA-1 integrin. Immunity. 2009;30(3):384–396. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19268609
Schenkel AR, Chew TW, Muller WA. Platelet Endothelial Cell Adhesion Molecule Deficiency or Blockade Significantly Reduces Leukocyte Emigration in a Majority of Mouse Strains. J Immunol. 2004 ;173(10):6403–6408. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15528380
Yilmaz G, Granger DN. Leukocyte Recruitment and Ischemic Brain Injury. NeuroMolecular Med 2010;12(2):193–204. Available from: http://link.springer.com/10.1007/s12017-009-8074-1
Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surg Neurol 2006;66(3):232–245.
Zlokovic B V. Remodeling after stroke. Nat Med 2006;12(4):390–391. Available from: http://www.nature.com/articles/nm0406-390
Cheng T, Petraglia AL, Li Z, et al. Activated protein C inhibits tissue plasminogen activator–induced brain hemorrhage. Nat Med 2006;12(11):1278–1285. Available from: http://www.nature.com/articles/nm1498
Zlokovic B V. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron. 2008;57(2):178–201.
Knowland D, Arac A, Sekiguchi KJ, et al. Stepwise Recruitment of Transcellular and Paracellular Pathways Underlies Blood-Brain Barrier Breakdown in Stroke. Neuron. 2014;82(3):603–617. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24746419
McColl BW, Rothwell NJ, Allan SM. Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J Neurosci. 2008;28(38):9451–9462. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18799677
Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic A V. Absence of the Chemokine Receptor CCR2 Protects Against Cerebral Ischemia/Reperfusion Injury in Mice. Stroke. 2007 38(4):1345–1353. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17332467
Gelderblom M, Leypoldt F, Steinbach K, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40(5):1849–1857. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19265055
Barone FC, Schmidt DB, Hillegass LM, Price WJ, White RF, Feuerstein GZ, et al. Reperfusion increases neutrophils and leukotriene B4 receptor binding in rat focal ischemia. Stroke. 1992 ;23(9):1337–1347; discussion 1347-8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1381529
Price CJS, Menon DK, Peters AM, et al. Cerebral neutrophil recruitment, histology, and outcome in acute ischemic stroke: an imaging-based study. Stroke. 2004;35(7):1659–1664. Available from: https://www.ahajournals.org/doi/10.1161/01.STR.0000130592.71028.92
Matsuo Y, Onodera H, Shiga Y, et al. Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat. Effects of neutrophil depletion. Stroke. 1994 ;25(7):1469–1475. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8023364
Hudome S, Palmer C, Roberts RL, Mauger D, Housman C, Towfighi J. The Role of Neutrophils in the Production of Hypoxic-Ischemic Brain Injury in the Neonatal Rat. Pediatr Res. 1997;41(5):607–616. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9128280
Connolly ES, Winfree CJ, Springer TA, et al. Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J Clin Invest. 1996;97(1):209–216. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8550836
Soriano SG, Lipton SA, Wang YF, et al. Intercellular adhesion molecule-1-deficient mice are less susceptible to cerebral ischemia-reperfusion lnjury. Ann Neurol 1996;39(5):618–624. Available from: http://doi.wiley.com/10.1002/ana.410390511
Hernandez LA, Grisham MB, Twohig B, Arfors KE, Harlan JM, Granger DN. Role of neutrophils in ischemia-reperfusion-induced microvascular injury. Am J Physiol Circ Physiol. 1987;253(3):H699–703. Available from: http://www.ncbi.nlm.nih.gov/pubmed/3631303
Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol 2006;6(3):173–182. Available from: http://www.nature.com/articles/nri1785
Barone FC, Feuerstein GZ. Inflammatory mediators and stroke. J Cereb Blood Flow Metab 1999;19:819–834.
Kriz J. Inflammation in ischemic brain injury: timing is important. Crit Rev Neurobiol 2006;18(1–2):145–157. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17725517
Harrison JK, Jiang Y, Chen S, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci. 1998;95(18):10896–10901. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9724801
Hughes PM, Allegrini PR, Rudin M, Perry VH, Mir AK, Wiessner C. Monocyte Chemoattractant Protein-1 Deficiency is Protective in a Murine Stroke Model. J Cereb Blood Flow Metab 2002;22(3):308–317. Available from: http://journals.sagepub.com/doi/10.1097/00004647-200203000-00008
Hammond MD, Ai Y, Sansing LH. Gr1+ Macrophages and Dendritic Cells Dominate the Inflammatory Infiltrate 12 h After Experimental Intracerebral Hemorrhage. Transl Stroke Res 2012;3(S1):125–131. Available from: http://link.springer.com/10.1007/s12975-012-0174-9
Brait VH, Rivera J, Broughton BRS, Lee S, Drummond GR, Sobey CG. Chemokine-related gene expression in the brain following ischemic stroke: No role for CXCR2 in outcome. Brain Res 2011;1372:169–179. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0006899310026004
Bennett ML, Bennett FC, Liddelow SA, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. 2016;113(12):E1738-E1746. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26884166
Wattananit S, Tornero D, Graubardt N, et al. Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J Neurosci 2016;36(15):4182–4195.
