Iron-Catalyzed Synthesis, Structure, and Photophysical Properties of Tetraarylnaphthidines




1
Faculty of Chemistry, Technische Universität Dresden, Bergstraße 66, 01069 Dresden, Germany
2
A. E. Arbuzov Institute of Organic and Physical Chemistry, FRC Kazan Scientific Center, Russian Academy of Sciences, Arbuzov Str. 8, 420088 Kazan, Russia
*
Author to whom correspondence should be addressed.
Molecules 2020, 25(7), 1608; https://doi.org/10.3390/molecules25071608
Submission received: 22 February 2020
/
Revised: 26 March 2020
/
Accepted: 29 March 2020
/
Published: 1 April 2020
(This article belongs to the Special Issue Recent Advances in Iron Catalysis)
Abstract
:We describe the synthesis and photophysical properties of tetraarylnaphthidines. Our synthetic approach is based on an iron-catalyzed oxidative C–C coupling reaction as the key step using a hexadecafluorinated iron–phthalocyanine complex as a catalyst and air as the sole oxidant. The N,N,N’,N’-tetraarylnaphthidines proved to be highly fluorescent with quantum yields of up to 68%.

1. Introduction
Triarylamines have been the subject of numerous studies due to their electron-rich character and the resulting low oxidation potentials [1,2,3]. These unique properties of triarylamine derivatives have induced extensive investigations towards their potential applications in organic films for optoelectronics [4] and as hole-transport materials in OLEDs (organic light emitting diodes) and solar cells [5,6,7,8]. Naphthidines (1,1′-binaphthalene-4,4′-diamines) have also been studied because of their ready oxidation to radical cations [9,10,11,12]. Starting from the corresponding 1-naphthylamines by an oxidative coupling, the synthesis of naphthidines can be induced electrochemically [9,10,13], by stoichiometric amounts of titanium tetrachloride [11], stoichiometric amounts of iron(III) chloride [14], chloranil [15], or (bis(trifluoroacetoxy)iodo)benzene (PIFA) [16]. It has been well-known for a long time that naphthidines can be separated into their stable atropisomers [17].
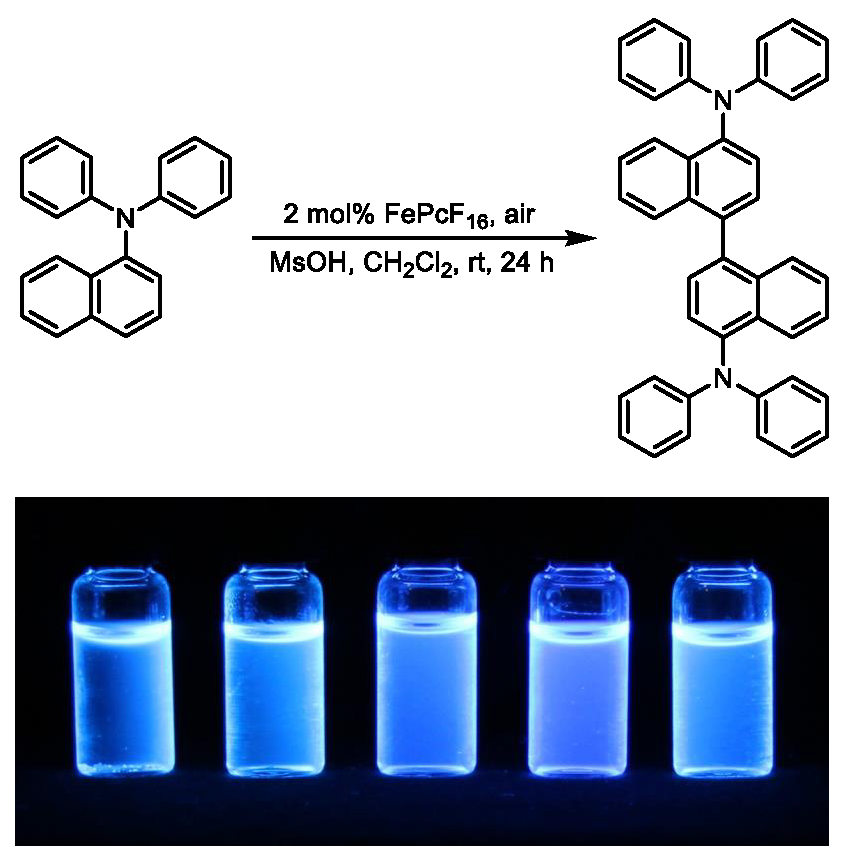
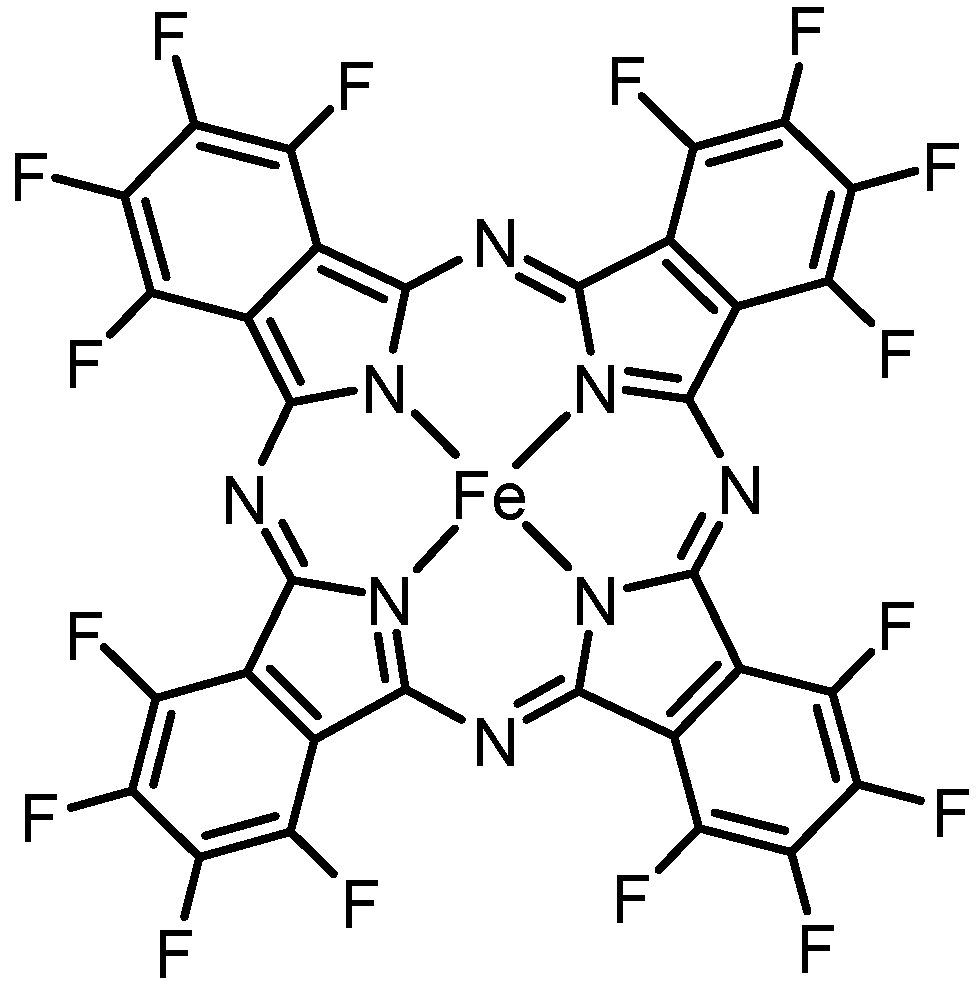
Herein, we report a simple synthesis of N,N,N’,N’-tetraarylnaphthidines by an iron-catalyzed oxidative coupling of the corresponding 1-(diphenylamino)naphthalene precursor using iron(II)–hexadecafluorophthalocyanine (FePcF16) [18] as the catalyst and air as the final oxidant (Figure 1). Moreover, we have investigated the photophysical properties of the obtained naphthidine derivatives.
