Delta-Integration of Single Gene Shapes the Whole Metabolomic Short-Term Response to Ethanol of Recombinant Saccharomyces cerevisiae Strains

 , ,
, ,  , ,
, ,
Abstract
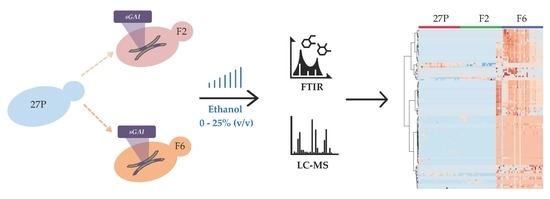
:
1. Introduction
2. Results and Discussion
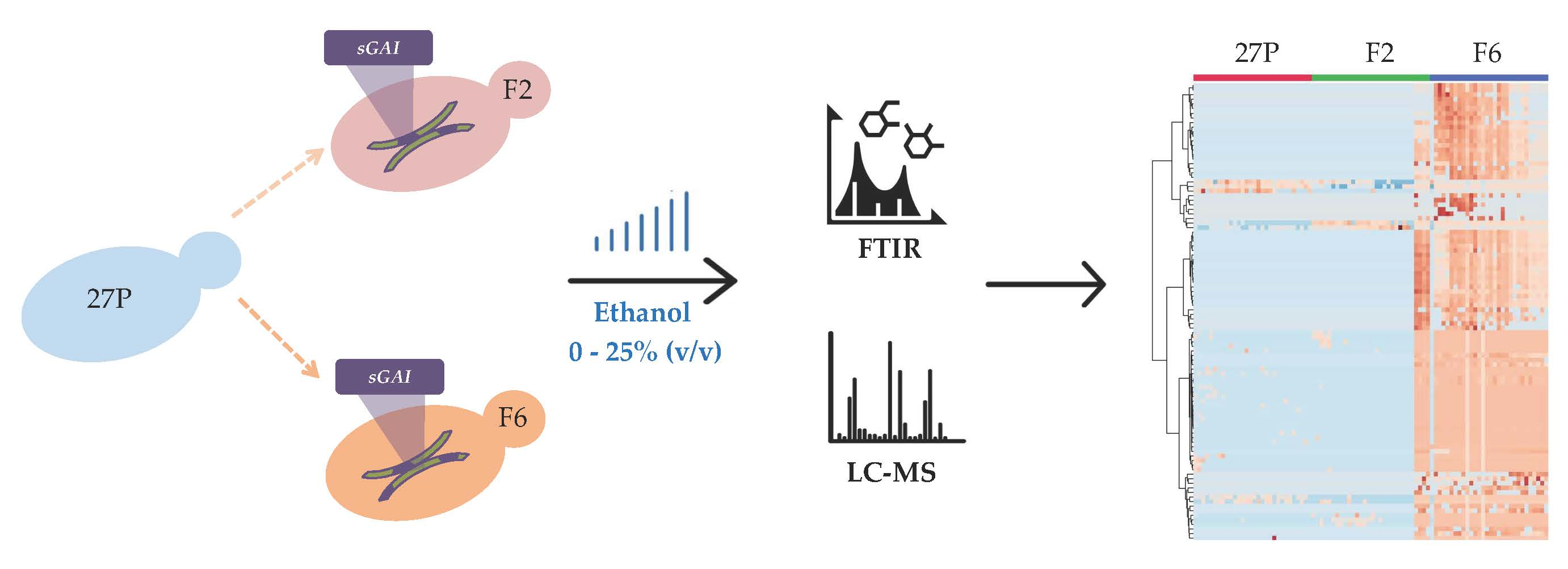
2.1. FTIR Fingerprints Under Ethanol Stress
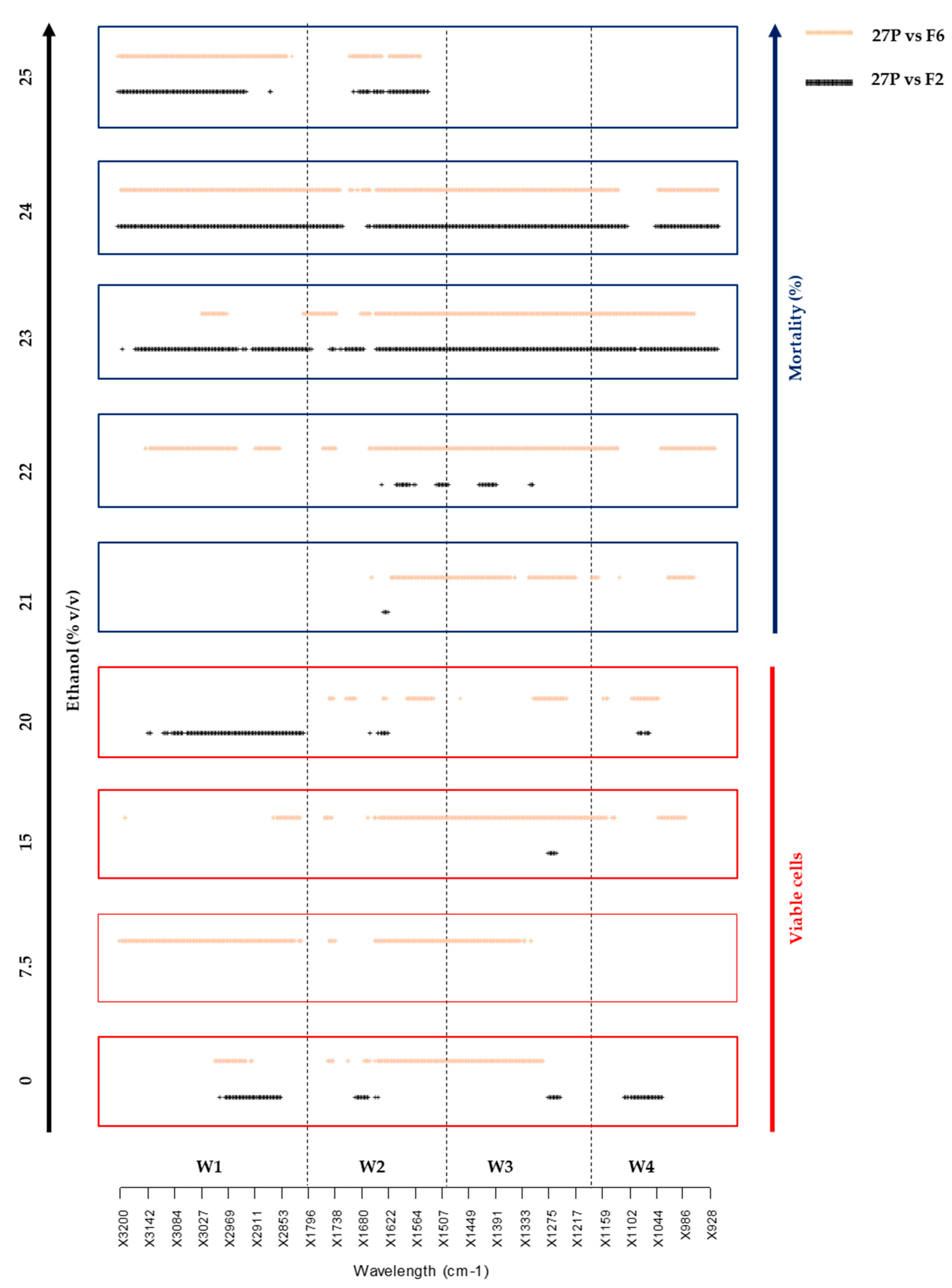
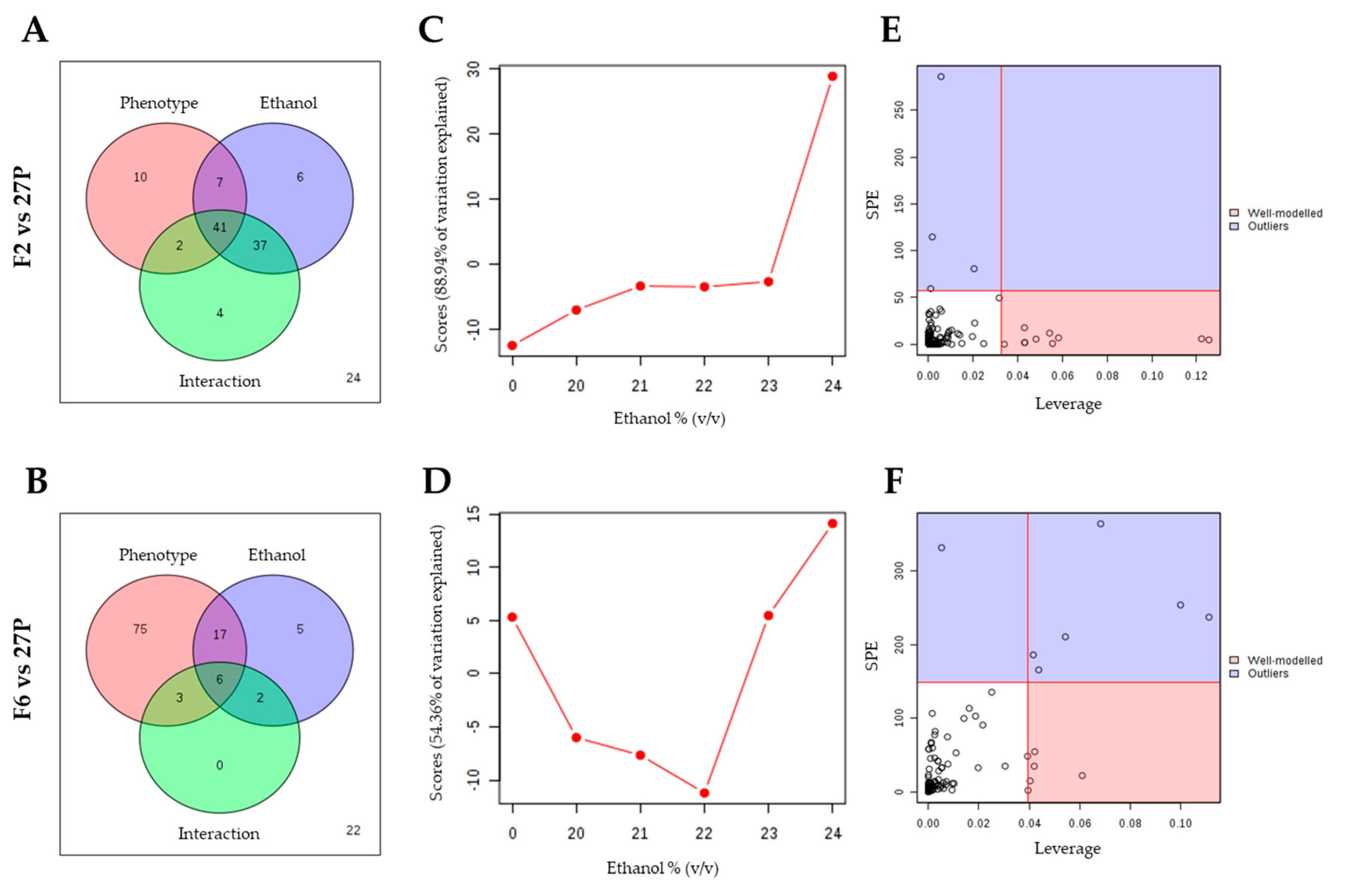
2.2. Metabolomic Changes Induced by δ-Integration
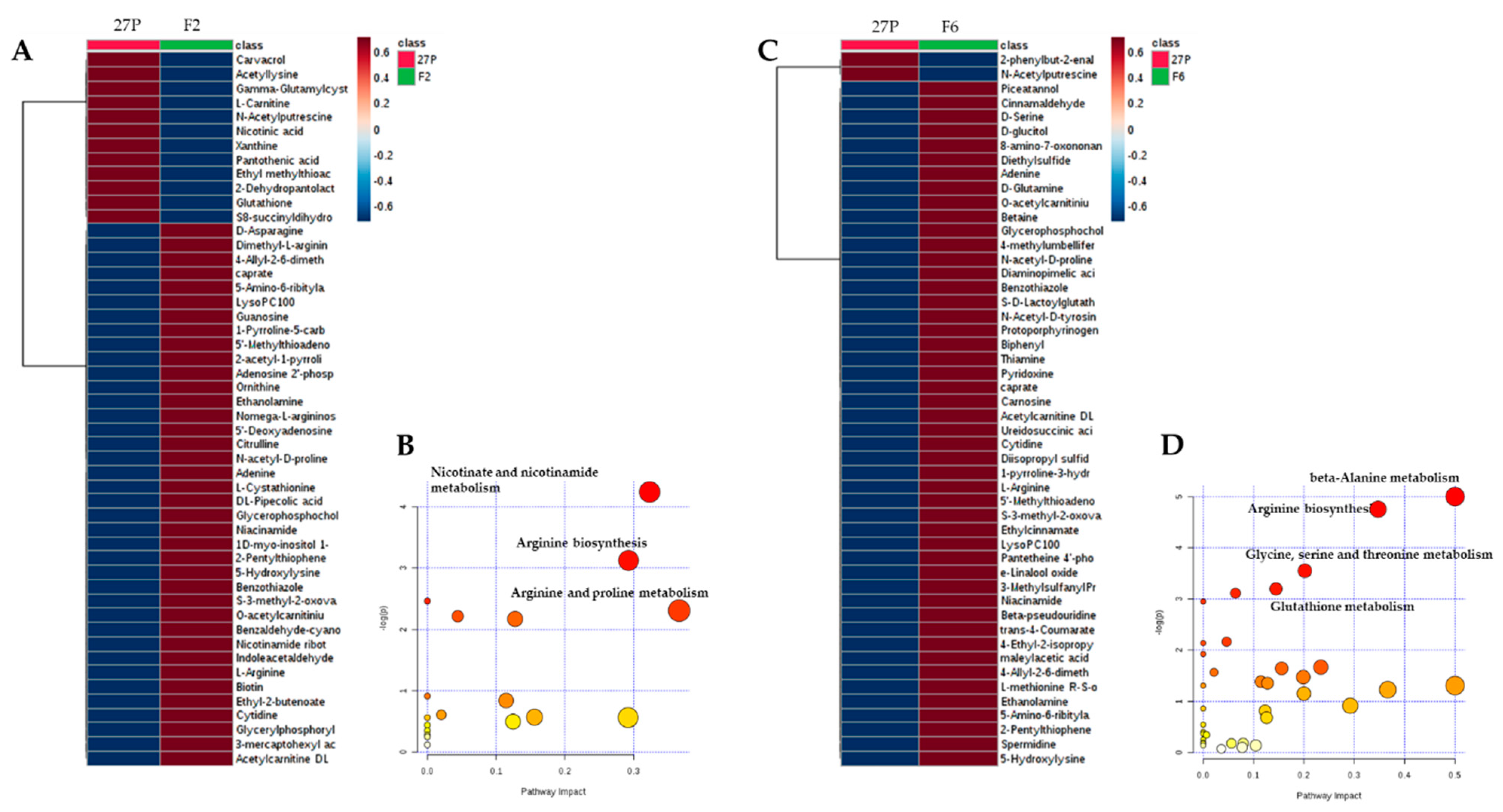
2.3. Changes in Intracellular Metabolites in the Short-Term Response to Ethanol of the Recombinant F2 and F6 S. Cerevisiae Strains
2.4. Metabolic Pathway Involved in the Response to Ethanol Stress of Engineered Strains F2 and F6.
3. Materials and Methods
3.1. Cultures and Growth Conditions
3.2. FTIR-Based Bioassay
3.3. Spectra Pre-Processing
3.4. Untargeted Metabolomics Profile Determination of Cells under Ethanol Stress by LC/MS Analysis
3.5. Data Analysis
3.5.1. Significant Wavelengths Analysis (SWA) Throughout the Spectra
3.5.2. Metabolite Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hamelinck, C.N.; Van Hooijdonk, G.; Faaij, A.P. Ethanol from lignocellulosic biomass: Techno-economic performance in short-, middle-and long-term. Biomass Bioenergy 2005, 28, 384–410. [Google Scholar] [CrossRef]
- Ishola, M.M.; Jahandideh, A.; Haidarian, B.; Brandberg, T.; Taherzadeh, M.J. Simultaneous saccharification, filtration and fermentation (SSFF): A novel method for bioethanol production from lignocellulosic biomass. Bioresour. Technol. 2013, 133, 68–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, P.K.; Singh, S.; Asthana, R.; Singh, S. Biohydrogen production from sugarcane bagasse by integrating dark-and photo-fermentation. Bioresour. Technol. 2014, 152, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Romanelli, M.G.; Povolo, S.; Favaro, L.; Fontana, F.; Basaglia, M.; Casella, S. Engineering Delftia acidovorans DSM39 to produce polyhydroxyalkanoates from slaughterhouse waste. Int. J. Biol. Macromol. 2014, 71, 21–27. [Google Scholar] [CrossRef]