Ritzel RM, Patel AR, Grenier JM, et al. Functional differences between microglia and monocytes after ischemic stroke. J Neuroinflammation 2015;12(1):106. Available from: http://www.jneuroinflammation.com/content/12/1/106
Fumagalli S, Perego C, Pischiutta F, Zanier ER, De Simoni M-G. The Ischemic Environment Drives Microglia and Macrophage Function. Front Neurol. 2015;6:81. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25904895
Otxoa-de-Amezaga A, Miró-Mur F, Pedragosa J, et al. Microglial cell loss after ischemic stroke favors brain neutrophil accumulation. Acta Neuropathol 2019;137(2):321–341. Available from: http://link.springer.com/10.1007/s00401-018-1954-4
Jin W-N, Shi SX-Y, Li Z, et al. Depletion of microglia exacerbates postischemic inflammation and brain injury. J Cereb Blood Flow Metab. 2017;37(6):2224–2236. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28273719
Zhao X, Wang H, Sun G, Zhang J, Edwards NJ, Aronowski J. Neuronal Interleukin-4 as a Modulator of Microglial Pathways and Ischemic Brain Damage. J Neurosci. 2015;35(32):11281–11291. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26269636
Zhao S, Wang C, Xu H, et al. Age-related differences in interferon regulatory factor-4 and -5 signaling in ischemic brains of mice. Acta Pharmacol Sin 2017;38(11):1425–1434. Available from: http://www.nature.com/articles/aps2017122
Bouhlel MA, Derudas B, Rigamonti E, et al. PPARγ Activation Primes Human Monocytes into Alternative M2 Macrophages with Anti-inflammatory Properties. Cell Metab 2007;6(2):137–143. Available from: https://www.sciencedirect.com/science/article/pii/S1550413107001660
Strle K, Zhou J-H, Broussard SR, et al. IL-10 promotes survival of microglia without activating Akt. J Neuroimmunol 2002;122(1–2):9–19. Available from: https://www.sciencedirect.com/science/article/abs/pii/S0165572801004441
Streit WJ. Microglial Response to Brain Injury: A Brief Synopsis. Toxicol Pathol 2000;28(1):28–30. Available from: http://journals.sagepub.com/doi/10.1177/019262330002800104
Wang J, Xing H, Wan L, Jiang X, Wang C, Wu Y. Treatment targets for M2 microglia polarization in ischemic stroke. Biomed Pharmacother. 2018;105:518–525.
Meng H-L, Li X-X, Chen Y-T, et al. Neuronal Soluble Fas Ligand Drives M1-Microglia Polarization after Cerebral Ischemia. CNS Neurosci Ther 2016;22(9):771–781. Available from: http://doi.wiley.com/10.1111/cns.12575
Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17981560
Gordon S, Martinez FO. Alternative Activation of Macrophages: Mechanism and Functions. Immunity. 2010;32(5):593–604.
Bokhari FA, Shakoori TA, Butt A, Ghafoor F. TNF-alpha: a risk factor for ischemic stroke. J Ayub Med Coll Abbottabad. 26(2):111–114. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25603656
Emsley HCA, Smith CJ, Georgiou RF, et al. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry. 2005;76(10):1366–1372. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16170078
Smith CJ, Hulme S, Vail A, et al. SCIL-STROKE (subcutaneous interleukin-1 receptor antagonist in ischemic stroke): a randomized controlled phase 2 trial. Stroke. 2018;49(5):1210–1216.
Boddaert J, Bielen K, Jongers B, et al. CD8 signaling in microglia/macrophage M1 polarization in a rat model of cerebral ischemia. PLoS One 2018; 13(1): e0186937. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29342151
Westendorp WF, Nederkoorn PJ, Vermeij J-D, Dijkgraaf MG, van de Beek D. Post-stroke infection: A systematic review and meta-analysis. BMC Neurol. 2011 ;11(1):110. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21933425
Fassbender K, Dempfle CE, Mielke O, et al. Proinflammatory cytokines: indicators of infection in high-risk patients. J Lab Clin Med. 1997;130(5):535–539. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9390642
Johnston KC, Li JY, Lyden PD, et al. Medical and neurological complications of ischemic stroke: experience from the RANTTAS trial. RANTTAS Investigators. Stroke. 1998 29(2):447–453. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9472888
Georgilis K, Plomaritoglou A, Dafni U, Bassiakos Y, Vemmos K. Aetiology of fever in patients with acute stroke. J Intern Med. 1999;246(2):203–209. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10447789
Grau AJ, Buggle F, Schnitzler P, Spiel M, Lichy C, Hacke W. Fever and infection early after ischemic stroke. J Neurol Sci. 1999;171(2):115–120. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10581377
Kammersgaard LP, Jørgensen HS, Reith J, et al. Early infection and prognosis after acute stroke: The Copenhagen Stroke Study. J Stroke Cerebrovasc Dis. 2001;10(5):217–221. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17903827
Hamidon BB, Raymond AA, Norlinah MI, Jefferelli SB. The predictors of early infection after an acute ischaemic stroke. Singap Med J. 2003;44(7):344–346. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14620725
Hilker R, Poetter C, Findeisen N, et al. Nosocomial pneumonia after acute stroke. Stroke. 2003;34(4):975–981. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12637700
Katzan IL, Cebul RD, Husak SH, Dawson N V, Baker DW. The effect of pneumonia on mortality among patients hospitalized for acute stroke. Neurology. 2003;60(4):620–625. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12601102
Aslanyan S, Weir CJ, Diener H-C, Kaste M, Lees KR, GAIN International Steering Committee and Investigators. Pneumonia and urinary tract infection after acute ischaemic stroke: a tertiary analysis of the GAIN International trial. Eur J Neurol. 2004;11(1):49–53. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14692888
Haeusler KG, Schmidt WUH, Föhring F, Meisel C, Helms T, Jungehulsing GJ, et al. Cellular Immunodepression Preceding Infectious Complications after Acute Ischemic Stroke in Humans. Cerebrovasc Dis. 2008;25(1–2):50–58. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18033958