2. Results and Discussion
2.1. Synthesis
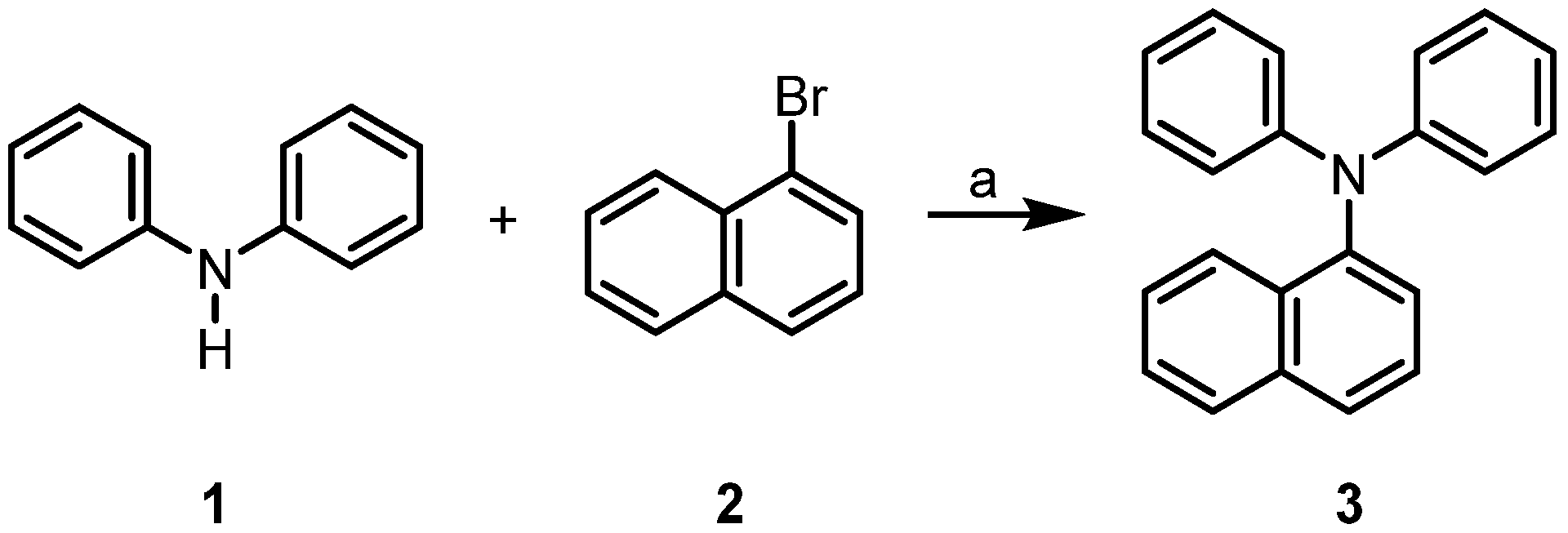
Following a procedure reported by Nechaev et al. for the Buchwald–Hartwig reaction under solvent-free conditions [19], we have achieved the coupling of diphenylamine (1) and 1-bromonaphthalene (2) to afford 1-(diphenylamino)naphthalene (3) in high yield (Scheme 1).
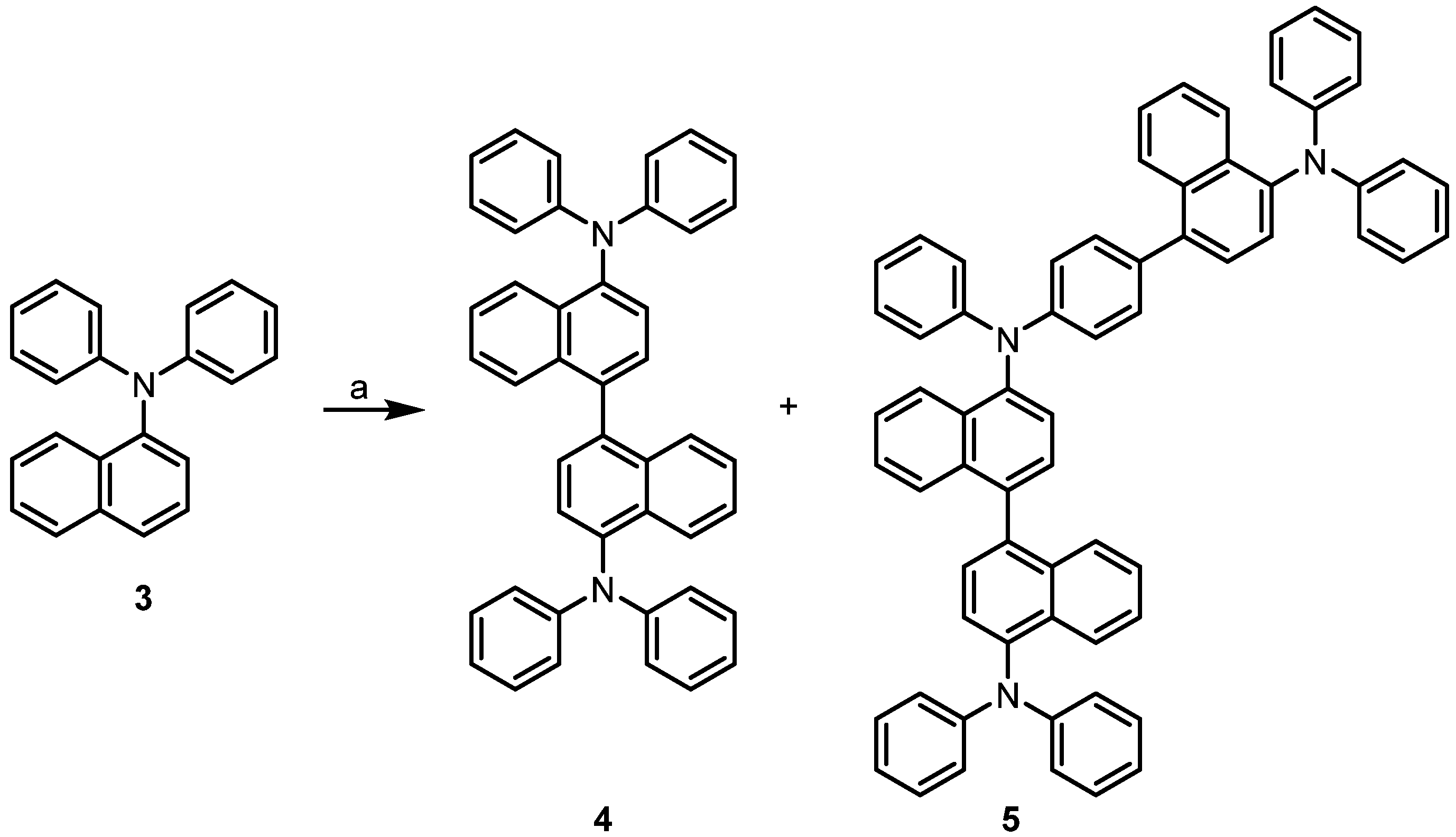
Then, we turned our attention to the projected iron-catalyzed oxidative homocoupling of 1-(diphenylamino)naphthalene (3). Recently, we applied our iron-catalyzed oxidative coupling methodology to the C–C homocoupling of diarylamines, 1-, and 2-hydroxycarbazoles, as well as to the cross coupling of tertiary anilines with hydroxyarenes [20,21,22]. Iron as a first-row transition metal is environmentally safe and has become a powerful tool in synthetic organic chemistry with many applications for selective C–H bond activation [23,24,25,26]. According to a previous report by Yang, N,N,N’,N’-tetraphenylnaphthidine (4) could not be prepared by oxidative coupling of compound 3 with stoichiometric amounts of iron(III) chloride [14]. The oxidation of compound 3 with chloranil provides compound 4 only as the minor isomer in an inseparable mixture with the corresponding benzidine derivative [15]. Based on our previous studies [20,21,22], we envisaged to develop an iron-catalyzed homocoupling of the triarylamine 3 with air as the sole oxidant. Using catalytic amounts (2 mol%) of FePcF16 and substoichiometric amounts (40 mol%) of methanesulfonic acid as an additive at room temperature provided N,N,N’,N’-tetraphenylnaphthidine (4) in 48% yield along with the N,N,N’,N’-tetraarylnaphthidine 5 as a by-product in 11% yield (Scheme 2). Obviously, compound 5 results from a further oxidative C–C coupling at the p-position of one of the phenyl rings of the initial coupling product compound 4. The structural assignments for compounds 4 and 5 were based on their 1H-NMR and 13C-NMR spectroscopic data and an X-ray analysis of compound 4 (see Section 2.2).