- Favaro, L.; Penna, A.L.B.; Todorov, S.D. Bacteriocinogenic LAB from cheeses–application in biopreservation? Trends Food Sci. Technol. 2015, 41, 37–48. [Google Scholar] [CrossRef]
- Shah, A.; Favaro, L.; Alibardi, L.; Cagnin, L.; Sandon, A.; Cossu, R.; Casella, S.; Basaglia, M. Bacillus sp. strains to produce bio-hydrogen from the organic fraction of municipal solid waste. Appl. Energy 2016, 176, 116–124. [Google Scholar] [CrossRef]
- Alibardi, L.; Green, K.; Favaro, L.; Vale, P.; Soares, A.; Cartmell, E.; Fernández, Y.B. Performance and stability of sewage sludge digestion under CO2 enrichment: A pilot study. Bioresour. Technol. 2017, 245, 581–589. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Kougias, P.G.; Treu, L.; Campanaro, S.; Angelidaki, I. Microbial community changes in methanogenic granules during the transition from mesophilic to thermophilic conditions. Appl. Microbiol. Biotechnol. 2017, 101, 1313–1322. [Google Scholar] [CrossRef]
- Myburgh, M.W.; Cripwell, R.A.; Favaro, L.; van Zyl, W.H. Application of industrial amylolytic yeast strains for the production of bioethanol from broken rice. Bioresour. Technol. 2019, 294, 122222. [Google Scholar] [CrossRef]
- Favaro, L.; Basaglia, M.; Casella, S. Improving polyhydroxyalkanoate production from inexpensive carbon sources by genetic approaches: A review. Biofuels Bioprod. Biorefining 2019, 13, 208–227. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Dale, B.E. Global potential bioethanol production from wasted crops and crop residues. Biomass Bioenergy 2004, 26, 361–375. [Google Scholar] [CrossRef]
- Cinelli, B.A.; Castilho, L.R.; Freire, D.M.; Castro, A.M. A brief review on the emerging technology of ethanol production by cold hydrolysis of raw starch. Fuel 2015, 150, 721–729. [Google Scholar] [CrossRef]
