
Abstract
We studied the characteristics of the provisional category de novo acute myeloid leukemia (AML) with mutated RUNX1 (AML-RUNX1mut) proposed by the World Health Organization (WHO). Until now, most published studies have combined de novo and secondary AML-RUNX1mut. We compared the clinicopathologic characteristics and outcomes of WHO-defined de novo AML-RUNX1mut with de novo AML without RUNX1 alterations (AML-RUNX1wt). We performed sequential NGS to assess RUNX1 mutation stability over disease course. We identified 46 de novo AML-RUNX1mut patients [32 (70%) men, 14 (30%) women; median age, 66.5 years] with 54 RUNX1 mutations [median VAF, 32% (2–97%)]. Point mutations clustered within the runt-homology-domain and frame-shift mutations within the transactivation domain. Compared with AML-RUNX1wt, AML-RUNX1mut showed male predominance (p = 0.02), higher frequency of SRSF2 (p = 0.02), and ASXL1 (p = 0.0004) mutations and normal karyotype (p = 0.01), and absent NPM1 mutations (p = 0.0002). De novo AML-RUNX1mut showed no significant difference in overall survival (OS) compared with AML-RUNX1wt (median: 26 vs. 32 months) (p = 0.71). AML-RUNX1mut with clonal RUNX1 mutation (≥20% VAF) had shorter OS than subclonal <20% VAF (23 months vs. undefined; p = 0.04). However, the difference was not significant when compared with AML-RUNX1wt (23 vs. 32 months; p = 0.23). No significant OS difference was noted between de novo AML-RUNX1mut and AML-NOS-RUNX1wt. By sequential multigene mutation profiling, RUNX1 mutation disappeared at relapse in one of ten patients. Overall, the findings support separate categorization of this entity. However, there is no significant outcome difference compared with AML-RUNX1wt.
Similar content being viewed by others

Introduction
The study of the molecular genetic alterations in acute myeloid leukemia (AML) has led to greater understanding of leukemogenesis. This knowledge has resulted in refinement of AML prognostic subgroups beyond standard cytogenetic findings, thereby confirming the complexities of the multistep leukemogenic process [1, 2]. The recent World Health Organization (WHO) revision has recognized de novo AML with RUNX1 mutation, in the absence of myelodysplasia-related changes (MRC) (including cytogenetic abnormalities), as a provisional category based on common biologic characteristics and adverse outcome [3]. AML cases that belong to this category should present de novo without a history of myeloid neoplasm, chemotherapy or radiation therapy, no MRC (either morphologic dysplasia or MDS-defining cytogenetic abnormalities) and should not arise in the setting of a familial germline predisposition.
Large-scale studies have shown poor prognostic effect of somatic RUNX1 mutations in AML [4,5,6,7]. However, majority of these studies have collectively studied the clinicopathologic findings of both de novo and secondary AML transformed from MDS; none have analyzed AML cases defined using the proposed criteria by the WHO. RUNX1 mutation is frequent in MDS (12–14%) and associated with poor outcome [7,8,9]. Hence, based on these studies, it is unclear if the poor prognosis of RUNX1 mutated AML is due to the mutation itself or is attributable to preexistent MDS [8,9,10,11,12]. Thus, the clinical importance of this new provisional of de novo AML with RUNX1 mutation remains to be explored. Furthermore, few studies have used next-generation sequencing (NGS)-based mutation analysis. Recent evidence demonstrates the effectiveness of BETP antagonists in inducing lethality of AML cell lines [13]. These prognostic implications and potential therapeutic opportunities highlight the importance of in-depth molecular analysis and clinical characterization of RUNX1 mutations in this provisional AML category [14, 15].
In this study, we present the clinicopathologic and genetic findings of a single institution series of 46 patients with de novo AML with RUNX1mut that met the proposed WHO criteria for the newly recognized provisional entity. Furthermore, using targeted NGS methods, we describe a characteristic pattern of concomitant gene mutations and clonal evolution over the course of the disease.
Materials and methods
We retrieved all cases of de novo AML-RUNX1mut that underwent NGS-based mutational profiling on bone marrow aspirate samples over a 2-year time period at our institution. The somatic nature of the mutation was inferred based on a combination of variant allele frequency, literature review, and information in databases such as COSMIC, dbSNP, and 1000 genome, etc. because we did not perform germline testing. We excluded cases with RUNX1 variants of unknown significance. We excluded cases of AML arising from an antecedent myeloid neoplasm (MDS or MDS/MPN), AML-MRC, therapy-related AML, cases with history of radiation, strong family history of leukemia and known familial RUNX1 mutations. The final study group included de novo AML cases with RUNX1 mutation that strictly met the criteria for the provisional entity as proposed by the WHO classification. For comparison, we selected 94 consecutive cases of de novo AML (excluding acute promyelocytic leukemia) that underwent NGS-based mutation analysis that lacked RUNX1 alteration (de novo AML-RUNX1wt). The study was approved by the Institutional Review Board. Informed consent was obtained in accordance with the Declaration of Helsinki.
