
Analysis of the Gas Phase Acidity of Substituted Benzoic Acids Using Density Functional Concepts

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
3. Methodology and Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hammett, L.P. Physical Organic Chemistry, 2nd ed.; McGraw-Hill: New York, NY, USA, 1970; pp. 146–177. ISBN 978-0070259058. [Google Scholar]
- Exner, O.; Böhm, S. Background of the Hammett Equation As Observed for Isolated Molecules: Meta- and Para-Substituted Benzoic Acids. J. Org. Chem. 2002, 67, 6320–6327. [Google Scholar] [CrossRef]
- Böhm, S.; Fiedller, P.; Exner, O. Analysis of the ortho effect: Acidity of 2-substituted benzoic acids. New J. Chem. 2004, 28, 67–74. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Zaitsau, D.H.; Emel’yanenko, V.N.; Stepurko, E.N.; Zherikova, K.V. Benzoic acid derivatives: Evaluation of thermochemical properties with complementary experimental and computational method. Thermochim. Acta 2015, 622, 18–30. [Google Scholar] [CrossRef]
- Taft, R.W.; Topsom, R.D. The Nature and Analysis of substitutent electronic effects Prog. Phys. Org. Chem. 1987, 16, 1–83. [Google Scholar] [CrossRef]
- Taft, R.W.; Koppel, I.A.; Topsom, R.D.; Anvia, F. Acidities of OH compounds, including alcohols, phenol, carboxylic acids, and mineral acids. J. Am. Chem. Soc. 1990, 112, 2047–2052. [Google Scholar] [CrossRef]
- Abboud, J.-L.M.; Catalán, J.; Elguero, J.; Taft, W. Polarizability effects on the aqueous solution basicity of substituted pyridines. J. Org. Chem. 1987, 53, 1137–1140. [Google Scholar] [CrossRef]
- Roithova, J.; Exner, O. Protonation of alkylpyridines: Polarizability and steric effects in the base and in the cation. J. Phys. Org. Chem. 2001, 14, 752–758. [Google Scholar] [CrossRef]
- Kulhánek, J.; Decouzon, M.; Gal, J.-F.; Maria, P.-C.; Fiedler, P.; Jiménez, P.; Roux, M.-V.; Exner, O. Steric effect and steric hindrance to resonance in tert-butylbenzoic acids in the gas phase and the solution. Eur. J. Org. Chem. 1999, 1999, 1589–1594. [Google Scholar] [CrossRef]
- De Proft, F.; Langenaeker, W.; Geerlings, P. Ab initio determination of substituent constants in a Density Functional Theory formalism: Calculation of intrinsic group electronegativity, hardness and softness. J. Phys. Chem. 1993, 97, 1826–1831. [Google Scholar] [CrossRef]
- De Proft, F.; Amira, S.; Choho, K.; Geerlings, P. Quantum Chemical Study of the acidity of substituted acetic acids with density functional theory based descriptors. J. Phys. Chem. 1994, 98, 5227–5233. [Google Scholar] [CrossRef]
- Gupta, K.; Giria, S.; Chattaraj, P.K. Acidity of meta- and para-substituted aromatic acids: A conceptual DFT study. New, J. Chem. 2008, 32, 1945–1952. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, L.; Liu, W.; Liu, S.; Liu, S. Modeling Molecular Acidity with Electronic Properties and Hammett Constants for Substituted Benzoic Acids. J. Phys. Chem. A 2011, 115, 14697–14707. [Google Scholar] [CrossRef] [PubMed]
- Vianello, R.; Maksic, Z.B. Gas-phase acidity of para-substituted benzoic acids—A triadic analysis of substituent effects. J. Phys. Org. Chem. 2005, 18, 699–705. [Google Scholar] [CrossRef]
- Hollingsworth, C.A.; Seybold, P.G.; Hadad, C.M. Substituent Effects on the Electronic Structure and pKa of Benzoic Acid. Int. J. Quantum Chem. 2002, 90, 1396–1403. [Google Scholar] [CrossRef]