- Favaro, L.; Cagnin, L.; Basaglia, M.; Pizzocchero, V.; van Zyl, W.H.; Casella, S. Production of bioethanol from multiple waste streams of rice milling. Bioresour. Technol. 2017, 244, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Van Zyl, W.; Bloom, M.; Viktor, M. Engineering yeasts for raw starch conversion. Appl. Microbiol. Biotechnol. 2012, 95, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Favaro, L.; Viktor, M.J.; Rose, S.H.; Viljoen-Bloom, M.; van Zyl, W.H.; Basaglia, M.; Cagnin, L.; Casella, S. Consolidated bioprocessing of starchy substrates into ethanol by industrial Saccharomyces cerevisiae strains secreting fungal amylases. Biotechnol. Bioeng. 2015, 112, 1751–1760. [Google Scholar] [CrossRef] [Green Version]
- Sakwa, L.; Cripwell, R.A.; Rose, S.H.; Viljoen-Bloom, M. Consolidated bioprocessing of raw starch with Saccharomyces cerevisiae strains expressing fungal alpha-amylase and glucoamylase combinations. FEMS Yeast Res. 2018, 18, foy085. [Google Scholar] [CrossRef] [Green Version]
- Cripwell, R.A.; Rose, S.H.; Favaro, L.; Van Zyl, W.H. Construction of industrial Saccharomyces cerevisiae strains for the efficient consolidated bioprocessing of raw starch. Biotechnol. Biofuels 2019, 12, 201. [Google Scholar] [CrossRef] [Green Version]
- Favaro, L.; Jansen, T.; van Zyl, W.H. Exploring industrial and natural Saccharomyces cerevisiae strains for the bio-based economy from biomass: The case of bioethanol. Crit. Rev. Biotechnol. 2019, 39, 800–816. [Google Scholar] [CrossRef]
- van Rensburg, E.; Den Haan, R.; Smith, J.; van Zyl, W.H.; Görgens, J.F. The metabolic burden of cellulase expression by recombinant Saccharomyces cerevisiae Y294 in aerobic batch culture. Appl. Microbiol. Biotechnol. 2012, 96, 197–209. [Google Scholar] [CrossRef]
- Ding, J.; Liang, G.; Zhang, K.; Hong, J.; Zou, S.; Lu, H.; Ma, Y.; Zhang, M. Extra metabolic burden by displaying over secreting: Growth, fermentation and enzymatic activity in cellobiose of recombinant yeast expressing β-glucosidase. Bioresour. Technol. 2018, 254, 107–114. [Google Scholar] [CrossRef]
- Favaro, L.; Jooste, T.; Basaglia, M.; Rose, S.H.; Saayman, M.; Görgens, J.F.; Casella, S.; van Zyl, W.H. Codon-optimized glucoamylase sGAI of Aspergillus awamori improves starch utilization in an industrial yeast. Appl. Microbiol. Biotechnol. 2012, 95, 957–968. [Google Scholar] [CrossRef]