All cases underwent morphologic review. Immunophenotypic analysis using multicolor flow cytometry was performed on BM aspirates using a FACScan instrument (Becton-Dickinson, San Jose, California) as described previously [16]. Conventional chromosomal analysis was performed using standard techniques described previously [6]. Targeted amplicon-based somatic mutation analysis was performed using DNA extracted from BM aspirates and a 28-gene panel as previously described [17, 18]. Within RUNX1, the Runt1 homology domain (RHD) was defined as spanning amino acids 77–204 and the transactivation domain (TAD) containing both activation and inhibitory domains was defined as spanning amino acids 269–438 [19]. Mutation analysis for SF3B1 (exons 14 and 15), SRSF2 (exon 1), and CEBPA genes were performed by PCR followed by Sanger sequencing. Internal tandem duplications (ITD) and point mutations in codon 835/836 of the FLT3 gene were assessed by PCR followed by capillary electrophoresis [17]. Additional details for methods are provided in Supplementary material.
The frequencies and percentages were calculated for all categorical variables; median and ranges were estimated for continuous variables. The unpaired t-test was used to compare independent values between the two groups. Overall survival (OS) was estimated from the time of initial AML diagnosis to either date of last follow-up or death using Kaplan–Meier curves. Event-free survival (EFS) was calculated with an event defined as first relapse or death, whichever came first. Univariate Cox proportional hazards regression analysis was used to determine the association between OS and several covariates of interest. Cut-off values at both 10% and 20% variant allele frequencies were assessed to determine any possible impact on survival, based on the study by Schnittger et al. [5]. If two or more RUNX1 mutations were present in the same patient, VAF of the larger RUNX1 mutant was selected.
Results
Study cohort
The final study cohort included 46 AML from a single institution that met the criteria for the provisional category “de novo AML with RUNX1 mutation”. These patients were identified from a total of 344 consecutive patients with various hematologic malignancies with RUNX1 alterations (mutations and variants) detected by NGS-based mutational profiling and filtered per proposed WHO criteria (Fig. 1). Of these, 39 patients were excluded because of the lack of evidence in the literature on the somatic nature of the variant; 3 patients were excluded because the RUNX1 variant was confirmed to be of germline origin; of the remaining, 169 patients had a diagnosis of AML, of which 68 patients with an antecedent diagnosis of MDS or MPN, 22 patients with a history of chemotherapy or radiation therapy were excluded; 6 patients with recurrent genetic abnormalities including 2 patients with t(15;17)/PML-RARA were excluded; further, 15 patients had multilineage dysplasia meeting criteria for AML-MRC by morphology and 12 patients who had MDS-related cytogenetic abnormalities were also excluded. After applying exclusion criteria, the final study group was composed of 46 patients that met the WHO criteria for the provisional category of de novo AML-RUNX1mut (Fig. 1). For comparison, we selected 94 consecutive patients with de novo AML (excluding acute promyelocytic leukemia) without RUNX1 alterations [AML-RUNX1wt] that underwent NGS-based sequencing. The distribution of these cases within WHO categories included: AML with NPM1 mutation (n = 20, 21%), AML-MRC (n = 16, 17%), AML with inv(16)/CBFB-MYH11 (n = 7, 7%), AML with minimal differentiation (n = 3, 3%), AML without maturation (n = 7, 7%), AML with maturation (n = 11, 12%), acute myelomonocytic leukemia (n = 1, 1%), acute monocytic/monoblastic leukemia (n = 5, 5%), AML with t(v;11q23)/KMT2A rearrangement (n = 4, 4%), AML with t(6;9)/DEK-NUP214 (n = 2, 2%), AML with t(8;21)/RUNX1-RUNX1T1 (n = 1, 1%), pure erythroid leukemia (n = 1, 1%), and relapsed AML, not otherwise specified (n = 16, 17%).

Horizontal arrows designate elimination criteria. AML acute myeloid leukemia, MDS myelodysplastic syndrome, MPN myeloproliferative neoplasm, MDS/MPN myelodysplastic/myeloproliferative neoplasms.
Clinical characteristics
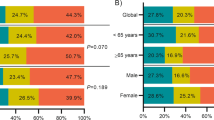
The cohort of patients with de novo AML-RUNX1mut included 32 (70%) men and 14 (30%) women with a median age of 66.5 years (range, 20–87). At the time of presentation, all 46 patients had anemia and 34 (74%) had thrombocytopenia. No patients had peripheral blood eosinophilia. Nineteen (41%) patients presented to our institution before receiving any treatment. The remaining 27 patients had relapsed or refractory AML and had received therapy prior to presenting to our institution. Based on the 2017 European LeukemiaNet (ELN) risk stratification by genetics, all 46 patients in the study cohort are included in the adverse risk category due to the presence of RUNX1 mutation [20]. We compared the clinical characteristics between the study and the control groups, the details of which are provided in Table 1. Compared with the control cohort, patients with AML-RUNX1mut showed a male predominance (p = 0.02), a significantly higher MCV (p = 0.009), and a trend towards older age (p = 0.06).
The study group patients were treated with multiple chemotherapeutic regimens. Fifteen (33%) patients underwent stem cell transplant (eight at first remission and seven after relapse); 39 (42%) patients in the control group underwent stem cell transplant (28 at first remission and 11 after relapse). These details are summarized in Supplementary Table 1.