Ovbiagele B, Hills NK, Saver JL, Johnston SC, California Acute Stroke Prototype Registry Investigators. Frequency and Determinants of Pneumonia and Urinary Tract Infection During Stroke Hospitalization. J Stroke Cerebrovasc Dis. 2006;15(5):209–213. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17904077
Vargas M, Horcajada JP, Obach V, Revilla M, Cervera Á, Torres F, et al. Clinical Consequences of Infection in Patients With Acute Stroke. Stroke. 2006;37(2):461–465. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16385093
Kwan J, Hand P. Infection after acute stroke is associated with poor short-term outcome. Acta Neurol Scand. 2007;115(5):331–338. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17489944
Sellars C, Bowie L, Bagg J, Sweeney MP, Miller H, Tilston J, et al. Risk Factors for Chest Infection in Acute Stroke. Stroke. 2007;38(8):2284–2291. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17569875
Walter U, Knoblich R, Steinhagen V, Donat M, Benecke R, Kloth A. Predictors of pneumonia in acute stroke patients admitted to a neurological intensive care unit. J Neurol. 2007;254(10):1323–1329. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17361338
Stott DJ, Falconer A, Miller H, Tilston JC, Langhorne P. Urinary tract infection after stroke. QJMed. 2009;102(4):243–249. Available from: https://academic.oup.com/qjmed/article-lookup/doi/10.1093/qjmed/hcp012
Vermeij FH, Scholte op Reimer WJM, de Man P, van Oostenbrugge RJ, Franke CL, de Jong G, et al. Stroke-Associated Infection Is an Independent Risk Factor for Poor Outcome after Acute Ischemic Stroke: Data from the Netherlands Stroke Survey. Cerebrovasc Dis. 2009;27(5):465–471. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19329851
Wartenberg KE, Stoll A, Funk A, Meyer A, Schmidt JM, Berrouschot J. Infection after Acute Ischemic Stroke: Risk Factors, Biomarkers, and Outcome. Stroke Res Treat. 2011;2011:1–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21789273
Popović N, Stefanović-Budimkić M, Mitrović N, Urošević A, Milošević B, Pelemiš M, et al. The Frequency of Poststroke Infections and Their Impact on Early Stroke Outcome. J Stroke Cerebrovasc Dis. 2013;22(4):424–429. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23540255
Matz K, Seyfang L, Dachenhausen A, Teuschl Y, Tuomilehto J, Brainin M, et al. Post-stroke pneumonia at the stroke unit – a registry based analysis of contributing and protective factors. BMC Neurol. 2016;16(1):107. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27430328
Prass K, Meisel C, Hoflich C, et al. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J Exp Med. 2003; 198(5): 725–736. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12939340
Chamorro Á, Amaro S, Vargas M, et al. Catecholamines, infection, and death in acute ischemic stroke. J Neurol Sci 2007;252(1):29–35.
Emsley HCA, Hopkins SJ. Acute ischaemic stroke and infection: recent and emerging concepts. Lancet Neurol. 2008;7(4):341–353. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18339349
Hug A, Dalpke A, Wieczorek N, et al. Infarct volume is a major determiner of post-stroke immune cell function and susceptibility to infection. Stroke. 2009;40(10):3226–3232.
Prass K, Braun JS, Dirnagl U, Meisel C, Meisel A. Stroke propagates bacterial aspiration to pneumonia in a model of cerebral ischemia. Stroke. 2006;37(10):2607–2612. Available from: https://www.ahajournals.org/doi/10.1161/01.STR.0000240409.68739.2b
Muhammad S, Haasbach E, Kotchourko M, et al. Influenza virus infection aggravates stroke outcome. Stroke. 2011;42(3):783–791. Available from: https://www.ahajournals.org/doi/10.1161/STROKEAHA.110.596783
Chamorro A, Horcajada JP, Obach V, Vargas M, Revilla M, Torres F, et al. The Early Systemic Prophylaxis of Infection After Stroke study: a randomized clinical trial. Stroke. 2005;36(7):1495–1500. Available from: https://www.ahajournals.org/doi/10.1161/01.STR.0000170644.15504.49
Harms H, Prass K, Meisel C, Klehmet J, Rogge W, Drenckhahn C, et al. Preventive Antibacterial Therapy in Acute Ischemic Stroke: A Randomized Controlled Trial. Chamorro A, editor. PLoS One. 2008;3(5):e2158. Available from: https://dx.plos.org/10.1371/journal.pone.0002158
Schwarz S, Al-Shajlawi F, Sick C, Meairs S, Hennerici MG. Effects of Prophylactic Antibiotic Therapy With Mezlocillin Plus Sulbactam on the Incidence and Height of Fever After Severe Acute Ischemic Stroke. Stroke. 2008;39(4):1220–1227. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18309164
Kalra L, Irshad S, Hodsoll J, Simpson M, Gulliford M, Smithard D, et al. Prophylactic antibiotics after acute stroke for reducing pneumonia in patients with dysphagia (STROKE-INF): a prospective, cluster-randomised, open-label, masked endpoint, controlled clinical trial. Lancet. 2015;386(10006):1835–1844. Available from: https://www.sciencedirect.com/science/article/pii/S0140673615001269
Westendorp WF, Vermeij J-D, Zock E, Hooijenga IJ, Kruyt ND, Bosboom HJLW, et al. The Preventive Antibiotics in Stroke Study (PASS): a pragmatic randomised open-label masked endpoint clinical trial. Lancet. 2015;385(9977):1519–1526. Available from: https://www.sciencedirect.com/science/article/pii/S0140673614624569
Zierath D, Kunze A, Fecteau L, Becker K. Effect of antibiotic class on stroke outcome. Stroke. 2015;46(8):2287–2292. Available from: https://www.ahajournals.org/doi/10.1161/STROKEAHA.115.008663
Chamorro A, Horcajada JP, Obach V, et al. The Early Systemic Prophylaxis of Infection After Stroke Study. Stroke. 2005;36(7):1495–1500. Available from: https://www.ahajournals.org/doi/10.1161/01.STR.0000170644.15504.49
Harms H, Prass K, Meisel C, et al. Preventive Antibacterial Therapy in Acute Ischemic Stroke: A Randomized Controlled Trial. Chamorro A, editor. PLoS One. 2008 ;3(5):e2158. https://doi.org/10.1371/journal.pone.0002158
Schwarz S, Al-Shajlawi F, Sick C, Meairs S, Hennerici MG. Effects of prophylactic antibiotic therapy with mezlocillin plus sulbactam on the incidence and height of fever after severe acute ischemic stroke: the Mannheim infection in stroke study (MISS). Stroke. 2008;39(4):1220–1227. Available from: https://www.ahajournals.org/doi/10.1161/STROKEAHA.107.499533