- Walker, G.M.; Walker, R.S. Enhancing yeast alcoholic fermentations. In Advances in Applied Microbiology; Elsevier: London, UK, 2018; Volume 105, pp. 87–129. [Google Scholar]
- Saini, P.; Beniwal, A.; Kokkiligadda, A.; Vij, S. Response and tolerance of yeast to changing environmental stress during ethanol fermentation. Process Biochem. 2018, 72, 1–12. [Google Scholar] [CrossRef]
- Auesukaree, C. Molecular mechanisms of the yeast adaptive response and tolerance to stresses encountered during ethanol fermentation. J. Biosci. Bioeng. 2017, 124, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Ming, M.; Wang, X.; Lian, L.; Zhang, H.; Gao, W.; Zhu, B.; Lou, D. Metabolic responses of Saccharomyces cerevisiae to ethanol stress using gas chromatography-mass spectrometry. Mol. Omics 2019, 15, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.-Z.; Li, B.-Z.; Cheng, J.-S.; Yuan, Y.-J. Metabolome analysis of differential responses of diploid and haploid yeast to ethanol stress. Omics J. Integr. Biol. 2010, 14, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ma, M.-L.; Luo, S.; Zhang, R.-M.; Han, P.; Hu, W. Metabolic responses to ethanol in Saccharomyces cerevisiae using a gas chromatography tandem mass spectrometry-based metabolomics approach. Int. J. Biochem. Cell Biol. 2012, 44, 1087–1096. [Google Scholar] [CrossRef]
- Moktaduzzaman, M.; Galafassi, S.; Vigentini, I.; Foschino, R.; Corte, L.; Cardinali, G.; Piškur, J.; Compagno, C. Strain-dependent tolerance to acetic acid in Dekkera bruxellensis. Ann. Microbiol. 2016, 66, 351–359. [Google Scholar] [CrossRef]
- Favaro, L.; Cagnin, L.; Corte, L.; Roscini, L.; De Pascale, F.; Treu, L.; Campanaro, S.; Basaglia, M.; van Zyl, W.H.; Casella, S.; et al. Metabolomic alterations do not induce metabolic burden in the industrial yeast M2n[pBKD2-Pccbgl1]-C1 engineered by multiple δ-integration of a fungal β-glucosidase gene. Front. Bioeng. Biotechnol. 