Types of RUNX1 mutations
There was a total of 54 RUNX1 mutations in 46 patients; seven (15%) patients harbored two or more mutations. Three RUNX1 mutations were splice site mutations; the remaining 51 mutations were exonic mutations (most frequent in exon 5, 31%). The median variant allele frequency was 32.4% (range, 1.97–96.6%); 34 patients had a VAF ≥ 20% and 12 had a VAF < 20%. Only four patients had a VAF < 5%. These mutations were clustered within the RHD (amino acids 77 through 204; n = 29, 57%) and the TAD (amino acids 269–438; n = 18, 35%) domains of the gene, although the mutations spanned the entire coding region [Fig. 2]. In total, there were 23 (45%) point mutations (15 missense; 8 nonsense), 13 (25%) duplications, 10 (20%) deletions, and 5 (10%) insertions. Within of the RHD, 19 of 29 (65.5%) mutations were point mutations [13 (68%) missense and 6 (32%) nonsense], in contrast to TAD where only 3 of 18 (17%; 2 nonsense and 1 missense mutation) were point mutations (p = 0.0022). Within the TAD, 15 of 18 (83%) were frame-shift mutations in contrast to the RHD where only 9 of 29 (31%) were frameshift mutations (p = 0.0008).

Fifty-one exonic mutations are shown. There are 29 (57%) mutations within the RHD. Point mutations are preferentially located within the RHD (19 point mutations and 9 frameshift) (p = 0.0022). There are 18 (39%) mutations within the transactivation domain (TAD). Frameshift mutations are preferentially located within the TAD (3 point mutations and 15 frameshift) (p = 0.0008). Circles represent single-nucleotide variations and squares represent insertions, duplications, or deletions. White color represents missense mutations, black represents nonsense mutations, dotted pattern represents frameshift mutations, and striped pattern represents in-frame deletion. The amino acid location for each exonic mutation is shown below the symbol.
Morphologic and flow cytometry immunophenotypic findings
All 46 cases were classified as de novo AML-RUNX1mut per 2017 WHO classification. There were no specific morphologic features. The median BM cellularity was 70% and median BM blast count was 58.5%. Megakaryocytes were decreased in number (78%). The blasts were nondescript (intermediate-sized, moderate cytoplasm, and occasional prominent nucleoli). Auer rods were noted in two cases. Four patients showed salmon pink cytoplasmic granulation similar to AML with t(8;21)(q22;q22)/RUNX1-RUNX1T1. In 60% of cases, the blasts were positive for myeloperoxidase (>3%). Based on selection criteria, cases with multilineage dysplasia were excluded. Ring sideroblasts were identified in eight (28%) cases (range, 1–7%). By flow cytometry immunophenotyping, the blasts expressed the following markers: CD13 (91%), CD33 (86%), CD34 (93%), CD38 (100%), CD117 (91%), CD123 (91%), HLA-DR (98%), and myeloperoxidase (62%). Twenty-six of 43 (60%) cases expressed CD4 at least to some extent. CD22 was positive in 10 of 39 (26%) cases, CD7 in 11 (26%), CD15 was expressed in 4 (9%), CD5 in 3 (7%), CD14 in 2 (5%), CD2 in 2 (5%), and CD19 in 2 (5%) cases.
Cytogenetic findings
The results of conventional cytogenetic analysis were available in all 46 patients. Thirty-three (72%) patients showed a normal karyotype. The remaining patients showed either one clonal abnormality (n = 8, 17%) or two abnormalities (n = 5, 11%). Frequent cytogenetic abnormalities included trisomy 8 (n = 4, 9%) and trisomy 13 (n = 3, 6.5%). No patients showed abnormalities of chromosome 21. The karyotype at the time of diagnosis was available in 90 patients in the control group. De novo AML-RUNX1mut group had a significantly higher frequency of normal karyotype (72% vs. 47%, p = 0.01) and a lower frequency of adverse karyotypic abnormalities with the control group. Trisomy 13 was more frequent in patients with AML-RUNX1mut compared with the control AML-RUNX1wt group (6.5% vs. 0%, p = 0.037). There was no significant difference in the frequencies of trisomy 8 or 21. Similarly, there was no difference in the frequency of combined trisomy 8 and 13. Only one patient had both trisomy 8 and trisomy 13. The results are summarized in Table 1.
Concurrent gene mutations
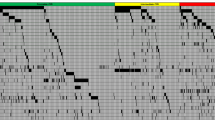
Using a combination of a targeted NGS panel and Sanger sequencing, the median number of mutations per patient (from a total of 31 tested genes) in de novo AML RUNX1mut was three (range, 1–11); all cases had at least one mutation in addition to RUNX1. The most frequent concomitant mutations in decreasing order of frequencies included the splicing factor genes SRSF2 (n = 9/20, 45%); FLT3 [n = 19/46, 41%, 12 ITDs with a median ITD ratio of 0.307 (range of 0.014–0.562)], ASXL1 (n = 14, 30%), NRAS (n = 13, 28%), DNMT3A (n = 10, 22%), IDH2 (n = 12, 26%), TET2 (n = 6, 13%) and EZH2 (n = 4, 9%). One of 43 cases (2%) showed a CEBPA mutation. By definition, NPM1 mutations were absent in all cases. Mutations in GATA2, JAK2, KIT, MPL, PTPN11, TP53, and WT1 were rare. Mutations in ABL1, BRAF, EGFR, GATA1, HRAS, IKZF2, MDM2, MLL, and MYD88 were mutually exclusive with RUNX1. The data are shown in Fig. 3.