2.2. Structure
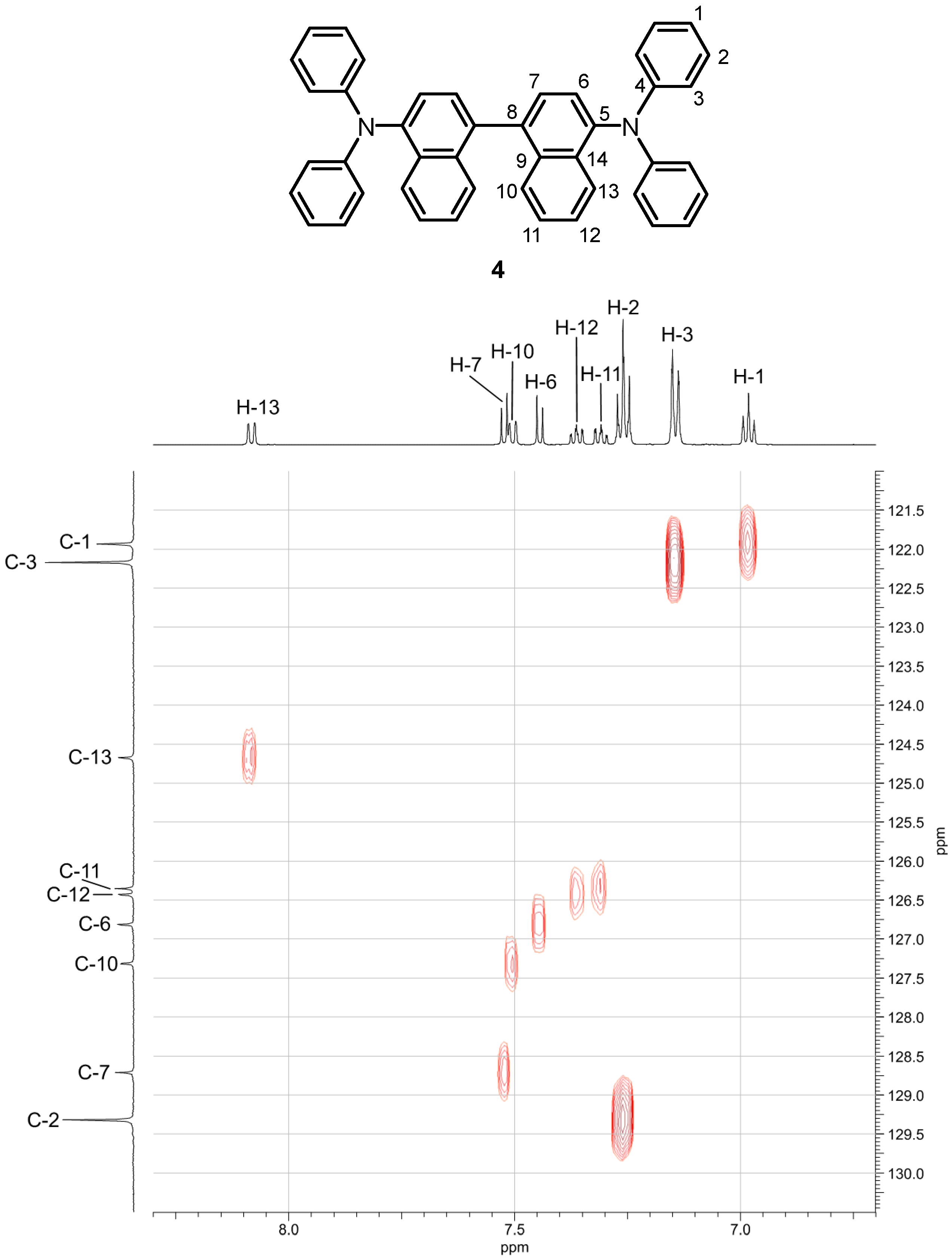
The 1H-NMR, 13C-NMR, and DEPT spectra of compound 4 displayed signals for a highly symmetrical compound. Signals for nine aromatic CH and five quaternary aromatic carbon atoms were identified. The COSY experiment revealed the presence of three spin systems (Supplementary Materials, COSY of compound 4). The first is caused by coupling of H-1 with H-2 and H-3, which belong to the phenyl fragment. The second spin system consists of H-6 and H-7. The third system is formed by H-10, H-11, H-12, and H-13. The assignment of all 13C-NMR signals to the respective 1H-NMR signals could be achieved by an HSQC measurement (Figure 2 and Table S1). The constitution of naphthidine 4 was confirmed by analysis of the HMBC spectrum (Supplementary Materials, HMBC of compound 4, Table S1). Characteristic HMBC signals (C-4/H-2 and C-4/H-3) led to elucidation of the quaternary carbon atom C-4 by connecting the proton spin systems. The position of C-5 was established based on the HMBC cross-peaks with H-6, H-7, and H-13. Accordingly, the quaternary aromatic carbon atom C-8 was assigned based on the HMBC interactions with H-6, H-7, and H-10. HMBC cross-peaks between C-9/H-7, H-11, H-13 and C-14/H-6, H-10, H-12 unambiguously clarified the location of both naphthyl-based quaternary carbon atoms. The NOE interactions were exploited to confirm the positions of H-3 and H-13 (Supplementary Materials, NOESY of compound 4). Due to the interactions of these fjord region protons, H-13 showed a substantial downfield shift to 8.08 ppm. Moreover, H-2, H-3, and H-6 exhibited an interaction with the 15N atom (Supplementary Materials, 1H/15N HMBC of compound 4).
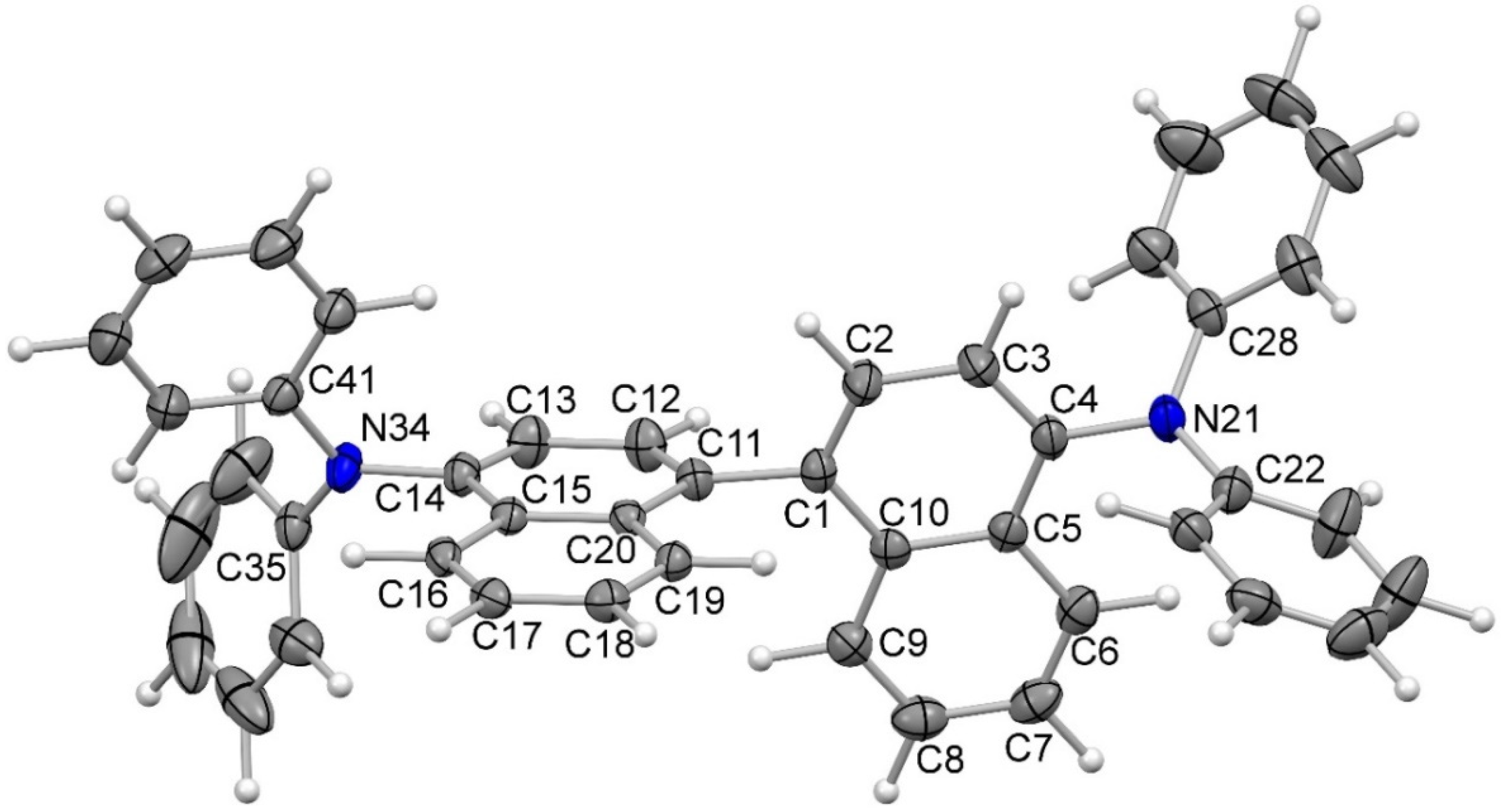
The structural assignment for N,N,N’,N’-tetraphenylnaphthidine (4) was confirmed by an X-ray crystal structure determination (Figure 3). The molecule of compound 4 adopts a trans conformation around the central biaryl bond (C1–C11) with a torsional angle of 99.4(2)° (Figure 4) according to the notation accepted for binaphthyl derivatives [27,28]. The geometry of the naphtho fragments exhibiting large variations of the bond lengths is in agreement with the crystal structure of naphthalene [29,30] and its structure obtained by quantum chemical calculations [31]. The orientation of the phenyl groups attached to the nitrogen atoms at the two sides of the molecule is different. The two phenyl rings at N21 are symmetrically oriented respective to the bisector plane drawn through the C4–N21 bond, while the two phenyl groups at N34 have different orientations. In the crystal, the molecules of compound 4 participate in multiple weak intermolecular C–H···π interactions between the naphtho groups (Figure 5). The distances between the hydrogen atoms and the sp2-carbon atoms range from 2.7 to 2.8 Å and the angles are typical for these weak hydrogen bonds [32]. This sort of packing with face-to-edge interactions has been observed also in naphthalene itself [29,30] and in binaphthyl derivatives [27,28]. It is noteworthy that the naphtho groups interact mostly with each other and the phenyl groups interact predominantly with phenyl groups, whereas naphtho–phenyl contacts are rare.