2019, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roscini, L.; Favaro, L.; Corte, L.; Cagnin, L.; Colabella, C.; Basaglia, M.; Cardinali, G.; Casella, S. A yeast metabolome-based model for an ecotoxicological approach in the management of lignocellulosic ethanol stillage. R. Soc. Open Sci. 2019, 6, 180718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapaval, V.; Møretrø, T.; Suso, H.P.; Åsli, A.W.; Schmitt, J.; Lillehaug, D.; Martens, H.; Böcker, U.; Kohler, A. A high-throughput microcultivation protocol for FTIR spectroscopic characterization and identification of fungi. J. Biophotonics 2010, 3, 512–521. [Google Scholar] [CrossRef]
- Shapaval, V.; Schmitt, J.; Møretrø, T.; Suso, H.P.; Skaar, I.; Åsli, A.W.; Lillehaug, D.; Kohler, A. Characterization of food spoilage fungi by FTIR spectroscopy. J. Appl. Microbiol. 2013, 114, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Favaro, L.; Corte, L.; Roscini, L.; Cagnin, L.; Tiecco, M.; Colabella, C.; Berti, A.; Basaglia, M.; Cardinali, G.; Casella, S. A novel FTIR-based approach to evaluate the interactions between lignocellulosic inhibitory compounds and their effect on yeast metabolism. RSC Adv. 2016, 6, 47981–47989. [Google Scholar] [CrossRef]
- Corte, L.; Rellini, P.; Roscini, L.; Fatichenti, F.; Cardinali, G. Development of a novel, FTIR (Fourier Transform InfraRed spectroscopy) based, yeast bioassay for toxicity testing and stress response study. Anal. Chim. Acta 2010, 659, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Guyot, S.; Pénicaud, C.; Passot, S.; Sandt, C.; Fonseca, F.; Saurel, R.; Husson, F. Understanding the responses of Saccharomyces cerevisiae yeast strain during dehydration processes using synchrotron infrared spectroscopy. Analyst 2017, 142, 3620–3628. [Google Scholar] [CrossRef] [PubMed]
- Barker, M.; Rayens, W. Partial least squares for discrimination. J. Chemom. 2003, 17, 166–173. [Google Scholar] [CrossRef]
- Wold, S.; Sjöström, M.; Eriksson, L. PLS-regression: A basic tool of chemometrics. Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar] [CrossRef]