Genes are segregated based on the biological function the left. The upper panels denote each patient represented by a unique patient number with all the mutations detected in each patient. Red, single mutation; dark red, two mutations; brown, three mutations; gray, wild type; white not tested.
The distribution of mutations within the control group was as follows: FLT3 [n = 23, 24%, 15 were ITDs with a median ITD ratio of 0.338 (range, 0.023–0.975)], NPM1 (n = 20, 21%), DNMT3A (n = 19, 20%), IDH2 (n = 18, 19%), NRAS (n = 14, 15%), CEBPA (n = 11, 12%), TET2 (n = 10, 11%), IDH1 (n = 9, 10%), TP53 (n = 7, 7%), KRAS (n = 6, 6%), PTPN11 (n = 6, 6%), GATA2 (n = 2, 2%), EZH2 (n = 1, 1%), and MPL (n = 1, 1%).
De novo AML RUNX1mut showed a significantly higher frequency of mutations in the SRSF2 (p = 0.02), FLT3 (p = 0.03), ASXL1 (p = 0.0004), EZH2 (p = 0.04), and compared with de novo AML RUNX1wt. In contrast, de novo AML RUNX1mut showed a significantly lower frequency of NPM1 mutations compared with de novo AML-RUNX1wt (n = 20, p = 0.0002) (Table 1). Single-CEBPA mutations were less common in AML-RUNX1mut; however, this finding did not achieve statistical significance. The distribution of the remaining gene mutations showed no significant differences between the two groups.
We found no difference in the distribution of concomitant mutations, including splicing factor mutations, with RUNX1 mutations of the RHD domain as compared with TAD domain. The median variant allelic frequency for the RUNX1 mutation was 32% (range, 2–97%). Within the tested genes using the NGS panel, RUNX1 gene mutation represented the major clone in 32 (70%) patients.
Sequential mutation profiling
We assessed the stability and clonal evolution patterns of RUNX1 and other concomitant gene mutations by performing sequential NGS-based mutation profiling in ten patients at the time of diagnosis and subsequently at the time of persistent AML (n = 3) or relapse (n = 7) (Fig. 4). Overall, these patients showed 12 RUNX1 mutations at the time of initial mutation profiling. At time of relapse or persistent disease the same RUNX1 mutations were retained in nine (90%) patients; this patient subgroup included two patients who had two different RUNX1 mutations, both of which were retained. In one (10%) patient with relapsed AML (blasts: 13%), RUNX1 mutation was present at initial diagnosis but absent at relapse. In this patient (UPN37), mutations in RUNX1, ASXL1, and DNMT3A genes were present at the time of diagnosis. NGS-based sequencing performed 16 months later for relapsed AML showed the absence of RUNX1 mutation and one ASXL1 mutation, whereas the other ASXL1 and DNMT3A mutations persisted (Fig. 4j).

a–j Sequential mutation analysis by next-generation sequencing (NGS) of de novo acute myeloid leukemia with mutated RUNX1. Each graph represents one patient, designated by the unique patient number (UPN). The mutated gene and protein change are shown along the horizontal axis. The variant allele frequency is shown along the vertical axis. Serial next-generation sequencing studies are shown by the different colors. The legend at the right of each chart indicates the number of months from initial mutational profiling of acute myeloid leukemia at our institution to the time of testing. The absence of bar, indicates that the mutation was not present at that time point. f FLT3-ITD mutations were detected in patient 46 by polymerase chain reaction but not NGS. The variant allele frequency is not available. i–j Patients 14 and 37 showed RUNX1 mutations on initial profiling which were no longer present at the time of repeat profiling.
NGS-based mutation analysis at the time of relapse or persistent disease also showed additional mutations in the following genes: ASXL1 (n = 1), NRAS (n = 1), IDH1 (n = 1), FLT3 D835H (n = 1), TET2 (n = 1), and TP53 (n = 2). Three patients received specific drugs targeting mutations in NRAS (n = 1), IDH1 (n = 1), and FLT3 (n = 1) with subsequent disappearance of IDH1 and FLT3 and reduction in the number of mutations of NRAS. In one patient, EZH2 mutation disappeared during relapse.
Survival analysis
At last follow-up, 27 (59%) patients with de novo AML-RUNX1mut had died. The median OS for this group was 26 months; median follow-up duration was 21.4 months. De novo AML-RUNX1mut patients had a shorter OS compared with de novo AML-RUNX1wt patients (median OS: 32 months, median follow-up: 24 months), although the difference was not statistically significant (p = 0.71) (Fig. 5a). There was no significant difference in median EFS between de novo AML-RUNX1mut and de novo AML-RUNX1WT (13.5 vs. 14 months, p = 0.44). No significant difference was noted in OS/EFS between the two groups among treatment naive patients. RUNX1-mutated AML patients with clonal RUNX1 mutation (defined here as ≥20% VAF) showed a significantly shorter median OS than those with subclonal <20% VAF (23 months vs. undefined; p = 0.04). However, there was no significant difference in OS/EFS when compared with wild-type RUNX1 (23 vs. 32 months; p = 0.23) (Fig. 5b, c). There was no statistically significant difference in the OS/EFS between the two patient groups (total and treatment naive subgroups) based on karyotype or 2017 ELN genetic risk (Table 2). Study group cases would be categorized as AML-NOS in the absence of RUNX1 mutation. So, we compared the OS/EFS with AML-NOS cases in the control cohort (essentially applying all the WHO elimination criteria proposed for this provisional entity to the control group, thus the only difference is the RUNX1 mutation status) and found no significant difference (26 vs. 38 months, p = 0.53). No significant difference in OS was noted when comparing de novo AML-RUNX1mut to patients with AML-MRC RUNX1wt (median OS, 26.3 vs. 19.4 months, p = 0.58) or AML patients with mutated NPM1 (26.3 vs. 42.5 months, p = 0.97).