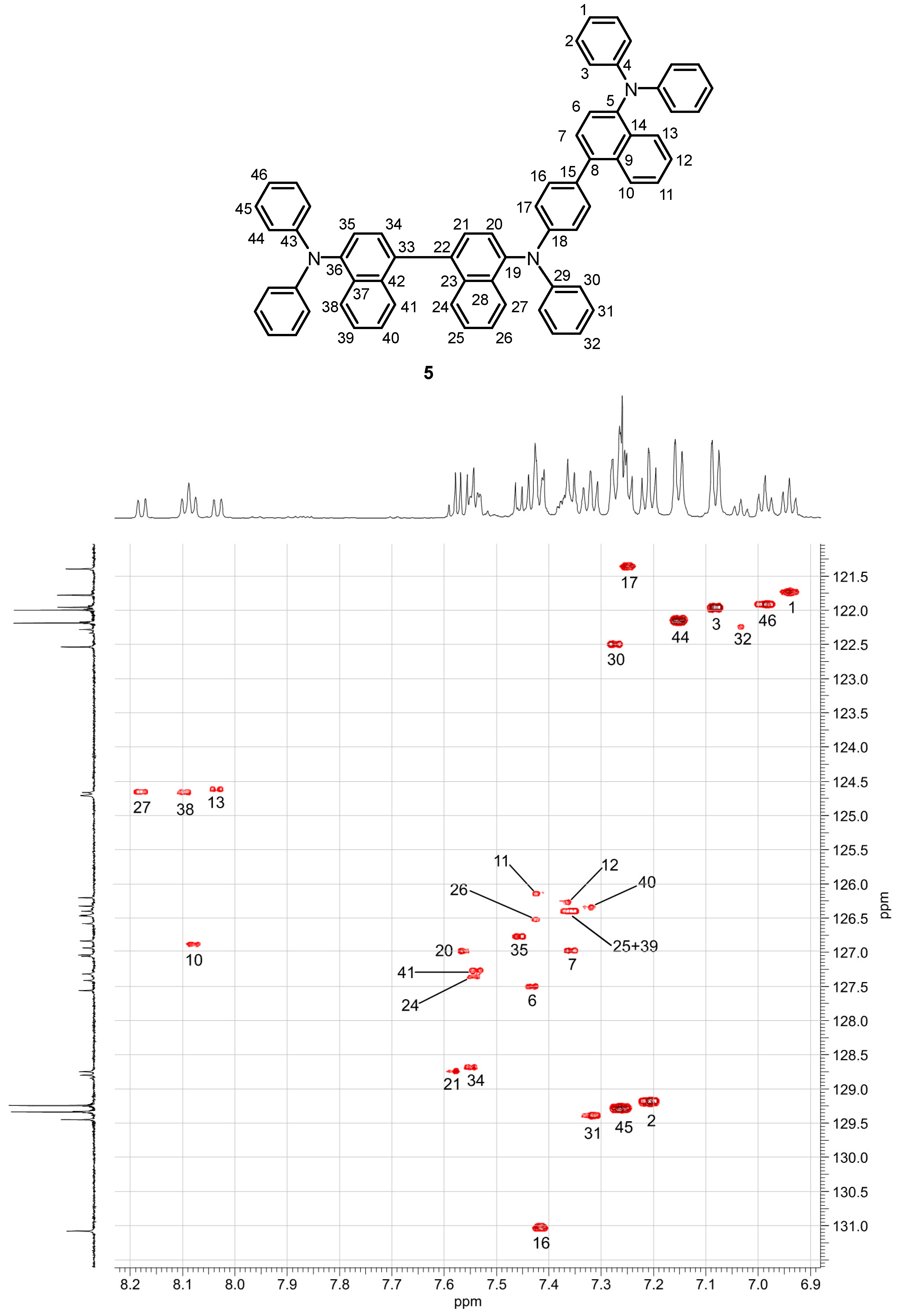
The connectivity in compound 5 has been secured by a full set of 2D-NMR spectra (see below and the copies of the 2D-NMR spectra in the Supplementary Materials). The NMR spectra of 5 display signals for a non-symmetrical aromatic compound. The JRES experiment revealed the absence of a singlet. Thus, the additional oxidative coupling of the naphthidine 4 with compound 3 did not occur at positions 2, 6, 7, 11, or 12 (Supplementary Materials, JRES of compound 5). The 1H-NMR spectrum in combination with the HSQC (Figure 6) and the JRES spectra displayed signals for four substantially downfield-shifted doublets. On the basis of the fjord region effect observed for compound 4, these four downfield-shifted signals in the 1H-NMR spectrum of compound 5 are the naphthyl protons H-10, H-13, H-27, and H-38. The signals of the four corresponding carbon atoms are detected at δC = 124.66 (C-13), 124.70 (C-27), 124.71 (C-38), and 126.93 (C-10) ppm in the 13C-NMR spectrum (HSQC in Figure 6, Table S2). The NOE interactions indicated the positions of the protons at the phenyl groups (Supplementary Materials, NOESY of compound 5). The positions of the protons at C-3, C-30, and C-44 have been identified unambiguously by NOE interactions with H-13, H-27, and H-38. Support for this assignment of the protons H-3, H-30, and H-44 is obtained from an HMBC interaction with 15N (Supplementary Materials, 1H/15N HMBC of compound 5). A strong NOE correlation of H-10 with H-16 and the presence of a spin interaction between H-16 and H-17 in the COSY (Supplementary Materials, COSY of compound 5) confirmed an oxidative coupling of compound 3 at C-1 of naphthidine 4. Further support was obtained from HMBC correlations of H-16 with the quaternary carbon atoms C-8 at δC = 138.64 ppm and C-18 at δC = 147.93 ppm and the HMBC correlation of H-17 with C-15 at δC = 133.51 ppm (Supplementary Materials, HMBC of compound 5). The obtained NMR data are in excellent agreement with the structure assigned for compound 5.
2.3. Photophysical Studies
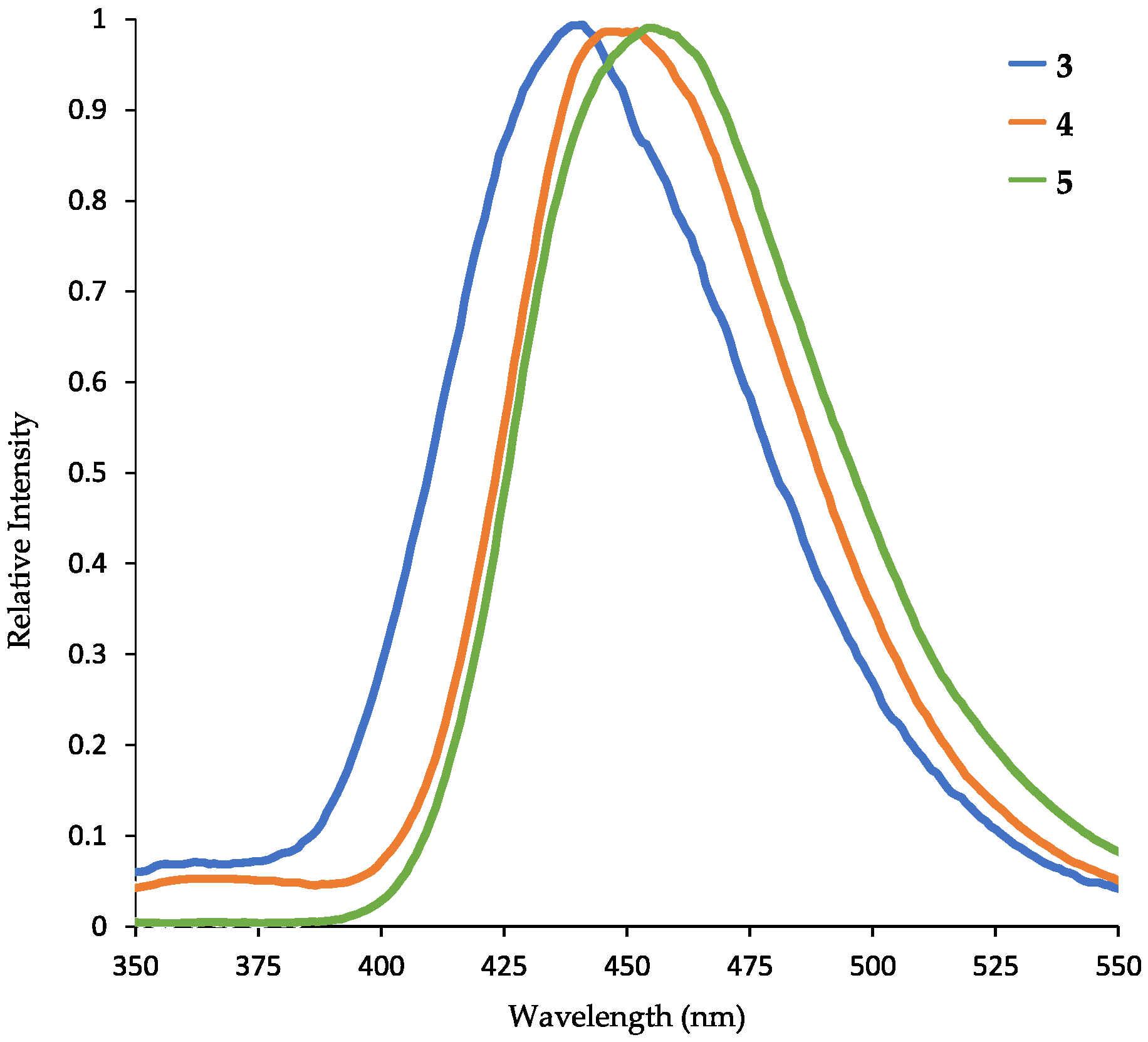
On irradiation with UV-light (λex = 254 nm) in methanolic solution, 1-(diphenylamino)naphthalene (3) and the resulting N,N,N’,N’-tetraarylnaphthidines 4 and 5 show a strong blue fluorescence (Figure 7). Therefore, we investigated the photophysical properties of compounds 3, 4, and 5 in more detail. The UV absorption and fluorescence emission data for all three compounds are listed in Table 1 and the normalized fluorescence emission spectra are displayed in Figure 8. The fluorophores 3, 4, and 5 exhibit large Stokes shifts of 149 to 162 nm. While the UV absorption maxima are identical, the fluorescence emission maxima show a bathochromic shift of only 7 nm for N,N,N’,N’-tetraphenylnaphthidine (4) and 14 nm for compound 5, as compared to 1-(diphenylamino)naphthalene (3). Previously, even larger Stokes shifts have been observed for N,N-dimethylaminonaphthalenes [33] and strong bathochromic shifts have been reported for substituted triarylamines [34].