Zemke D, Majid A. The potential of minocycline for neuroprotection in human neurologic disease. Clin Neuropharmacol. 27(6):293–298. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15613934
Zierath D, Tanzi P, Shibata D, Becker KJ. Cortisol is More Important than Metanephrines in Driving Changes in Leukocyte Counts after Stroke. J Stroke Cerebrovasc Dis 2018;27(3):555–562. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1052305717305323
Haeusler KG, Schmidt WUH, Föhring F, et al. Cellular Immunodepression Preceding Infectious Complications after Acute Ischemic Stroke in Humans. Cerebrovasc Dis. 2008;25(1–2):50–58. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18033958
Haeusler KG, Schmidt WUH, Foehring F, et al. Immune responses after acute ischemic stroke or myocardial infarction. Int J Cardiol 2012;155(3):372–377. Available from: https://www.sciencedirect.com/science/article/pii/S0167527310009010
Urra X, Cervera Á, Villamor N, Planas AM, Chamorro Á. Harms and benefits of lymphocyte subpopulations in patients with acute stroke. Neuroscience. 2009;158(3):1174–1183. Available from: https://www.sciencedirect.com/science/article/pii/S0306452208009032
Vogelgesang A, May VEL, Grunwald U, et al. Functional status of peripheral blood T-cells in ischemic stroke patients. PLoS One. 2010;5(1):e8718. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20090932
Urra X, Cervera A, Obach V, Climent N, Planas AM, Chamorro A. Monocytes are major players in the prognosis and risk of infection after acute stroke. Stroke. 2009;40(4):1262–1268. Available from: https://www.ahajournals.org/doi/10.1161/STROKEAHA.108.532085
Ma S, Zhao H, Ji X, Luo Y. Peripheral to central: Organ interactions in stroke pathophysiology. Exp Neurol 2015;272:41–49. https://doi.org/10.1016/j.expneurol.2015.05.014
Tanzi P, Cain K, Kalil A, et al. Post-Stroke Infection: A Role for IL-1ra? Neurocrit Care 2011;14(2):244–252. Available from: http://link.springer.com/10.1007/s12028-010-9490-7
Zaremba J, Losy J. Early TNF-alpha levels correlate with ischaemic stroke severity. Acta Neurol Scand. 2001;104(5):288–295. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11696023
Intiso D, Zarrelli MM, Lagioia G, et al. Tumor necrosis factor alpha serum levels and inflammatory response in acute ischemic stroke patients. Neurol Sci 2004;24(6):390–396. Available from: http://link.springer.com/10.1007/s10072-003-0194-z
Sotgiu S, Zanda B, Marchetti B, et al. Inflammatory biomarkers in blood of patients with acute brain ischemia. Eur J Neurol 2006;13(5):505–513. Available from: http://doi.wiley.com/10.1111/j.1468-1331.2006.01280.x
Dorrance AM, Fink G. Effects of Stroke on the Autonomic Nervous System. In: Comprehensive Physiology. Hoboken: John Wiley & Sons, Inc.; 2015. p. 1241–1263. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26140717
Haspula D, Clark MA. Neuroinflammation and sympathetic overactivity: Mechanisms and implications in hypertension. Auton Neurosci Basic Clin. 2018;210(2017):10–17. https://doi.org/10.1016/j.autneu.2018.01.002
Ajmo CT, Collier LA, Leonardo CC, et al. Blockade of adrenoreceptors inhibits the splenic response to stroke. Exp Neurol 2009;218(1):47–55. Available from: https://www.sciencedirect.com/science/article/pii/S0014488609001320
An C, Shi Y, Li P, et al. Molecular dialogs between the ischemic brain and the peripheral immune system: Dualistic roles in injury and repair. Prog Neurobiol 2014;115(C):6–24. https://doi.org/10.1016/j.pneurobio.2013.12.002
Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci 2005;6(10):775–786. Available from: http://www.nature.com/articles/nrn1765
Fathali N, Ostrowski RP, Hasegawa Y, Lekic T, Tang J, Zhang JH. Splenic Immune Cells in Experimental Neonatal Hypoxia–Ischemia. Transl Stroke Res. 2013;4(2):208–219. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23626659
Ajmo CT, Vernon DOL, Collier L, et al. The spleen contributes to stroke-induced neurodegeneration. J Neurosci Res. 2008;86(10):2227–2234. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18381759
Ostrowski RP, Schulte RW, Nie Y, et al. Acute Splenic Irradiation Reduces Brain Injury in the Rat Focal Ischemic Stroke Model. Transl Stroke Res 2012;3(4):473–481. Available from: http://link.springer.com/10.1007/s12975-012-0206-5
Dirnagl U, Klehmet J, Braun JS, et al. Stroke-induced immunodepression: Experimental evidence and clinical relevance. Stroke. 2007;38(2 PART 2):770–773.
Kin NW, Sanders VM. It takes nerve to tell T and B cells what to do. J Leukoc Biol. 2006;79(6):1093–1104. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16531560
Offner H, Subramanian S, Parker SM, et al. Splenic Atrophy in Experimental Stroke Is Accompanied by Increased Regulatory T Cells and Circulating Macrophages. J Immunol. 2006;176(11):6523–6531. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16709809
Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental Stroke Induces Massive, Rapid Activation of the Peripheral Immune System. J Cereb Blood Flow Metab. 2006;26(5):654–665. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16121126
Straub RH. Complexity of the bi-directional neuroimmune junction in the spleen. Trends Pharmacol Sci. 2004;25(12):640–646. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15530642
Feng G, Xu X, Wang Q, Liu Z, Li Z, Liu G. The protective effects of calcitonin gene-related peptide on gastric mucosa injury after cerebral ischemia reperfusion in rats. Regul Pept 2010;160(1–3):121–128. Available from: https://www.sciencedirect.com/science/article/abs/pii/S0167011509002304
Stanley D, Mason LJ, Mackin KE, et al. Translocation and dissemination of commensal bacteria in post-stroke infection. Nat Med 2016;22(11):1277–1284. Available from: http://www.nature.com/articles/nm.4194
Shi K, Wood K, Shi FD, Wang X, Liu Q. Stroke-induced immunosuppression and poststroke infection. Stroke Vasc Neurol 2018;3(1):34–41.