a Overall survival (OS) for de novo AML patients with mutated RUNX1 (27/46 died, median OS, 26 months) showed no significant difference compared to de novo AML patients with wild-type RUNX1 (54/94 died, median OS, 32 months; p = 0.71). b OS for de novo AML patients with mutated RUNX1 with a variant allele frequency of >20% (24/34 died, median OS, 23 months) was significantly different compared to those with variant allele frequency <20% (3/12 died, median OS, undefined months; p = 0.04). c De novo AML patients with mutated RUNX1 with a variant allele frequency of >20% (median OS, 23 months) showed no difference in OS compared to de novo AML patients with wild-type RUNX1 (median OS, 32 months; p = 0.23).
Among de novo AML-RUNX1mut patients, 31 patients received chemotherapy alone and 15 patients received stem cell transplant. In the control group, 55 patients received chemotherapy alone and 39 patients received stem cell transplant. There was no significant difference in OS in the de novo AML-RUNX1mut and de novo AML-RUNX1WT patients treated with chemotherapy alone (20.4 months vs. 17.3 months, p = 0.94). There was no difference in OS between de novo AML-RUNX1mut and de novo AML-RUNX1WT patients who received stem cell transplant (45 months vs. 57.5 months, p = 0.62). However, we acknowledge that the number of cases is small.
Within the study cohort, we explored the prognostic effect of the location and type of RUNX1 mutations on outcome. There was no difference in OS for patients with RUNX1 RHD mutations as compared with RUNX1 TAD mutations (25.9 vs. 26.3 months, p = 0.72). There was no difference in OS for patients with RUNX1 missense mutations as compared to those with deleterious RUNX1 mutations (defined as nonsense and frameshift) mutations (25.9 vs. 28 months, p = 0.60).
Finally, to explore the effect of multilineage dysplasia, we compared 15 patients de novo AML-RUNX1mut cases that were originally excluded based on the presence of multilineage dysplasia (Fig. 1). These patients showed no differences in OS compared to de novo AML RUNX1mut, de novo AML RUNX1WT, de novo AML RUNX1WT not otherwise specified, de novo AML RUNX1WT NOS, de novo AML RUNX1WT with myelodysplasia related changes, or de novo AML RUNX1WT with NPM1 mutation (Supplementary Fig. 1 and Supplementary Table 3).
By univariate analysis, RUNX1 mutation was neither associated with OS nor EFS. OS significantly associated with age and ELN risk, and EFS with age and stem cell transplant (Supplementary Table 4).
Discussion
The recent WHO revision to the AML classification has recognized de novo AML with RUNX1 mutation, in the absence of MRC (including cytogenetic abnormalities), as a provisional category based on common biologic characteristics and adverse outcome [3]. However, the clinical relevance of this new AML subcategory compared with de novo AML without RUNX1 mutations has not been assessed. Multiple large studies that have analyzed the clinical implications of RUNX1 mutations [4,5,6,7, 21, 22], including the largest series to date by Gaidzik et al., have included a mixture of de novo and secondary AML from multicenter clinical trials [4, 7]. Few studies on de novo AML have not used the criteria proposed by the WHO and include cases with MRCs [5, 6, 21, 23]. Furthermore, none of the above studies utilized NGS-based technologies for mutation assessment.
In our study, trisomy 13 was more frequent in patients with AML-RUNX1mut compared to the control AML-RUNX1wt group, consistent with other studies [7, 23,24,25]. Trisomy 8 was noted in four (9%) and trisomy 13 in three (6.5%) patients with AML-RUNX1mut. Trisomy 13 is a very rare event in myeloid neoplasms, but is associated with mutations in RUNX1 and SRSF2 in AML [26]. Trisomy 13 was implicated as a mechanism of overexpression of FLT3 and the authors hypothesized cooperative effects between mutated RUNX1 and FLT3 [24, 25]. In this study, de novo AML-RUNX1mut group had a significantly higher frequency of normal karyotype compared with the control group, consistent with other studies. However, there was no significant difference in OS between the two groups among patients with normal cytogenetics (p = 0.55). The low frequency of adverse karyotypic abnormalities [4, 6] may be partly due to the WHO criteria to eliminate MDS-related cytogenetic abnormalities.