- Romero María de, L.; Méndez, F. Is the Hydrogen Atomic Charge Representative of the Acidity of Parasubstituted Phenols? J. Phys. Chem. A 2003, 107, 4526–4530. [Google Scholar] [CrossRef]
- Méndez, F.; Romero, M.L.; De Proft, F.; Geerlings, P. The basicity of p-substituted phenolates and the elimination-substitution ratio in p-nitrophenethyl bromide: A HSAB theoretical study. J. Org. Chem. 1998, 63, 5774–5778. [Google Scholar] [CrossRef]
- Romero, M.L.; Méndez, F. The local HSAB principle and the bond dissociation energy of p-substituted phenols. J. Phys. Chem. A 2003, 107, 5874–5875. [Google Scholar] [CrossRef]
- Ramírez, E.R.; García-Martínez, C.; Méndez, F. Influence of fluorine atoms and aromatic rings on the acidity of ethanol. J. Phys. Chem. A 2009, 113, 10753–10758. [Google Scholar] [CrossRef]
- Ramírez, E.R.; García-Martínez, C.; Méndez, F. Understanding the Nucleophilic Character and Stability of the Carbanions and Alkoxides of 1-(9-Anthryl)ethanol and Derivatives. Molecules 2013, 18, 10254–10265. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989; pp. 47–67, 978-0195092769. [Google Scholar]
- Pearson, R.G. Hard and soft acids and bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Méndez, F.; Gázquez, J.L. Chemical reactivity of enolate ions: The local hard and soft acids and bases principle viewpoint. J. Am. Chem. Soc. 1994, 116, 9298–9301. [Google Scholar] [CrossRef]
- Vela, A.; Gázquez, J.L. A relationship between the static dipole polarizability, the global softness, and the fukui function. J. Am. Chem. Soc. 1990, 12, 1490–1492. [Google Scholar] [CrossRef]
- Roy, R.; Chandra, A.K.; Pal, S. Correlation of Polarizability, Hardness, and Electronegativity: Polyatomic Molecules. J. Phys. Chem. 1994, 98, 10447–10450. [Google Scholar] [CrossRef]
- Boisdenghien, Z.; Fias, S.; Da Pieve, F.; De Proft, F.; Geerlings, P. The polarizability of atoms and molecules: A comparison between a conceptual density functional theory approach and time-dependent density functional theory. Mol. Phys. 2015, 113, 1890–1898. [Google Scholar] [CrossRef]
- Komorowski, L.; Lipinski, J.; Szarek, P.; Ordon, P. Polarization justified Fukui functions: The theory and applications for molecules. J. Chem. Phys. 2011, 135, 014109. [Google Scholar] [CrossRef]
- Krishtal, A.; Senet, P.; Van Alsenoy, C. Local softness, softness dipole and polarizabilities of functional groups: Application to the side chains of the twenty amino acids. J. Chem. Phys. 2009, 131, 044312. [Google Scholar] [CrossRef] [Green Version]
- López, P.; Méndez, F. Fukui function as a descriptor of the imidazolium protonated cation resonance hybrid structure. Org. Lett. 2004, 6, 1781–1783. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- NIST Chemistry Web Book. Available online: http://webbook.nist.gov/chemistry/ (accessed on 12 May 2019).
- Yang, W.; Mortier, W.J. The use of global and local molecular parameters for the analysis of the gas phase basicity of amines. J. Am. Chem. Soc. 1986, 108, 5708–5711. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theoret. Chim. Acta (Berl.) 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Levine, I. Quantum Chemistry, 5th ed.; Prentice Hall: Upper Saddle River, NJ, USA, 2001; pp. 154–196, 414–415, 0136855121. [Google Scholar]
- Laaksonen, L. gOpenMol, Center for Scientific Computing: Espoo, Finland.