Durgan DJ, Lee J, McCullough LD, Bryan RM. Examining the Role of the Microbiota-Gut-Brain Axis in Stroke. Stroke. 2019;50(8):2270–2277. Available from: https://www.ahajournals.org/doi/10.1161/STROKEAHA.119.025140
Durgan DJ, Ganesh BP, Cope JL, et al. Role of the Gut Microbiome in Obstructive Sleep Apnea–Induced Hypertension. Hypertension 2016;67(2):469–474. Available from: https://www.ahajournals.org/doi/10.1161/HYPERTENSIONAHA.115.06672
Singh V, Roth S, Llovera G, et al. Microbiota dysbiosis controls the neuroinflammatory response after stroke. J Neurosci 2016;36(28):7428–7440.
Houlden A, Goldrick M, Brough D, et al. Brain injury induces specific changes in the caecal microbiota of mice via altered autonomic activity and mucoprotein production. Brain Behav Immun 2016;57:10–20. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0889159116300940
Gan Y, Liu Q, Wu W, et al. Ischemic neurons recruit natural killer cells that accelerate brain infarction. Proc Natl Acad Sci U S A. 2014;111(7):2704–2709. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24550298
Zhang Y, James SJ, Teh H-Y, Takahashi H, Flavell RA, Dong C. 212: MAP kinase phosphatase 5 expression induced by influenza virus infection negatively regulates IRF3 activation and type I interferon response. Cytokine 2014;70(1):79. Available from: https://www.sciencedirect.com/science/article/abs/pii/S1043466614004293
Tsai AS, Berry K, Beneyto MM, et al. A year-long immune profile of the systemic response in acute stroke survivors. Brain. 2019;142(4).
Schulze J, Vogelgesang A, Dressel A. Catecholamines, steroids and immune alterations in ischemic stroke and other acute diseases. Aging Dis. 2014;5(5):327–339. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25276491
Wang J, Asensio VC, Campbell IL. Cytokines and chemokines as mediators of protection and injury in the central nervous system assessed in transgenic mice. Curr Top Microbiol Immunol 2002;265:23–48. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12014193
TAarkowski E, Rosengren L, Blomstrand C, et al. Intrathecal release of pro- and anti-inflammatory cytokines during stroke. Clin Exp Immunol. 1997;110(3):492–499. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9409656
Lehrmann E, Kiefer R, Finsen B, Diemer NH, Zimmer J, Hartung HP. Cytokines in cerebral ischemia: expression of transforming growth factor beta-1 (TGF-beta 1) mRNA in the postischemic adult rat hippocampus. Exp Neurol 1995;131(1):114–123. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7895806
Clausen BH, Lambertsen KL, Meldgaard M, Finsen B. A quantitative in situ hybridization and polymerase chain reaction study of microglial-macrophage expression of interleukin-1β mRNA following permanent middle cerebral artery occlusion in mice. Neuroscience 2005;132(4):879–892. Available from: https://www.sciencedirect.com/science/article/pii/S0306452205000953
Haddad JJ, Saadé NE, Safieh-Garabedian B. Cytokines and neuro-immune-endocrine interactions: a role for the hypothalamic-pituitary-adrenal revolving axis. J Neuroimmunol. 2002 133(1–2):1–19. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12446003
Turnbull A V., Rivier CL. Regulation of the Hypothalamic-Pituitary-Adrenal Axis by Cytokines: Actions and Mechanisms of Action. Physiol Rev. 1999;79(1):1–71. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9922367
Kohm AP, Sanders VM. Norepinephrine and beta 2-adrenergic receptor stimulation regulate CD4+ T and B lymphocyte function in vitro and in vivo. Pharmacol Rev. 2001;53(4):487–525. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11734616
Jara LJ, Navarro C, Medina G, Vera-Lastra O, Blanco F. Immune-Neuroendocrine Interactions and Autoimmune Diseases. Clin Dev Immunol. 2006;13(2–4):109–123. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17162354
Campanella M, Sciorati C, Tarozzo G, Beltramo M. Flow Cytometric Analysis of Inflammatory Cells in Ischemic Rat Brain. Stroke. 2002;33(2):586–592. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11823674
Yilmaz G, Arumugam T V, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation. 2006;113(17):2105–2112. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16636173
Stoll G, Jander S, Schroeter M. Cytokines in CNS disorders: Neurotoxicity versus neuroprotection. J Neural Transm Suppl 2000;59:81–89.