For the naphthidines 4 and 5, the UV absorption and fluorescence emission maxima along with fluorescence quantum yields, fluorescence lifetimes, and CIE-coordinates were determined (Table 2).
The UV absorption maximum for the naphthidines 4 and 5 is at the same wavelength (294 nm), whereas the fluorescence emission maximum of compound 5 shows a bathochromic shift of 4 nm as compared with compound 4. The fluorescence quantum yields of compounds 4 and 5 are relatively high and in the same range (68% and 65%, respectively, by excitation at 310 nm). Compound 5 exhibits a shorter fluorescence lifetime of 3.6 ns as compared with 4.2 ns for N,N,N’,N’-tetraphenylnaphthidine (4). The CIE-coordinates for the blue light emissions of compounds 4 and 5 are indicative of a blue light which could be useful for applications in OLEDs [35,36,37,38,39]. The fluorescence quantum yields are very comparable with compounds already investigated as blue fluorescent OLEDs [37,38,39]. It has been demonstrated for other naphthalene fluorophores that a bathochromic shift of the emission can be achieved by increasing the size of the π-system and by the introduction of appropriate substituents [40,41]. Thus, application of these tools should easily allow an optimization of the present fluorophoric system.
In addition, we investigated the absorption and fluorescence properties of N,N,N’,N’-tetraphenylnaphthidine (4) in various nonpolar and polar solvents. The fluorescence behavior of compound 4 on excitation at 254 nm was demonstrated in isohexane, dichloromethane, ethyl acetate, THF, and methanol (Figure 9). The corresponding UV absorption and fluorescence emission spectra are shown in the Supplementary Materials. The corresponding photophysical data of compound 4 are summarized in Table 3. For a series of N,N-dimethylaminonaphthalene fluorophores, Brummond et al. observed a significant red shift of the fluorescence emission maxima by increasing the solvent polarity [33], whereas in other systems the solvent dependency of the emission was significantly lower [37]. For N,N,N’,N’-tetraphenylnaphthidine (4), this solvatochromic shift is much less pronounced (about 21 nm in dichloromethane as compared with the corresponding value in isohexane) (Table 3).
3. Materials and Methods
3.1. General
All reactions were performed in oven-dried glassware using anhydrous solvents under argon, unless stated otherwise. Pd(OAc)2 was recrystallized from glacial AcOH. All other chemicals were used as received from commercial sources. The iron(III)-catalyzed reactions were carried out in non-dried solvents under air. Iron(II)–hexadecafluorophthalocyanine was prepared following a procedure reported previously [18]. Flash chromatography was performed using silica gel from Acros Organics (0.035 to 0.070 mm). Automated flash chromatography was performed on a Büchi Sepacore system equipped with an UV monitor on silica gel (Acros Organics, 0.035 to 0.070 mm). The TLC was performed with TLC plates from Merck (60 F254) using UV light for visualization. The melting points were measured on a Gallenkamp MPD 350 melting point apparatus. Ultraviolet spectra were recorded on a PerkinElmer 25 UV/Vis spectrometer. The fluorescence spectra were measured on a Varian Cary Eclipse spectrophotometer. The IR spectra were recorded on a Thermo Nicolet Avatar 360 FT-IR spectrometer using the ATR method (attenuated total reflectance). The NMR spectra were recorded on Bruker DRX 500 and Avance III 600 spectrometers. The chemical shifts δ were reported in ppm with the solvent signal as internal standard. Standard abbreviations were used to denote the multiplicities of the signals. EI mass spectra were recorded by GC/MS-coupling using an Agilent Technologies 6890 N GC System equipped with a 5973 Mass Selective Detector (70 eV). The ESI-MS spectra were recorded on an Esquire LC using an ion trap detector from Bruker. Positive and negative ions were detected. Elemental analyses were measured on a EuroVector EuroEA3000 elemental analyzer. The X-ray crystal structure analyses were performed with a Bruker–Nonius Kappa CCD and with a Bruker AXS Smart APEX diffractometer equipped with a 700 series Cryostream low temperature device from Oxford Cryosystems. SHELXS-97 [42], SADABS version 2.10 [43], SHELXL-97 [44], POV-Ray for Windows version 3.7.0.msvc10.win64, and ORTEP-3 for Windows [45] were used as software.
3.2. Photoluminescence Measurements
The 2 wt% emitter films were prepared by doctor blading a solution of an emitter in a 10 wt% poly(methyl methacrylate) solution in dichloromethane on a quartz substrate with a 60 μm doctor blade. The film was dried, and the emission measured under nitrogen atmosphere. Excitation was conducted in a wavelength range of 250–400 nm (Xe lamp with monochromator), and the emission was detected with a calibrated quantum yield detection system (Hamamatsu, model C11347-01). The fluorescence lifetime was measured with an Edinburgh Instruments mini-τ by excitation with pulses of an EPLED (Edinburgh picosecond Pulsed Light Emitting Diode) (360 nm, 20 kHz) and time-resolved photon counting (TCSPC).
3.3. Procedures
3.3.1. N,N-Diphenylnaphthalen-1-amine (1-(Diphenylamino)naphthalene) (3)
Compound 3 was prepared following a literature procedure [19]. To a screw-cap vial were added 1-bromonaphthalene (2) (307 µL, 455 mg, 2.20 mmol), diphenylamine (1) (338 mg, 2.00 mmol), Pd(OAc)2 (5.4 mg, 24 µmol), RuPhos (19 mg, 41 µmol), and powdered NaOtBu (236 mg, 2.46 mmol). The reaction mixture was stirred at 110 °C for 12 h. After cooling to room temperature, the mixture was dissolved in CH2Cl2/H2O (1:1). The aqueous layer was extracted twice with dichloromethane (5 mL). The combined organic layers were dried over MgSO4, the solvent was evaporated in vacuo, and the crude product was purified by chromatography on silica gel with isohexane/dichloromethane (30:1) to afford compound 3 (523 mg, 1.77 mmol, 89%) as a colorless solid. M.p. 132.5 °C; UV (MeOH) λ = 219, 290, and 336 nm; fluorescence (MeOH) λex = 291 and λem = 439 nm; IR (ATR) ν = 3036, 1934, 1740, 1586, 1563, 1488, 1389, 1341, 1290, 1272, 1176, 1087, 1026, 1014, 891, 798, 773, 749, 693, and 624 cm−1; 1H-NMR (500 MHz, CDCl3) δ = 6.93 (t, J = 7.5 Hz, 2H), 7.03 (dd, J = 8.7, 0.8 Hz, 4H), 7.15–7.22 (m, 4H), 7.30–7.38 (m, 2H), 7.42–7.50 (m, 2H), 7.77 (d, J = 8.2 Hz, 1H), 7.88 (d, J = 8.2 Hz, 1H), and 7.94 (d, J = 8.5 Hz, 1H); 13C-NMR and DEPT at θ = 135° (125 MHz, CDCl3) δ = 121.76 (2 CH), 121.97 (4 CH), 124.41 (CH), 126.25 (CH), 126.48 (CH), 126.51 (CH), 126.56 (CH), 127.39 (CH), 128.51 (CH), 129.21 (4 CH), 131.41 (C), 135.41 (C), 143.71 (C), 148.58 (2 C); MS (EI): m/z (%) = 295 (100, [M]+), 294 (45), 293 (11), 217 (23), and 77 (10); elemental analysis calcd. for C22H17N, C 89.46, H 5.80, and N 4.74; found, C 89.61, H 6.05, and N 4.76.
3.3.2. Iron-Catalyzed Oxidative C−C Coupling
Dichloromethane (9 mL) was added to a mixture of iron(II)–hexadecafluorophthalocyanine (15.7 mg, 18.3 μmol, 2 mol%), methanesulfonic acid (34.8 mg, 362 μmol) and 1-(diphenylamino)naphthalene (3) (265 mg, 897 μmol). A 100 mL splash head was attached on the flask to prevent evaporation of the solvent, while ensuring sufficient gas exchange. The resulting solution was stirred at room temperature for 24 h under air. Then, 10 mL of a saturated aqueous solution of sodium hydrogen carbonate was added. The aqueous layer was extracted three times with dichloromethane. The combined organic layers were dried (magnesium sulfate). The solvent was evaporated and the residue was purified by automated flash chromatography (silica gel, isohexane/dichloromethane, 20% to 50% in 1.5 h) to provide N,N,N′,N′-tetraphenylnaphthidine (4) (128 mg, 217 µmol, 48%) as a colorless solid (less polar fraction) and naphthidine 5 (29.5 mg, 33.4 µmol, 11%) as a colorless solid (more polar fraction). Crystallization of compound 4 from isohexane afforded colorless crystals which were suitable for X-ray analysis.