RUNX1 mutations clustered within the RHD and TAD domains of the gene, although the mutations spanned the entire coding region. Hence, for clinical testing, sequencing the entire coding region of the RUNX1 gene is recommended. The types of mutations identified in this study are in accord with the function of the protein. Within the RHD, point mutations accounted for most of the mutations versus the TAD where frameshift mutations were more common. The RHD contains the DNA-binding site of RUNX1 protein and hence, a point missense mutation is likely to disrupt protein function. In contrast, the TAD is responsible for the in vivo function of the RUNX1 protein, and these functions are affected substantially by truncation of large segments due to frameshift and nonsense mutations [6, 27].
NGS-based mutation analysis using a multigene panel facilitated assessment of concomitant cooperating gene mutations. RUNX1 mutation alone is insufficient for leukemogenesis [10]. We noted concomitant mutations in FLT3 (41%), DNMT3A (22%), ASXL1 (30%), IDH2 (26%), and NRAS (28%). However, only mutations in SRSF2, FLT3, ASXL1, EZH2, and NPM1 were significantly different from the AML with RUNX1wt group. When grouped into functional categories as described by the Cancer Genome Atlas Research Network [1], de novo AML with RUNX1 mutation cases show a significant association with mutations in spliceosome-related genes, specifically SRSF2 and in chromatin and histone modifiers, such as ASXL1 and EZH2 compared with de novo AML RUNX1wt cases. Gaidzaik et al. also noted significant differences in mutation frequencies of IDH2 and SF3B1 genes; the reasons could be related to selection of cases based on the proposed WHO criteria and the smaller number of cases in our study [7]. The precise role of each of these concomitant mutations is unknown, but there is ample evidence supporting the idea of a multistep process of leukemogenesis.
SRSF2 mutation was the most frequent mutation in this entity. Haferlach et al. showed SRSF2 mutation in 36% of de novo AML with RUNX1 [23]. While RUNX1 is a pan-AML mutation, SRSF2 mutation is almost (>95%) specific for AML arising in a setting of an antecedent myeloid malignancy [28]. This suggests that these cases may arise from a previously unrecognized MDS, despite de novo presentation. Splicing factor mutations were also noted in up to 11% of de novo AML and associated with mutations in RUNX1, ASXL1, IDH2, and TET2 genes in a study by Hou et al.; the presence of splicing factor mutations in de novo AML was an independent predictor of OS and disease-free survival [29]. EZH2 mutations were observed at a higher frequency (~10%) in de novo AML-RUNX1mut, compared with the control group. EZH2 mutations are uncommon (0–2%) in de novo AML [30, 31]. EZH2 is located on chromosome 7q36 and is an epigenetic regulator of transcription that encodes for the catalytic subunit of a histone methyltransferase, polycomb repressive complex 2. Inactivating mutations in EZH2 may result in myeloid proliferation that may be a driving factor for a small subset of de novo AML-RUNX1mut [32]. None of the 12 de novo AML-RUNX1mut patients with an SRSF2 mutation had a concurrent EZH2 mutation. EZH2 is further downstream of SRSF2-dependent splicing, thereby leading to mutual exclusivity.
By sequential NGS-based multigene mutation profiling in ten patients at the time of diagnosis and subsequently at time of persistent or relapsed disease, we show that RUNX1 mutation disappeared at time of relapse in one patient while mutations in DNMT3A and ASXL1 persisted. Similar finding was shown by Tang et al. by using less sensitive sequencing technique [6]. “Founder” mutations tend to persist at the time of remission, but mutations implicated in disease progression tend to disappear [28]. This finding raises the possibility that RUNX1 mutation may not be a “founder” or “driver” mutation for AML, at least in a small subset of patients [5, 6]. However, this possibility is based on the results of only one patient, and a larger cohort with sequential NGS testing is necessary for confirmation. Based on the mutation pattern, likely founder or driver mutations in de novo AML-RUNX1mut in this study include splicing factor genes, ASXL1, DNMT3A, and TET2. In aggregate, about 65% of patients had at least one of these mutations in addition to RUNX1. Whereas DNMT3A and TET2 mutations were frequent in both AML-RUNX1mut and AML-RUNX1WT, mutations in splicing factors and ASXL1 were significantly more frequent in the AML-RUNX1mut group, suggesting that these mutations play a larger role as drivers in de novo AML-RUNX1mut.
There was no statistically significant difference in OS between de novo AML with mutated RUNX1 patients versus control group patients. Based on our study findings, the outcome of RUNX1 mutated AML depends on the disease context, de novo versus secondary. In multiple large-scale studies, RUNX1 mutation has been shown to be an independent predictor of poor OS [5, 6, 21] and EFS [4, 7, 21]. However, most of these studies have included patients with both de novo AML and AML transformed from MDS (secondary AML). None of these studies have focused on de novo AML with mutated RUNX1 using new WHO criteria. Recent studies that included only de novo AML or those tested using NGS-sequencing OS [23, 33,34,35] have not observed a significant effect on OS. Only mutations in RUNX1 and three or more additional gene mutations were independently associated with poor OS [23].