- Farrés, M.; Piña, B.; Tauler, R. Chemometric evaluation of Saccharomyces cerevisiae metabolic profiles using LC–MS. Metabolomics 2015, 11, 210–224. [Google Scholar] [CrossRef] [Green Version]
- Chong, I.-G.; Jun, C.-H. Performance of some variable selection methods when multicollinearity is present. Chemom. Intell. Lab. Syst. 2005, 78, 103–112. [Google Scholar] [CrossRef]
- Mandelstam, J. Protein turnover and its function in economy of cell. Ann. N. Y. Acad. Sci. 1963, 102, 621–636. [Google Scholar] [CrossRef]
- Vanegas, J.M.; Contreras, M.F.; Faller, R.; Longo, M.L. Role of unsaturated lipid and ergosterol in ethanol tolerance of model yeast biomembranes. Biophys. J. 2012, 102, 507–516. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Du, Z.; Zhu, H.; Guo, X.; He, X. Protective effects of arginine on Saccharomyces cerevisiae against ethanol stress. Sci. Rep. 2016, 6, 31311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourenco, A.B.; Roque, F.C.; Teixeira, M.C.; Ascenso, J.R.; Sá-Correia, I. Quantitative 1H-NMR-metabolomics reveals extensive metabolic reprogramming and the effect of the aquaglyceroporin FPS1 in ethanol-stressed yeast cells. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-K.; Jo, J.-H.; Jin, Y.-S.; Seo, J.-H. Enhanced ethanol fermentation by engineered Saccharomyces cerevisiae strains with high spermidine contents. Bioprocess Biosyst. Eng. 2017, 40, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Morita, Y.; Nakamori, S.; Takagi, H. Effect of proline and arginine metabolism on freezing stress of Saccharomyces cerevisiae. J. Biosci. Bioeng. 2002, 94, 390–394. [Google Scholar] [CrossRef]
- Takagi, H.; Taguchi, J.; Kaino, T. Proline accumulation protects Saccharomyces cerevisiae cells in stationary phase from ethanol stress by reducing reactive oxygen species levels. Yeast 2016, 33, 355–363. [Google Scholar] [CrossRef] [Green Version]
- Walters, D.; Cowley, T. Polyamine metabolism in Saccharomyces cerevisiae exposed to ethanol. Microbiol. Res. 1998, 153, 179–184. [Google Scholar] [CrossRef]