N4,N4,N4’,N4’-Tetraphenyl-[1,1’-binaphthalene]-4,4’-diamine (N,N,N′,N′-Tetraphenylnaphthidine) (4): M.p. 249–250 °C; UV (MeOH) λ = 217, 291, and 361 nm; fluorescence (MeOH) λex = 291 and λem = 446 nm; IR (ATR) ν = 3058, 3034, 1931, 1740, 1582, 1488, 1458, 1420, 1397, 1376, 1266, 1153, 1075, 1053, 1025, 920, 836, 746, 691, and 624 cm−1; 1H-NMR (600 MHz, CDCl3) δ = 6.98 (t, J = 7.3 Hz, 4H), 7.14 (dd, J = 8.7, 1.1 Hz, 8H), 7.23–7.28 (m, 8H), 7.31 (ddd, J = 8.4, 6.9, 1.3 Hz, 2H), 7.36 (ddd, J = 8.4, 6.9, 1.3 Hz, 2H), 7.44 (d, J = 7.3 Hz, 2H), 7.50 (d, J = 7.9 Hz, 2H), 7.52 (d, J = 7.3 Hz, 2H), and 8.08 (d, J = 8.1 Hz, 2H); 13C-NMR and DEPT at θ = 135° (150 MHz, CDCl3) δ = 121.93 (4 CH), 122.17 (8 CH), 124.67 (2 CH), 126.35 (2 CH), 126.43 (2 CH), 126.81 (2 CH), 127.32 (2 CH), 128.71 (2 CH), 129.32 (8 CH), 131.40 (2 C), 134.72 (2 C), 136.76 (2 C), 143.55 (2 C), and 148.70 (4 C); MS (ESI, +10 V) m/z = 589.3 [M + H]+; elemental analysis calcd. for C44H32N2, C 89.76, H 5.48, and N 4.76; found, C 89.61, H 5.63, and N 4.73.
Crystallographic data for N,N,N’,N’-tetraphenylnaphthidine (4): C44H32N2, crystal size 0.100 × 0.102 × 0.522 mm3, M = 588.71 g mol−1, orthorhombic, space group P212121, a = 7.6835(4), b = 12.5020(6), c = 33.1785(18) Å, V = 3187(3) Å3, Z = 4, ρcalcd. = 1.227 g cm−3, µ = 0.071 mm−1, T = 150(2) K, λ = 0.71073 Å, θ range 2.04–28.34°, 50987 reflections collected, 7939 independent (Rint = 0.0473), and 415 parameters. The structure was solved by direct methods and refined by full-matrix least-squares on F2; R1 = 0.0391 and wR2 = 0.0890 [I > 2σ(I)]; maximal residual electron density 0.172 e Å−3. The absolute structure was not determined. CCDC 1983355.
N4-(4-(4-(Diphenylamino)naphthalen-1-yl)phenyl)-N4,N4’,N4’-triphenyl-[1,1’-binaphthalene]-4,4’-diamine (5): M.p. 272–275 °C; UV (MeOH) λ = 215, 291, and 367 nm; fluorescence (MeOH) λex = 291 and λem = 453 nm; IR (ATR) ν = 3061, 3035, 2952, 2920, 2851, 1727, 1585, 1488, 1457, 1420, 1377, 1265, 1174, 1154, 1074, 1026, 952, 920, 827, 748, 692, and 625 cm−1; 1H-NMR (600 MHz, CDCl3) δ = 6.94 (t, J = 7.3 Hz, 2H), 6.99 (t, J = 7.3 Hz, 2H), 7.03 (t, J = 7.3 Hz, 1H), 7.06–7.11 (m, 4H), 7.13–7.17 (m, 4H), 7.19–7.30 (m, 12H), 7.32 (t, J = 8.1 Hz, 3H), 7.34–7.39 (m, 4H), 7.40–7.48 (m, 6H), 7.49–7.61 (m, 5H), 8.03 (d, J = 8.3 Hz, 1H), 8.09 (t, J = 7.7 Hz, 2H), and 8.18 (d, J = 8.7 Hz, 1H); 13C-NMR and DEPT at θ = 135° (150 MHz, CDCl3) δ = 121.40 (2 CH), 121.78 (2 CH), 121.95 (2 CH), 122.00 (4 CH), 122.19 (4 CH), 122.28 (CH), 122.54 (2 CH), 124.66 (CH), 124.70 (CH), 124.71 (CH), 126.19 (CH), 126.31 (CH), 126.39 (CH), 126.45 (CH), 126.46 (CH), 126.57 (CH), 126.82 (CH), 126.93 (CH), 127.03 (CH), 127.05 (CH), 127.31 (CH), 127.41 (CH), 127.55 (CH), 128.74 (CH), 128.79 (CH), 129.23 (4 CH), 129.33 (4 CH), 129.44 (2 CH), 131.07 (2 CH), 131.41 (C), 131.51 (C), 131.67 (C), 133.51 (C), 133.72 (C), 134.72 (C), 134.80 (C), 136.72 (C), 137.03 (C), 138.64 (C), 142.80 (C), 143.39 (C), 143.61 (C), 147.93 (C), 148.46 (C), 148.61 (2 C), and 148.70 (2 C); MS (ESI, +10 V) m/z = 882.5 [M + H]+; elemental analysis calcd. for C66H47N3, C 89.87, H 5.37, and N 4.76; found, C 89.56, H 5.31, and N 4.79.
4. Conclusions
In conclusion, we have developed a two-step synthesis of N,N,N’,N’-tetraphenylnaphthidine (4). Starting from diphenylamine (1), Buchwald–Hartwig coupling with 1-bromonaphthalene (2) and subsequent iron-catalyzed oxidative homocoupling of the resulting 1-(diphenylamino)naphthalene (3) provides compound 4 as the major product and as a minor product compound 5, resulting from an additional oxidative C–C coupling. Thus, we could demonstrate that our method of iron-catalyzed oxidative C–C coupling with air as the final oxidant can be applied to the regioselective homocoupling of triarylamines. Compounds 4 and 5 exhibit a strong blue-light fluorescence with quantum yields of up to 68% and fluorescence lifetimes of 4.2 and 3.6 ns, for compounds 4 and 5, respectively. Further structural changes of the N,N,N’,N’-tetraarylnaphthidines by extension of the π-system or modification of the substitution pattern could lead to improved photophysical properties and fluorophores for various potential applications.
Supplementary Materials
The following data are available online. Copies of the 1H-NMR, 13C-NMR, DEPT (θ = 135°), UV, and fluorescence spectra of the compounds 3, 4, and 5; copies of the 2D-NMR spectra (COSY, HSQC, HMBC, NOESY, 1H/15N-HMBC, JRES) of the compounds 4 and 5.
Author Contributions
A.P. performed the chemical synthesis and characterized the compounds; A.P. and H.-J.K. designed the experiments and analyzed the data; S.S. determined the fluorescence quantum yields and the fluorescence lifetimes; O.K. performed the X-ray analysis; A.P. and H.-J.K. wrote the paper. All authors have read and agreed to the final version of the manuscript.
Funding
We thank the Deutsche Forschungsgemeinschaft (DFG) for the financial support of our project “Green and Sustainable Catalysts for Synthesis of Organic Building Blocks” (DFG grant KN 240/19-2).