Per 2016 WHO criteria, the presence of multilineage dysplasia (at least 50% dysplastic cells in at least two lineages) in de novo AML-RUNX1mut categorizes the case as AML-MRC and excludes from the de novo AML-RUNX1mut category. To explore the effect of multilineage dysplasia, we evaluated the characteristics of 15 de novo AML-RUNX1mut cases that we had originally excluded (Fig. 1). Overall, there were no significant differences in the parameters assessed except for fewer circulating blasts compared to the WHO-defined study cohort composed of de novo AML with mutated RUNX1 (Supplementary Table 2). In addition, there were no differences in OS compared to de novo AML RUNX1mut, de novo AML RUNX1WT, de novo AML RUNX1WT, not otherwise specified, de novo AML RUNX1WT with myelodysplasia related changes, or de novo AML RUNX1WT with NPM1 mutation (Supplementary Fig. 1 and Supplementary Table 3). This suggests that de novo AML-RUNX1mut with multilineage dysplasia in the absence of history of myeloid neoplasms or MDS-related cytogenetic changes may not be significantly different from WHO-defined de novo AML-RUNX1mut. Hence, the importance of multilineage dysplasia in de novo RUNX1 mutated AML is unclear and needs to be evaluated further for future AML classifications.
We acknowledge limitations in our study: it is retrospective with a relatively small number of cases and variation in treatment regimens. Our institution is a referral center and a significant proportion of patients present after already having received induction chemotherapy. We did not find significant differences in gene mutation profiles between treatment naive AML patients and relapsed AML patients following treatment in this cohort. However, since new mutations are known to arise following chemotherapy, caution must be used when analyzing and interpreting mutational profiles following chemotherapy. As such, validation with larger cohorts and prospective studies are necessary and efforts to this end are ongoing.
In conclusion, de novo AML patients with RUNX1 mutation show characteristic morphologic findings, a high frequency of a normal karyotype, a high frequency of concurrent mutations in ASXL1 and SRSF2 genes, and absent NPM1 mutations. Cases of de novo AML with RUNX1 mutation have minimal overlap with other unique subtypes of AML, such as those cases associated with t(8;21), inv(16), or NPM1 mutations. The data supports the separate categorization of de novo AML with RUNX1 mutation as proposed in the current WHO classification based on distinctive clinicopathologic features. However, these patients show no significant outcome difference compared with de novo AML patients without RUNX1 alterations.
References
Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74.
Gilliland DG. Hematologic malignancies. Curr Opin Hematol. 2001;8:189–91.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Gaidzik VI, Bullinger L, Schlenk RF, Zimmermann AS, Rock J, Paschka P, et al. RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J Clin Oncol. 2011;29:1364–72.
Schnittger S, Dicker F, Kern W, Wendland N, Sundermann J, Alpermann T, et al. RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis. Blood. 2011;117:2348–57.
Tang JL, Hou HA, Chen CY, Liu CY, Chou WC, Tseng MH, et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood. 2009;114:5352–61.
Gaidzik VI, Teleanu V, Papaemmanuil E, Weber D, Paschka P, Hahn J, et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia. 2016;30:2160–68.
Harada H, Harada Y, Niimi H, Kyo T, Kimura A, Inaba T. High incidence of somatic mutations in the AML1/RUNX1 gene in myelodysplastic syndrome and low blast percentage myeloid leukemia with myelodysplasia. Blood. 2004;103:2316–24.
Chen CY, Lin LI, Tang JL, Ko BS, Tsay W, Chou WC, et al. RUNX1 gene mutation in primary myelodysplastic syndrome-the mutation can be detected early at diagnosis or acquired during disease progression and is associated with poor outcome. Br J Haematol. 2007;139:405–14.
Dicker F, Haferlach C, Sundermann J, Wendland N, Weiss T, Kern W, et al. Mutation analysis for RUNX1, MLL-PTD, FLT3-ITD, NPM1 and NRAS in 269 patients with MDS or secondary AML. Leukemia. 2010;24:1528–32.
Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Mutations of AML1 are common in therapy-related myelodysplasia following therapy with alkylating agents and are significantly associated with deletion or loss of chromosome arm 7q and with subsequent leukemic transformation. Blood. 2004;104:1474–81.
Niimi H, Harada H, Harada Y, Ding Y, Imagawa J, Inaba T, et al. Hyperactivation of the RAS signaling pathway in myelodysplastic syndrome with AML1/RUNX1 point mutations. Leukemia. 2006;20:635–44.
Mill CP, Fiskus W, DiNardo CD, Qian Y, Raina K, Rajapakshe K, et al. RUNX1-targeted therapy for AML expressing somatic or germline mutation in RUNX1. Blood. 2019;134:59–73.
Illendula A, Gilmour J, Grembecka J, Tirumala VS, Boulton A, Kuntimaddi A, et al. Small molecule inhibitor of CBFbeta-RUNX binding for RUNX transcription factor driven cancers. EBioMedicine. 2016;8:117–31.
Dohner K, Paschka P. Intermediate-risk acute myeloid leukemia therapy: current and future. Hematol Am Soc Hematol Educ Program. 2014;2014:34–43.
Kanagal-Shamanna R, Bueso-Ramos CE, Barkoh B, Lu G, Wang S, Garcia-Manero G, et al. Myeloid neoplasms with isolated isochromosome 17q represent a clinicopathologic entity associated with myelodysplastic/myeloproliferative features, a high risk of leukemic transformation, and wild-type TP53. Cancer. 2011;118:2879–88.