- Kim, S.-K.; Jin, Y.-S.; Choi, I.-G.; Park, Y.-C.; Seo, J.-H. Enhanced tolerance of Saccharomyces cerevisiae to multiple lignocellulose-derived inhibitors through modulation of spermidine contents. Metab. Eng. 2015, 29, 46–55. [Google Scholar] [CrossRef]
- Espindola, A.D.S.; Gomes, D.S.; Panek, A.D.; Eleutherio, E.C.A. The role of glutathione in yeast dehydration tolerance. Cryobiology 2003, 47, 236–241. [Google Scholar] [CrossRef]
- Ayer, A.; Gourlay, C.W.; Dawes, I.W. Cellular redox homeostasis, reactive oxygen species and replicative ageing in Saccharomyces cerevisiae. FEMS Yeast Res. 2013, 14, 60–72. [Google Scholar] [CrossRef] [Green Version]
- Kieliszek, M.; Błażejak, S.; Bzducha-Wróbel, A.; Kot, A.M. Effect of selenium on growth and antioxidative system of yeast cells. Mol. Biol. Rep. 2019, 46, 1797–1808. [Google Scholar] [CrossRef] [Green Version]
- Grant, C.M. Role of the glutathione/glutaredoxin and thioredoxin systems in yeast growth and response to stress conditions. Mol. Microbiol. 2001, 39, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Câmara, A.A.; Nguyen, T.D.; Jossier, A.; Endrizzi, A.; Saurel, R.; Simonin, H.; Husson, F. Improving total glutathione and trehalose contents in Saccharomyces cerevisiae cells to enhance their resistance to fluidized bed drying. Process Biochem. 2018, 69, 45–51. [Google Scholar] [CrossRef]
- Saharan, R.K.; Kanwal, S.; Sharma, S.C. Role of glutathione in ethanol stress tolerance in yeast Pachysolen tannophilus. Biochem. Biophys. Res. Commun. 2010, 397, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Stephen, D.W.; Jamieson, D.J. Glutathione is an important antioxidant molecule in the yeast Saccharomyces cerevisiae. FEMS Microbiol. Lett. 1996, 141, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.-Z.; Wang, X.; Yang, Y.; Yuan, Y.-J. Comparative metabolic profiling of parental and inhibitors-tolerant yeasts during lignocellulosic ethanol fermentation. Metabolomics 2012, 8, 232–243. [Google Scholar] [CrossRef]