- Domingo, L.R.; Pérez, P.; Sáezc, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr function. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Georgousaki, K.; Tsafantakis, N.; Gumeni, S.; Lambrinidis, G.; González-Menéndez, V.; Tormo, J.R.; Genilloud, O.; Trougakos, I.P.; Fokialakis, N. Biological Evaluation and In Silico Study of Benzoic Acid Derivatives from Bjerkandera adusta Targeting Proteostasis Network Modules. Molecules 2020, 25, 666. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
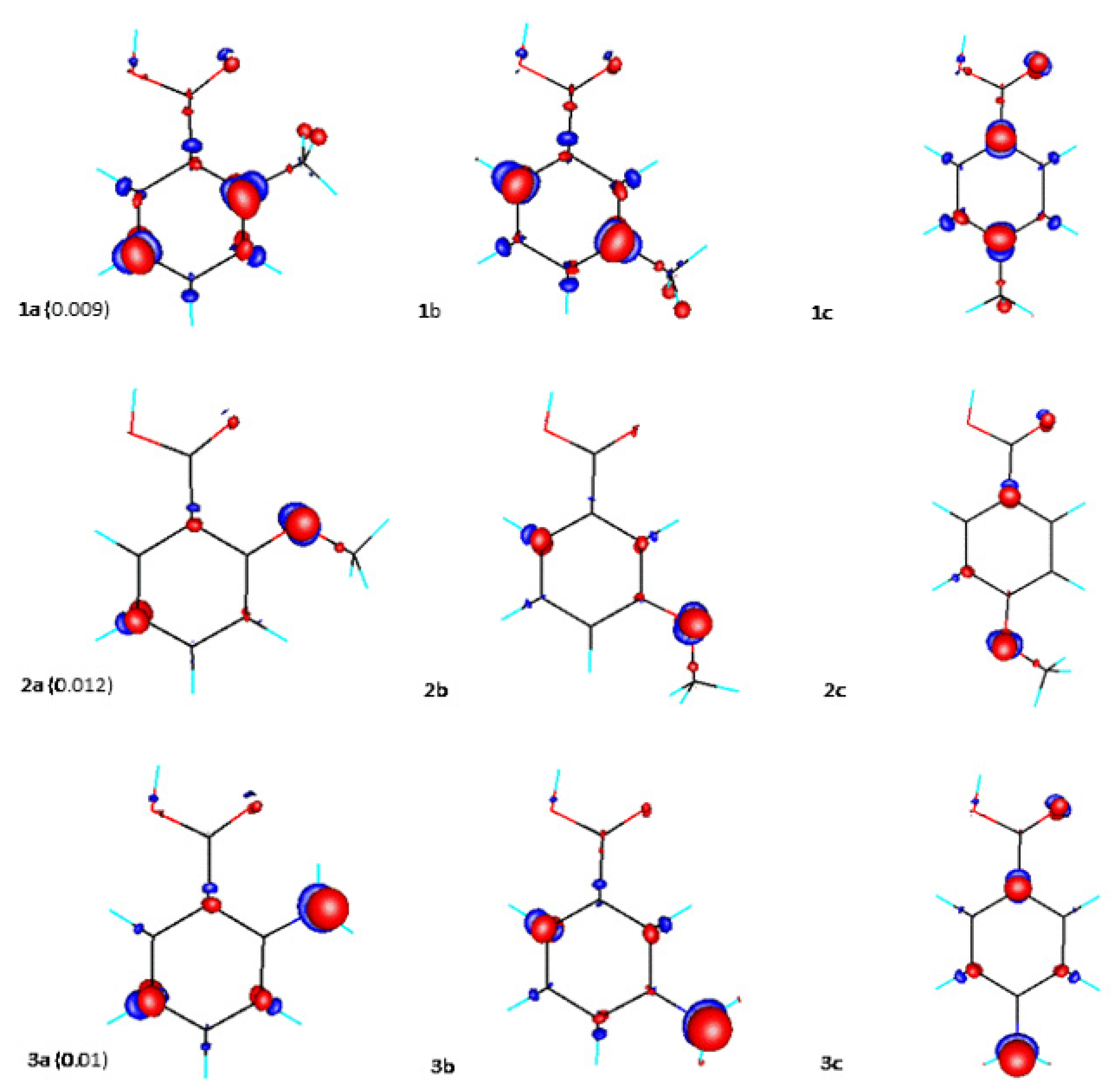{kind=link}
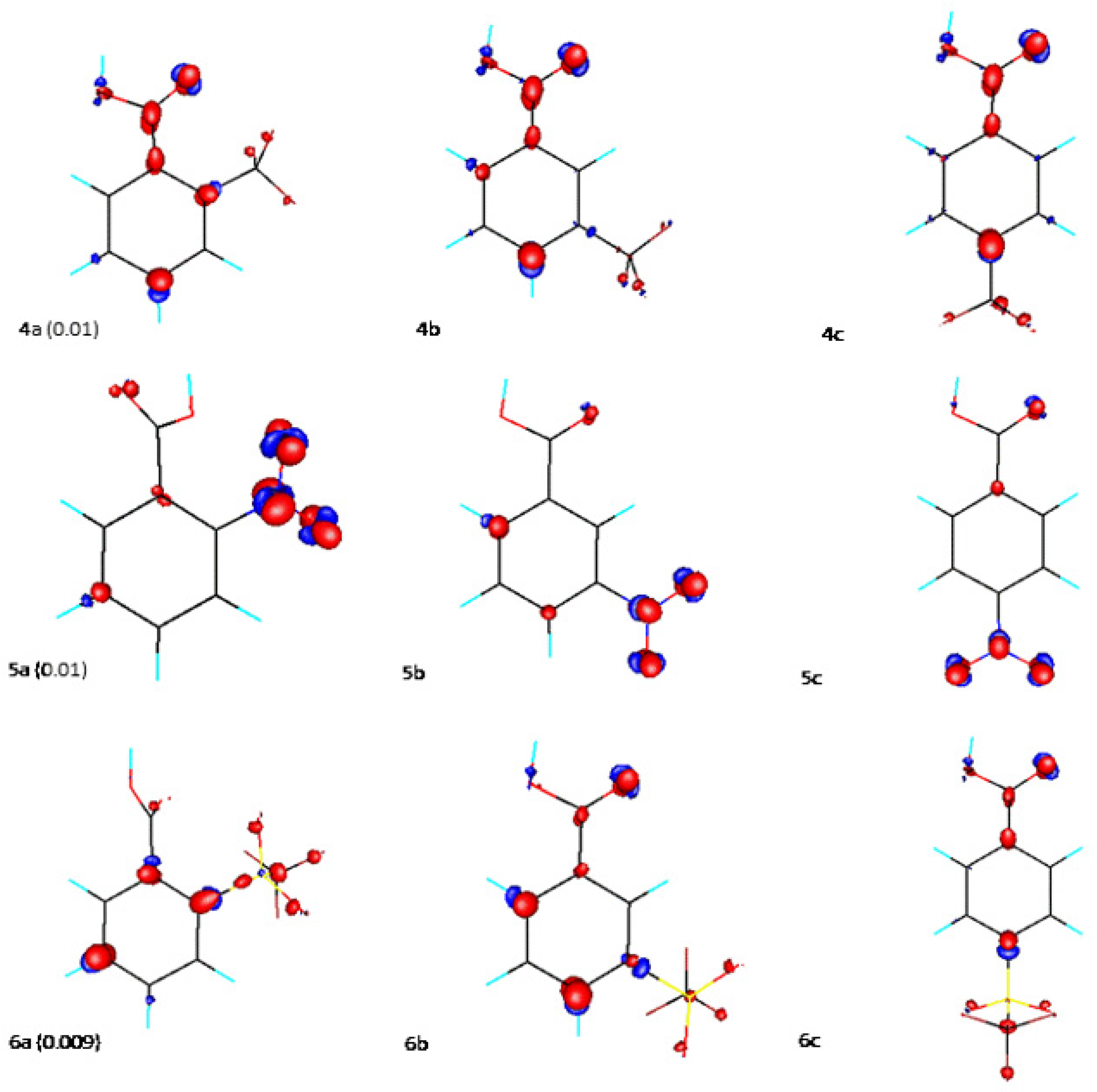{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| a |
331.09 (332.46 ± 2.01) |
330.21 (332.46 ± 2.01) |
331.03 (330.31 ± 2.01) |
325.99 (-------) |
321.86 (324.33 ± 2.01) |
319.06 (-----) |
| b |
332.76 (333.65 ± 2.01) |
332.47 (332.46 ± 2.01) |
333.62 (334.61 ± 2.01) |
324.34 (325.29 ± 2.01) |
320.51 (322.18 ± 2.01) |
318.19 (-----) |
| c |
333.35 (333.89 ± 2.01) |
333.97 (333.89 ± 2.01) |
336.24 (336.28 ± 2.01) |