Acknowledgments
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lambert, C.; Nöll, G. One- and two-dimensional electron transfer processes in triarylamines with multiple redox centers. Angew. Chem. Int. Ed. 1998, 37, 2107–2110. [Google Scholar] [CrossRef]
- Lambert, C.; Nöll, G. The class II/III transition in triarylamine redox systems. J. Am. Chem. Soc. 1999, 121, 8434–8442. [Google Scholar] [CrossRef]
- Low, P.J.; Paterson, M.A.J.; Puschmann, H.; Goeta, A.E.; Howard, J.A.K.; Lambert, C.; Cherryman, J.C.; Tackley, D.R.; Leeming, S.; Brown, B. Crystal, molecular and electronic structure of N,N’-diphenyl-N,N’-bis(2,4-dimethylphenyl)-(1,1′-biphenyl)-4,4′-diamine and the corresponding radical cation. Chem. Eur. J. 2004, 10, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-J.; Seo, E.-M.; Vak, D.; Kim, D.-Y. Photodynamic properties of azobenzene molecular films with triphenylamines. Chem. Mater. 2003, 15, 4021–4027. [Google Scholar] [CrossRef]
- Schumann, J.; Kanitz, A.; Hartmann, H. Synthesis and characterization of some heterocyclic analogues of N,N’-perarylated phenylene-1,4-diamines and benzidines as a new class of hole transport materials. Synthesis 2002, 9, 1268–1276. [Google Scholar] [CrossRef]
- Meng, H.; Herron, N. Organic small molecule materials for organic light-emitting diodes. In Organic Light-Emitting Materials and Devices, 1st ed.; Li, Z., Meng, H., Eds.; CRC Press, Taylor & Francis: Boca Raton, FL, USA, 2007; pp. 295–412. [Google Scholar]
- Calió, L.; Kazim, S.; Grätzel, M.; Ahmad, S. Hole-transport materials for perovskite solar cells. Angew. Chem. Int. Ed. 2016, 55, 14522–14545. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, K.; Ma, L.; Zhan, X. Triarylamine: Versatile platform for organic, dye-sensitized, and perovskite solar cells. Chem. Rev. 2016, 116, 14675–14725. [Google Scholar] [CrossRef] [PubMed]
- Miras, M.C.; Silber, J.J.; Sereno, L. Two-electron oxidation of N,N,N‘,N‘-tetramethylnaphthidine in acetonitrile–The reactivity of its dication to aromatic nucleophilic substitution by pyridine. J. Electroanal. Chem. 1986, 201, 367–386. [Google Scholar] [CrossRef]
- Zón, M.A.; Fernández, H. Study of the bielectronic electro-oxidation of N,N,N’,N’-tetramethylnaphthidine (TMN) in non-aqueous medium–Determination of the individual kinetic parameters by a Laplace space analysis. J. Electroanal. Chem. 1990, 295, 41–58. [Google Scholar] [CrossRef]
- Desmarets, C.; Champagne, B.; Walcarius, A.; Bellouard, C.; Omar-Amrani, R.; Ahajji, A.; Fort, Y.; Schneider, R. Facile synthesis and characterization of naphthidines as a new class of highly nonplanar electron donors giving robust radical cations. J. Org. Chem. 2006, 71, 1351–1361. [Google Scholar] [CrossRef]
- Desmarets, C.; Omar-Amrani, R.; Walcarius, A.; Lambert, J.; Champagne, B.; Fort, Y.; Schneider, R. Naphthidine di(radical cation)s-stabilized palladium nanoparticles for efficient catalytic Suzuki–Miyaura cross-coupling reactions. Tetrahedron 2008, 64, 372–381. [Google Scholar] [CrossRef]
- Hornback, J.M.; Gossage, H.E. Electrochemical oxidative dehydrodimerization of naphthylamines. An efficient synthesis of 8,8’-dianilino-5,5’-binaphthalene-1,1’-disulfonate. J. Org. Chem. 1985, 50, 541–543. [Google Scholar] [CrossRef]
- Li, X.-L.; Huang, J.-H.; Yang, L.-M. Iron(III)-promoted oxidative coupling of naphthylamines: Synthetic and mechanistic investigations. Org. Lett. 2011, 13, 4950–4953. [Google Scholar] [CrossRef] [PubMed]
- Maddala, S.; Mallick, S.; Venkatakrishnan, P. Metal-free oxidative C–C coupling of arylamines using a quinone-based organic oxidant. J. Org. Chem. 2017, 82, 8958–8972. [Google Scholar] [CrossRef]
- Morimoto, K.; Koseki, D.; Dohi, T.; Kita, Y. Oxidative biaryl coupling of N-aryl anilines by using a hypervalent iodine(III) reagent. Synlett 2017, 28, 2941–2945. [Google Scholar] [CrossRef]
- Theilacker, W.; Hopp, R. Spaltung des Naphthidins und des 2,3,2′,3′-Tetramethylbenzidins in optische Antipoden. Chem. Ber. 1959, 92, 2293–2301. [Google Scholar] [CrossRef]
- Jones, J.G.; Twigg, M.V. A fluorinated iron phthalocyanine. Inorg. Chem. 1969, 8, 2018–2019. [Google Scholar] [CrossRef]
- Topchiy, M.A.; Asachenko, A.F.; Nechaev, M.S. Solvent-free Buchwald–Hartwig reaction of aryl and heteroaryl halides with secondary amines. Eur. J. Org. Chem. 2014, 2014, 3319–3322. [Google Scholar] [CrossRef]
- Fritsche, R.F.; Theumer, G.; Kataeva, O.; Knölker, H.-J. Iron-catalyzed oxidative C–C and N–N coupling of diarylamines and synthesis of spiroacridines. Angew. Chem. Int. Ed. 2017, 56, 549–553. [Google Scholar] [CrossRef]
- Brütting, C.; Fritsche, R.F.; Kutz, S.K.; Börger, C.; Schmidt, A.W.; Kataeva, O.; Knölker, H.-J. Synthesis of 1,1′- and 2,2′-bicarbazole alkaloids by iron(III)-catalyzed oxidative coupling of 2- and 1-hydroxycarbazoles. Chem. Eur. J. 2018, 24, 458–470. [Google Scholar] [CrossRef]
- Purtsas, A.; Kataeva, O.; Knölker, H.-J. Iron-catalyzed oxidative C–C cross-coupling reaction of tertiary anilines with hydroxyarenes by using air as sole oxidant. Chem. Eur. J. 2020, 26, 2499–2508. [Google Scholar] [CrossRef] [PubMed]
- Bolm, C.; Legros, J.; Le Paih, J.; Zani, L. Iron-catalyzed reactions in organic synthesis. Chem. Rev. 2004, 104, 6217–6254. [Google Scholar] [CrossRef] [PubMed]
- Bauer, I.; Knölker, H.-J. Iron catalysis in organic synthesis. Chem. Rev. 2015, 115, 3170–3387. [Google Scholar] [CrossRef] [PubMed]
- Fürstner, A. Iron catalysis in organic synthesis: A critical assessment of what it takes to make this base metal a multitasking champion. ACS Cent. Sci. 2016, 2, 778–789. [Google Scholar] [CrossRef]
- Shang, R.; Ilies, L.; Nakamura, E. Iron-catalyzed C–H bond activation. Chem. Rev. 2017, 117, 9086–9139. [Google Scholar] [CrossRef]
- Kress, R.B.; Duesler, E.N.; Etter, M.C.; Paul, I.C.; Curtin, D.Y. Solid-state resolution of binaphthyl: Crystal and molecular structures of the chiral (A) form and racemic (B) form and the study of the rearrangement of single crystals. Requirements for development of hemihedral faces for enantiomer identification. J. Am. Chem. Soc. 1980, 102, 7709–7714. [Google Scholar] [CrossRef]
- Roszak, K.; Katrusiak, A. High-pressure and temperature dependence of the spontaneous resolution of 1,1′-binaphthyl enantiomers. Phys. Chem. Chem. Phys. 2018, 20, 5305–5311. [Google Scholar] [CrossRef]
- Brock, C.P.; Dunitz, J.D. Temperature dependence of thermal motion in crystalline naphthalene. Acta Cryst. 1982, B38, 2218–2228. [Google Scholar] [CrossRef]
- Capelli, S.C.; Albinati, A.; Mason, S.A.; Willis, B.T.M. Molecular motion in crystalline naphthalene: Analysis of multi-temperature X-ray and neutron diffraction data. J. Phys. Chem. A 2006, 110, 11695–11703. [Google Scholar] [CrossRef]
- Baba, M.; Kowaka, Y.; Nagashima, U.; Ishimoto, T.; Goto, H.; Nakayama, N. Geometrical structure of benzene and naphthalene: Ultrahigh-resolution laser spectroscopy and ab initio calculation. J. Chem. Phys. 2011, 135, 054305. [Google Scholar] [CrossRef] [Green Version]
- Nishio, M.; Umezawa, Y.; Honda, K.; Tsuboyama, S.; Suezawa, H. CH/π hydrogen bonds in organic and organometallic chemistry. CrystEngComm 2009, 11, 1757–1788. [Google Scholar] [CrossRef]
- Benedetti, E.; Kocsis, L.S.; Brummond, K.M. Synthesis and photophysical properties of a series of cyclopenta[b]naphthalene solvatochromic fluorophores. J. Am. Chem. Soc. 2012, 134, 12418–12421. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Gong, Z.-L.; Tang, J.-H.; Sun, M.-J.; Shao, J.-Y.; Zhong, Y.-W.; Yao, J. Triarylamines with branched multi-pyridine groups: Modulation of emission properties by structural variation, solvents, and tris(pentafluorophenyl)borane. Sci. China Chem. 2018, 61, 545–556. [Google Scholar] [CrossRef]
- Lee, M.-T.; Chen, H.-H.; Liao, C.-H.; Tsai, C.-H.; Chen, C.H. Stable styrylamine-doped blue organic electroluminescent device based on 2-methyl-9,10-di(2-naphthyl)anthracene. Appl. Phys. Lett. 2004, 85, 3301–3303. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.Q.; Mi, B.X.; Chen, C.H.; Cheah, K.W.; Cheng, Y.K.; Wen, W.-S. High-efficiency deep blue host for organic light-emitting devices. Appl. Phys. Lett. 2007, 90, 123506. [Google Scholar] [CrossRef]
- Wu, T.-L.; Chou, H.-H.; Huang, P.-Y.; Cheng, C.-H.; Liu, R.-S. 3,6,9,12-Tetrasubstituted chrysenes: Synthesis, photophysical properties, and application as blue fluorescent OLED. J. Org. Chem. 2014, 79, 267–274. [Google Scholar] [CrossRef]
- Cho, S.; Kim, C.; Lee, S.E.; Kim, Y.K.; Yoon, S.S. Naphthalene derivatives end-capped with 2-(diphenylamino)-9,9-diethylfluorenes for blue organic light-emitting diodes. J. Nanosci. Nanotechnol. 2018, 18, 1251–1255. [Google Scholar] [CrossRef]
- Lee, K.H.; Kang, L.K.; Lee, J.Y.; Kang, S.; Jeon, S.O.; Yook, K.S.; Lee, J.Y.; Yoon, S.S. Molecular engineering of blue fluorescent molecules based on silicon end-capped diphenylaminofluorene derivatives for efficient organic light-emitting materials. Adv. Funct. Mater. 2010, 20, 1345–1358. [Google Scholar] [CrossRef]
- Maeda, H.; Maeda, T.; Mizuno, K. Absorption and fluorescence spectroscopic properties of 1- and 1,4-silyl-substituted naphthalene derivatives. Molecules 2012, 17, 5108–5125. [Google Scholar] [CrossRef] [Green Version]
- Koo, J.Y.; Heo, C.H.; Shin, Y.-H.; Kim, D.; Lim, C.S.; Cho, B.R.; Kim, H.M.; Park, S.B. Readily accessible and predictable naphthalene-based two-photon fluorophore with full visible-color coverage. Chem. Eur. J. 2016, 22, 14166–14170. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS-97, Programs for Crystal Structure Solution; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SADABS, v. 2.10, Bruker/Siemens Area Detector Absorption Correction Program; Bruker AXS Inc.: Madison, WI, USA, 2002. [Google Scholar]
- Sheldrick, G.M. SHELXL-97, Programs for Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Farrugia, L.J. ORTEP-3 for Windows—A version of Ortep-III with a graphical user interface (GUI). J. Appl. Cryst. 1997, 30, 565. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 3–5 are available from the authors. |
Figure 1.