Liesz A, Zhou W, Na S-Y, et al. Boosting regulatory T cells limits neuroinflammation in permanent cortical stroke. J Neurosci. 2013;33(44):17350–17362. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24174668
Stubbe T, Ebner F, Richter D, et al. Regulatory T Cells Accumulate and Proliferate in the Ischemic Hemisphere for up to 30 Days after MCAO. J Cereb Blood Flow Metab. 2013;33(1):37–47. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22968321
Li P, Mao L, Zhou G, et al. Adoptive regulatory T cell therapy preserves systemic immune homeostasis following cerebral ischemia. Stroke. 2013;44(12):3509. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3895539/
Liesz A, Suri-Payer E, Veltkamp C, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009; 15(2): 192–199. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19169263
Liesz A, Zhou W, Mracsko E, et al. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain. 2011;134(Pt 3):704–720. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21354973
Becker K, Kindrick D, McCarron R, Hallenbeck J, Winn R. Adoptive transfer of myelin basic protein-tolerized splenocytes to naive animals reduces infarct size: a role for lymphocytes in ischemic brain injury? Stroke. 2003;34(7):1809–1815. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12791945
Kleinschnitz C, Kraft P, Dreykluft A, et al. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood. 2013 121(4):679–691. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23160472
Xu X, Li M, Jiang Y. The paradox role of regulatory T cells in ischemic stroke. ScientificWorldJournal. 2013;2013:174373. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24288462
Waje-Andreassen U, Krakenes J, Ulvestad E, et al. IL-6: an early marker for outcome in acute ischemic stroke. Acta Neurol Scand. 2005;111(6):360–365. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15876336
Clarkson BDS, Ling C, Shi Y, et al. T cell-derived interleukin (IL)-21 promotes brain injury following stroke in mice. J Exp Med. 2014;211(4):595–604. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24616379
Seifert HA, Pennypacker KR. Molecular and Cellular Immune Responses to Ischemic Brain Injury. Transl Stroke Res 2014;5(5):543–553. Available from: http://link.springer.com/10.1007/s12975-014-0349-7
Trinchieri G. Interleukin-10 production by effector T cells: Th1 cells show self control. J Exp Med. 2007;204(2):239–243. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17296790
Terao S, Yilmaz G, Stokes KY, et al. Blood cell-derived RANTES mediates cerebral microvascular dysfunction, inflammation, and tissue injury after focal ischemia-reperfusion. Stroke 2008;39(9):2560–2570. Available from: https://www.ahajournals.org/doi/10.1161/STROKEAHA.107.513150
Ren X, Akiyoshi K, Dziennis S, et al. Regulatory B cells limit CNS inflammation and neurologic deficits in murine experimental stroke. J Neurosci. 2011; Available from: http://www.ncbi.nlm.nih.gov/pubmed/21653859.
Doyle KP, Quach LN, Sole M, et al. B-lymphocyte-mediated delayed cognitive impairment following stroke. J Neurosci. 2015;35(5):2133–2145. Available from: https://www.ncbi.nlm.nih.gov/pubmed/25653369.
Schuhmann MK, Langhauser F, Kraft P, Kleinschnitz C. B cells do not have a major pathophysiologic role in acute ischemic stroke in mice. J Neuroinflammation. 2017;14(1):112. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28576128
Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. IL-10-producing B-cells limit CNS inflammation and infarct volume in experimental stroke. Metab Brain Dis 2013;28(3):375–386. Available from: http://link.springer.com/10.1007/s11011-013-9413-3
Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 2008;118(10):3420–3430. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18802481
Planas AM, Gomez-Choco M, Urra X, Gorina R, Caballero M, Chamorro A. Brain-derived antigens in lymphoid tissue of patients with acute stroke. J Immunol. 2012;188(5):2156–2163. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22287710
Popovich PG, Stokes BT, Whitacre CC. Concept of autoimmunity following spinal cord injury: Possible roles for T lymphocytes in the traumatized central nervous system. J Neurosci Res. 1996;45(4):349–363. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8872895
Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8(4):279–289. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18340345
Iadecola C. Brain-Immune Interactions and Ischemic Stroke. Arch Neurol 2012;69(5):576.
Sun H, Gong S, Carmody RJ, et al. TIPE2, a negative regulator of innate and adaptive immunity that maintains immune homeostasis. Cell. 2008;133(3):415–426. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18455983
Kennedy AD, DeLeo FR. Neutrophil apoptosis and the resolution of infection. Immunol Res. 2009;43(1–3):25–61. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19066741
Nathan C, Ding A. Nonresolving Inflammation. Cell. 2010;140(6):871–882. https://doi.org/10.1016/j.cell.2010.02.029
Meszaros AJ, Reichner JS, Albina JE. Macrophage-induced neutrophil apoptosis. J Immunol. 2000;165(1):435–441. Available from: http://www.ncbi.nlm.nih.gov/pubmed/3492537
Ehrchen J, Steinmuller L, Barczyk K, et al. Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood. 2006;109(3):1265–1274. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17018861
Perretti M, D’Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. 2009;9(1):62–70. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19104500
Luzina IG, Keegan AD, Heller NM, Rook GAW, Shea-Donohue T, Atamas SP. Regulation of inflammation by interleukin-4: a review. J Leukoc Biol. 2012;92(4):753–764. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22782966
Chung AG, Frye JB, Zbesko JC, et al. Liquefaction of the brain following stroke shares a similar molecular and morphological profile with atherosclerosis and mediates secondary neurodegeneration in an osteopontin-dependent mechanism. eNeuro. 2018;5(5):ENEURO.0076-18.2018. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30417081
Leys D, Henon H, Mackowiak-Cordoliani MA, Pasquier F. Poststroke dementia. Lancet Neurol. 2005;4(11):752–759. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16239182
Levine DA, Galecki AT, Langa KM, et al. Trajectory of cognitive decline after incident stroke. JAMA. 2015;314(1):41–51. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26151265
Cuartero MI, de la Parra J, Pérez-Ruiz A, et al. Abolition of aberrant neurogenesis ameliorates cognitive impairment after stroke in mice. J Clin Invest 2019;129(4):1536–1550. Available from: https://www.jci.org/articles/view/120412
Dambinova S, Khounteev G, Izykenova G, Zavolokov I, Ilyukhina A, Skoromets A. Blood test detecting autoantibodies to N-methyl-D-aspartate neuroreceptors for evaluation of patients with transient ischemic attack and stroke. Clin Chem. 2003;49(10):1752–1762. Available from: https://academic.oup.com/clinchem/article/49/10/1752/5642162
Kalev-Zylinska ML, Symes W, Little KCE, et al. Stroke patients develop antibodies that react with components of n-methyl-d-aspartate receptor subunit 1 in proportion to lesion size. Stroke 2013;44(8):2212–2219.