- Essendoubi, M.; Toubas, D.; Bouzaggou, M.; Pinon, J.-M.; Manfait, M.; Sockalingum, G.D. Rapid identification of Candida species by FT-IR microspectroscopy. Biochim. Biophys. Acta BBA Gen. Subj. 2005, 1724, 239–247. [Google Scholar] [CrossRef]
- Ramirez-Gaona, M.; Marcu, A.; Pon, A.; Guo, A.C.; Sajed, T.; Wishart, N.A.; Karu, N.; Djoumbou Feunang, Y.; Arndt, D.; Wishart, D.S. YMDB 2.0: A significantly expanded version of the yeast metabolome database. Nucleic Acids Res. 2017, 45, D440–D445. [Google Scholar] [CrossRef]
- Chong, J.; Wishart, D.S.; Xia, J. Using metaboanalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mortality (%) | |||
|---|---|---|---|
| Ethanol% (v/v) | P27 | F2 | F6 |
| 0 | 0 | 0 | 0 |
| 7.5 | 0 | 0 | 0 |
| 15 | 0 | 0 | 0 |
| 20 | 0 | 0 | 0 |
| 21 | 13 | 22 | 18 |
| 22 | 33 | 36 | 34 |
| 23 | 49 | 54 | 50 |
| 24 | 78 | 82 | 72 |
| 25 | 100 | 100 | 100 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corte, L.; Roscini, L.; Pierantoni, D.C.; Pellegrino, R.M.; Emiliani, C.; Basaglia, M.; Favaro, L.; Casella, S.; Cardinali, G. Delta-Integration of Single Gene Shapes the Whole Metabolomic Short-Term Response to Ethanol of Recombinant Saccharomyces cerevisiae Strains. Metabolites 2020, 10, 140. https://doi.org/10.3390/metabo10040140
Corte L, Roscini L, Pierantoni DC, Pellegrino RM, Emiliani C, Basaglia M, Favaro L, Casella S, Cardinali G. Delta-Integration of Single Gene Shapes the Whole Metabolomic Short-Term Response to Ethanol of Recombinant Saccharomyces cerevisiae Strains. Metabolites. 2020; 10(4):140. https://doi.org/10.3390/metabo10040140
Chicago/Turabian StyleCorte, Laura, Luca Roscini, Debora Casagrande Pierantoni, Roberto Maria Pellegrino, Carla Emiliani, Marina Basaglia, Lorenzo Favaro, Sergio Casella, and Gianluigi Cardinali. 2020. "Delta-Integration of Single Gene Shapes the Whole Metabolomic Short-Term Response to Ethanol of Recombinant Saccharomyces cerevisiae Strains" Metabolites 10, no. 4: 140. https://doi.org/10.3390/metabo10040140