323.61 (325.29 ± 2.01) |
319.13 (320.98 ± 2.01) |
316.87 (-----) |
| Fragment | Position | R2 |
|---|---|---|
| Z | o | 0.1974 |
| m | 0.0004 | |
| p | 0.0493 | |
| C6H4 | o | 0.5038 |
| m | 0.855 | |
| p | 0.8021 | |
| ZC6H4 | o | 0.0011 |
| m | 0.4267 | |
| p | 0.227 | |
| ZC6H4COO | o | 0.0089 |
| m | 0.31 | |
| p | 0.0026 | |
| ZC6H4COOH | o | 0.0034 |
| m | 0.2933 | |
| p | 0.0008 | |
| qH | o | 0.9239 |
| m | 0.9938 | |
| p | 0.9903 |
| Fragment | Position | R2 |
|---|---|---|
| Z | o | 0.7333 |
| m | 0.5409 | |
| p | 0.0125 | |
| C6H4 | o | 0.4642 |
| m | 0.6352 | |
| p | 0.6654 | |
| ZC6H4 | o | 0.9415 |
| m | 0.8498 | |
| p | 0.8699 | |
| ZC6H4COO | o | 0.8422 |
| m | 0.920 | |
| p | 0.9746 | |
| ZC6H4COOH | o | 0.8266 |
| m | 0.9206 | |
| p | 0.9711 |
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| a | 18.79% (81.21%) | 31.26% (68.74%) | 43.31% (56.69%) | 19.78% (80.22%) | 22.69% (77.31%) | 18.20% (81.80%) |
| b | 15.51% (84.49%) | 25.80% (74.20%) | 35.76% (64.24%) | 16.33% (83.67%) | 18.74% (81.26%) | 15.03% (84.97%) |
| c | 13.71% (86.29%) | 22.82% (77.18%) | 31.61% (68.39%) | 14.44% (85.56%) | 16.57% (83.43%) | 13.29% (86.71%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amador-Balderas, J.A.; Martínez-Sánchez, M.-A.; Ramírez, R.E.; Méndez, F.; Meléndez, F.J. Analysis of the Gas Phase Acidity of Substituted Benzoic Acids Using Density Functional Concepts. Molecules 2020, 25, 1631. https://doi.org/10.3390/molecules25071631
Amador-Balderas JA, Martínez-Sánchez M-A, Ramírez RE, Méndez F, Meléndez FJ. Analysis of the Gas Phase Acidity of Substituted Benzoic Acids Using Density Functional Concepts. Molecules. 2020; 25(7):1631. https://doi.org/10.3390/molecules25071631
Chicago/Turabian StyleAmador-Balderas, Jorge A., Michael-Adán Martínez-Sánchez, Ramsés E. Ramírez, Francisco Méndez, and Francisco J. Meléndez. 2020. "Analysis of the Gas Phase Acidity of Substituted Benzoic Acids Using Density Functional Concepts" Molecules 25, no. 7: 1631. https://doi.org/10.3390/molecules25071631