Ortega SB, Noorbhai I, Poinsatte K, et al. Stroke induces a rapid adaptive autoimmune response to novel neuronal antigens. Discov Med. 2015;19(106):381–392. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26105701
Royl G, Fokou TJ, Chunder R, et al. Antibodies against neural antigens in patients with acute stroke: joint results of three independent cohort studies. J Neurol 2019;266(11):2772–2779. Available from: http://link.springer.com/10.1007/s00415-019-09470-2
Mitsdoerffer M, Lee Y, Jäger A, et al. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci U S A. 2010;107(32):14292–14297. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20660725
Doyle KP, Quach LN, Solé M, et al. B-lymphocyte-mediated delayed cognitive impairment following stroke. J Neurosci. 2015;35(5).
Pruss H, Iggena D, Baldinger T, et al. Evidence of intrathecal immunoglobulin synthesis in stroke: a cohort study. Arch Neurol. 2012;69(6):714–717. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22371852
Barba R, Martinez-Espinosa S, Rodriguez-Garcia E, Pondal M, Vivancos J, Del Ser T. Poststroke dementia : clinical features and risk factors. Stroke. 2000;31(7):1494–1501. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10884443
Bejot Y, Aboa-Eboule C, Durier J, et al. Prevalence of early dementia after first-ever stroke: a 24-year population-based study. Stroke. 2011;42(3):607–612. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=21233464
Becker KJ, Buckwalter M. Stroke, inflammation and the immune response: dawn of a new era. Neurotherapeutics. 2016;13(4).
Becker KJ, Tanzi P, Zierath D, Buckwalter MS. Antibodies to myelin basic protein are associated with cognitive decline after stroke. J Neuroimmunol. 2016;295–296:9–11. Available from: https://www.ncbi.nlm.nih.gov/pubmed/27235342
Bornstein NM, Aronovich B, Korczyn AD, Shavit S, Michaelson DM, Chapman J. Antibodies to brain antigens following stroke. Neurology. 2001;56(4):529–530. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11222800
Mecocci P, Parnetti L, Romano G, et al. Serum anti-GFAP and anti-S100 autoantibodies in brain aging, Alzheimer’s disease and vascular dementia. J Neuroimmunol. 1995;57(1–2):165–170. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7706432
Shibata D, Cain K, Tanzi P, Zierath D, Becker K. Myelin basic protein autoantibodies, white matter disease and stroke outcome. J Neuroimmunol. 2012;252(1–2):106–112. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22939639
Schneider D, Berrouschot J, Brandt T, Hacke W, Ferbert A, Norris SH, et al. Safety, Pharmacokinetics and Biological Activity of Enlimomab (Anti-ICAM-1 Antibody): An Open-Label, Dose Escalation Study in Patients Hospitalized for Acute Stroke. Eur Neurol. 1998;40(2):78–83. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9693236
Enlimomab Acute Stroke Trial I. Use of anti-ICAM-1 therapy in ischemic stroke: results of the Enlimomab Acute Stroke Trial. Neurology. 2001;57(8):1428–1434. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11673584
Fu Y, Hao J, Zhang N, et al. Fingolimod for the treatment of intracerebral hemorrhage. JAMA Neurol 2014;71(9):1092. Available from: http://archneur.jamanetwork.com/article.aspx?doi=10.1001/jamaneurol.2014.1065
Fu Y, Zhang N, Ren L, et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc Natl Acad Sci U S A. 2014;111(51):18315–18320. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25489101
Zhu Z, Fu Y, Tian D, et al. Combination of the immune modulator fingolimod with alteplase in acute ischemic stroke. Circulation. 2015;132(12):1104–1112. Available from: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.115.016371
Elkins J, Veltkamp R, Montaner J, et al. Safety and efficacy of natalizumab in patients with acute ischaemic stroke (ACTION): a randomised, placebo-controlled, double-blind phase 2 trial. Lancet Neurol 2017;16(3):217–226. https://doi.org/10.1016/S1474-4422(16)30357-X
Krams M, Lees KR, Hacke W, et al. Acute Stroke Therapy by Inhibition of Neutrophils (ASTIN): an adaptive dose-response study of UK-279,276 in acute ischemic stroke. Stroke. 2003;34(11):2543–2548. Available from: https://www.ahajournals.org/doi/10.1161/01.STR.0000092527.33910.89
Shibuya M, Hirai S, Seto M, Satoh S, Ohtomo E. Effects of fasudil in acute ischemic stroke: Results of a prospective placebo-controlled double-blind trial. J Neurol Sci 2005;238(1–2):31–39. Available from: https://www.sciencedirect.com/science/article/abs/pii/S0022510X05002212
Lampl Y, Boaz M, Gilad R, Lorberboym M, Dabby R, Rapoport A, et al. Minocycline treatment in acute stroke: An open-label, evaluator-blinded study. Neurology. 2007;69(14):1404–1410. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17909152
Fagan SC, Waller JL, Nichols FT, Edwards DJ, Pettigrew LC, Clark WM, et al. Minocycline to Improve Neurologic Outcome in Stroke (MINOS). Stroke. 2010;41(10):2283–2287. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20705929
Singh R, Augustin SJ, Jane M, et al. Does Minocycline Improve Recovery After Acute Ischemic Stroke? J Stroke Med 2019;2(1):40–46. Available from: http://journals.sagepub.com/doi/10.1177/2516608519856263
Montaner J, Chacón P, Krupinski J, et al. Simvastatin in the acute phase of ischemic stroke: a safety and efficacy pilot trial. Eur J Neurol. 2007;15(1):82–90. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18070096