Kanagal-Shamanna R, Singh RR, Routbort MJ, Patel KP, Medeiros LJ, Luthra R. Principles of analytical validation of next-generation sequencing based mutational analysis for hematologic neoplasms in a CLIA-certified laboratory. Expert Rev Mol Diagn. 2016;16:461–72.
Kanagal-Shamanna R, Luthra R, Yin CC, Patel KP, Takahashi K, Lu X, et al. Myeloid neoplasms with isolated isochromosome 17q demonstrate a high frequency of mutations in SETBP1, SRSF2, ASXL1 and NRAS. Oncotarget. 2016;7:14251–8.
Sood R, Kamikubo Y, Liu P. Role of RUNX1 in hematological malignancies. Blood. 2017;129:2070–82.
Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–47.
Mendler JH, Maharry K, Radmacher MD, Mrozek K, Becker H, Metzeler KH, et al. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and MicroRNA expression signatures. J Clin Oncol. 2012;30:3109–18.
Khan M, Cortes J, Kadia T, Naqvi K, Brandt M, Pierce S, et al. Clinical outcomes and co-occurring mutations in patients with RUNX1-mutated acute myeloid leukemia. Int J Mol Sci. 2017;18.
Haferlach T, Stengel A, Eckstein S, Perglerova K, Alpermann T, Kern W, et al. The new provisional WHO entity ‘RUNX1 mutated AML’ shows specific genetics but no prognostic influence of dysplasia. Leukemia. 2016;30:2109–12.
Dicker F, Haferlach C, Kern W, Haferlach T, Schnittger S. Trisomy 13 is strongly associated with AML1/RUNX1 mutations and increased FLT3 expression in acute myeloid leukemia. Blood. 2007;110:1308–16.
Silva FP, Lind A, Brouwer-Mandema G, Valk PJ, Giphart-Gassler M. Trisomy 13 correlates with RUNX1 mutation and increased FLT3 expression in AML-M0 patients. Haematologica. 2007;92:1123–6.
Herold T, Metzeler KH, Vosberg S, Hartmann L, Röllig C, Stölzel F, et al. Isolated trisomy 13 defines a homogeneous AML subgroup with high frequency of mutations in spliceosome genes and poor prognosis. Blood. 2014;124:1304–11.
Kuo M, Liang D, Huang C, Shih Y, Wu J, Lin T, et al. RUNX1 mutations are frequent in chronic myelomonocytic leukemia and mutations at the C-terminal region might predict acute myeloid leukemia transformation. Leukemia. 2009;23:1426–31.
Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125:1367–76.
Hou HA, Liu CY, Kuo YY, Chou WC, Tsai CH, Lin CC, et al. Splicing factor mutations predict poor prognosis in patients with de novo acute myeloid leukemia. Oncotarget. 2016;7:9084–101.
Greenblatt SM, Nimer SD. Chromatin modifiers and the promise of epigenetic therapy in acute leukemia. Leukemia. 2014;28:1396–406.
Wang X, Dai H, Wang Q, Wang Q, Xu Y, Wang Y, et al. EZH2 mutations are related to low blast percentage in bone marrow and -7/del(7q) in de novo acute myeloid leukemia. PLoS One. 2013;8:e61341.
Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42:722–6.
Weinberg OK, Gibson CJ, Blonquist TM, Neuberg D, Pozdnyakova O, Kuo F, et al. NPM1 mutation but not RUNX1 mutation or multilineage dysplasia defines a prognostic subgroup within de novo acute myeloid leukemia lacking recurrent cytogenetic abnormalities in the revised 2016 WHO classification. Am J Hematol. 2017;92:E123–E124.
Shin S-Y, Lee S-T, Kim H-J, Cho EH, Kim J-W, Park S, et al. Mutation profiling of 19 candidate genes in acute myeloid leukemia suggests significance of DNMT3A mutations. Oncotarget. 2016;7:54825.
Lin PH, Li HY, Fan SC, Yuan TH, Chen M, Hsu YH, et al. A targeted next‐generation sequencing in the molecular risk stratification of adult acute myeloid leukemia: implications for clinical practice. Cancer Med. 2017;6:349–60.
Funding
This study was partly supported by institutional start-up funds awarded to RK-S.
Author information
Authors and Affiliations
Contributions
RK-S designed the study. AEQ and RK-S collected and interpreted the data; AEQ and RK-S wrote and revised the paper; AEQ, RK-S, RB and KS performed statistical analysis; GMB, RL, KPP, KS, CB-R, JDK, MJR, JH-L, CZ, PL, SL, CYO, TK, CND, HK, GG-M, and RK-S provided study patients and related data; all authors contributed to scientific discussions, revised, and approved the final paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Quesada, A.E., Montalban-Bravo, G., Luthra, R. et al. Clinico-pathologic characteristics and outcomes of the World Health Organization (WHO) provisional entity de novo acute myeloid leukemia with mutated RUNX1. Mod Pathol 33, 1678–1689 (2020). https://doi.org/10.1038/s41379-020-0531-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-0531-2
This article is cited by
-
RUNX1 mutation has no prognostic significance in paediatric AML: a retrospective study of the AML-BFM study group
Leukemia (2023)
-
The Emerging Role of Hematopathologists and Molecular Pathologists in Detection, Monitoring, and Management of Myeloid Neoplasms with Germline Predisposition
Current Hematologic Malignancy Reports (2021)