Lees KR, Zivin JA, Ashwood T, et al. NXY-059 for Acute Ischemic Stroke. N Engl J Med 2006;354(6):588–600. Available from: http://www.nejm.org/doi/abs/10.1056/NEJMoa052980
Diener H-C, Lees KR, Lyden P, et al. NXY-059 for the Treatment of Acute Stroke. Stroke. 2008;39(6):1751–1758. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18369171
Bath PM, Iddenden R, Bath FJ, Orgogozo JM, Tirilazad International Steering Committee. Tirilazad for acute ischaemic stroke. Cochrane Database Syst Rev. 2001:(4):CD002087. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11687138
Yamaguchi T, Sano K, Takakura K, et al. Ebselen in acute ischemic stroke: a placebo-controlled, double-blind clinical trial. Ebselen Study Group. Stroke. 1998;29(1):12–17. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9445321
Margaill I, Plotkine M, Lerouet D. Antioxidant strategies in the treatment of stroke. Free Radic Biol Med 2005;39(4):429–443. Available from: https://www.sciencedirect.com/science/article/pii/S0891584905002662
Chamorro Á, Amaro S, Castellanos M, et al. Safety and efficacy of uric acid in patients with acute stroke (URICO-ICTUS): a randomised, double-blind phase 2b/3 trial. Lancet Neurol 2014;13(5):453–460. Available from: https://www.sciencedirect.com/science/article/abs/pii/S1474442214700547
Yagita Y, Kitagawa K, Sasaki T, et al. Rho-kinase activation in endothelial cells contributes to expansion of infarction after focal cerebral ischemia. J Neurosci Res. 2007;85(11):2460–2469. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17526023
Jiang BH, Tawara S, Abe K, Takaki A, Fukumoto Y, Shimokawa H. Acute vasodilator effect of fasudil, a Rho-kinase inhibitor, in monocrotaline-induced pulmonary hypertension in rats. J Cardiovasc Pharmacol 2007
Zhang Y, Wu S. Effects of fasudil on pulmonary hypertension in clinical practice. Pulm Pharmacol Ther 2017.
Jiang N, Chopp M, Chahwala S. Neutrophil inhibitory factor treatment of focal cerebral ischemia in the rat. Brain Res. 1998 788(1–2):25–34. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9554941
Vuorte J, Lindsberg PJ, Kaste M, et al. Anti-ICAM-1 monoclonal antibody R6.5 (Enlimomab) promotes activation of neutrophils in whole blood. J Immunol 1999;162(4):2353–2357. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9973515
Justicia C, Martín A, Rojas S, et al. Anti-VCAM-1 antibodies did not protect against ischemic damage either in rats or in mice. J Cereb Blood Flow Metab 2006;26(3):421–432. Available from: http://journals.sagepub.com/doi/10.1038/sj.jcbfm.9600198
Biogen. Biogen Reports Top-Line Results from Phase 2b Study of Natalizumab in Acute Ischemic Stroke | Biogen [Internet]. 2018. Available from: http://media.biogen.com/news-releases/news-release-details/biogen-reports-top-line-results-phase-2b-study-natalizumab-acute.
Langhauser F, Kraft P, Göb E, et al. Blocking of α4 integrin does not protect from acute ischemic stroke in mice. Stroke. 2014;45(6):1799–1806.
Llovera G, Hofmann K, Roth S, et al. Results of a preclinical randomized controlled multicenter trial (pRCT): anti-CD49d treatment for acute brain ischemia. Sci Transl Med. 2015;7(299).
Becker K, Kindrick D, Relton J, Harlan J, Winn R. Antibody to the α4 integrin decreases infarct size in transient focal cerebral ischemia in rats. Stroke. 2001;32(1):206–211. Available from: https://www.ahajournals.org/doi/10.1161/01.STR.32.1.206
Relton JK, Sloan KE, Frew EM, Whalley ET, Adams SP, Lobb RR. Inhibition of α4 integrin protects against transient focal cerebral ischemia in normotensive and hypertensive rats. Stroke 2001;32(1):199–205.
Liu J, Zhang C, Tao W, Liu M. Systematic review and meta-analysis of the efficacy of sphingosine-1-phosphate (S1P) receptor agonist FTY720 (Fingolimod) in animal models of stroke. Int J Neurosci 2013;123(3):163–169. Available from: http://www.tandfonline.com/doi/full/10.3109/00207454.2012.749255
Jickling GC, Sharp FR. Improving the translation of animal ischemic stroke studies to humans. Metab Brain Dis 2015;30(2):461–467.
Maysami S, Haley MJ, Gorenkova N, Krishnan S, McColl BW, Lawrence CB. Prolonged diet-induced obesity in mice modifies the inflammatory response and leads to worse outcome after stroke. J Neuroinflammation 2015;12(1):140. Available from: http://www.jneuroinflammation.com/content/12/1/140
Ergul A, Hafez S, Fouda A, Fagan SC. Impact of comorbidities on acute injury and recovery in preclinical stroke research: focus on hypertension and diabetes. Transl Stroke Res 2016;7(4):248–260. Available from: http://link.springer.com/10.1007/s12975-016-0464-8
Xu M, Wang MM, Gao Y, Keep RF, Shi Y. The effect of age-related risk factors and comorbidities on white matter injury and repair after ischemic stroke. Neurobiol Dis. 2019;126(July):13–22. https://doi.org/10.1016/j.nbd.2018.07.008
Acknowledgments
We thank Amy Robinson for help with editing the manuscript. This work was supported by funding from the Weston-Havens Foundation and the American Heart Association/Allen Initiative in Brain Health and Cognitive Impairment 9PABHI34580007.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 1337 kb)
Rights and permissions
About this article

Cite this article
Zera, K.A., Buckwalter, M.S. The Local and Peripheral Immune Responses to Stroke: Implications for Therapeutic Development. Neurotherapeutics 17, 414–435 (2020). https://doi.org/10.1007/s13311-020-00844-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-020-00844-3