Structural formula of iron(II)–hexadecafluorophthalocyanine (FePcF16).

Scheme 1.
Synthesis of 1-(diphenylamino)naphthalene (3). Reagents and conditions: (a) 1 mol% Pd(OAc)2, 2 mol% RuPhos, 1.2 equivalents NaOtBu, air, 110 °C, 12 h, 89% compound 3.
Scheme 1.
Synthesis of 1-(diphenylamino)naphthalene (3). Reagents and conditions: (a) 1 mol% Pd(OAc)2, 2 mol% RuPhos, 1.2 equivalents NaOtBu, air, 110 °C, 12 h, 89% compound 3.

Scheme 2.
Iron-catalyzed oxidative C–C coupling of 1-(diphenylamino)naphthalene (3). Reagents and conditions: (a) 2 mol% FePcF16, 40 mol% MsOH, CH2Cl2, air, room temperature, 24 h, 48% compound 4, 11% compound 5.
Scheme 2.
Iron-catalyzed oxidative C–C coupling of 1-(diphenylamino)naphthalene (3). Reagents and conditions: (a) 2 mol% FePcF16, 40 mol% MsOH, CH2Cl2, air, room temperature, 24 h, 48% compound 4, 11% compound 5.

Figure 2.
Assignment of the 13C-NMR signals of the N,N,N’,N’-tetraphenylnaphthidine (4) to the respective 1H-NMR signals by the HSQC spectrum.
Figure 2.
Assignment of the 13C-NMR signals of the N,N,N’,N’-tetraphenylnaphthidine (4) to the respective 1H-NMR signals by the HSQC spectrum.

Figure 3.
Molecular structure of N,N,N’,N’-tetraphenylnaphthidine (4) in the crystal (ORTEP plot showing thermal ellipsoids at the 50% probability level). Selected bond lengths (Å): C1–C2 1.370(3), C1–C10 1.424(2), C5–C10 1.431(2), C4–N21 1.436(2), C1–C11 1.500(2), C11–C12 1.370(2), C11–C20 1.428(2), C15–C20 1.432(2), C14–N34 1.430(2). Selected bond angles (°): C10–C1–C11 121.4(2), C2–C1–C11 118.8(2), C2–C1–C10 119.7(2), C1–C10–C9 122.2(2), C3–C4–N21 120.6(2), C5–C4–N21 120.0(2), C4–N21–C22 117.1(2), C4–N21–C28 118.3(2), and C22–N21–C28 120.6(2).
Figure 3.
Molecular structure of N,N,N’,N’-tetraphenylnaphthidine (4) in the crystal (ORTEP plot showing thermal ellipsoids at the 50% probability level). Selected bond lengths (Å): C1–C2 1.370(3), C1–C10 1.424(2), C5–C10 1.431(2), C4–N21 1.436(2), C1–C11 1.500(2), C11–C12 1.370(2), C11–C20 1.428(2), C15–C20 1.432(2), C14–N34 1.430(2). Selected bond angles (°): C10–C1–C11 121.4(2), C2–C1–C11 118.8(2), C2–C1–C10 119.7(2), C1–C10–C9 122.2(2), C3–C4–N21 120.6(2), C5–C4–N21 120.0(2), C4–N21–C22 117.1(2), C4–N21–C28 118.3(2), and C22–N21–C28 120.6(2).

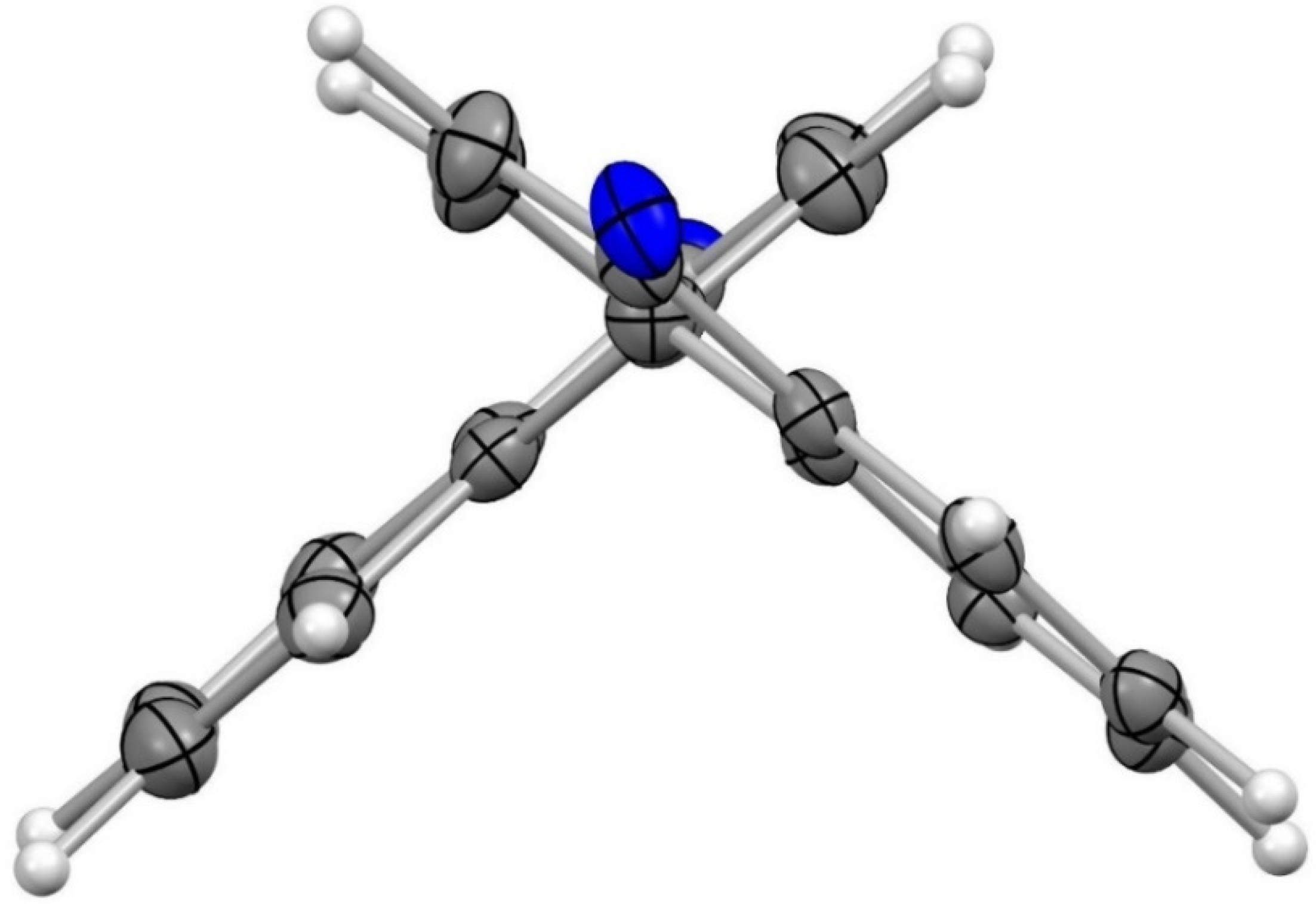
Figure 4.
Crystal structure of compound 4 with a view along the N21–C4/C1–C11/C14–N34 axis (ORTEP plot showing thermal ellipsoids at the 50% probability level, the N–phenyl substituents have been omitted for clarity). Torsional angle (C2–C1–C11–C12) 99.4(2)°.
Figure 4.
Crystal structure of compound 4 with a view along the N21–C4/C1–C11/C14–N34 axis (ORTEP plot showing thermal ellipsoids at the 50% probability level, the N–phenyl substituents have been omitted for clarity). Torsional angle (C2–C1–C11–C12) 99.4(2)°.

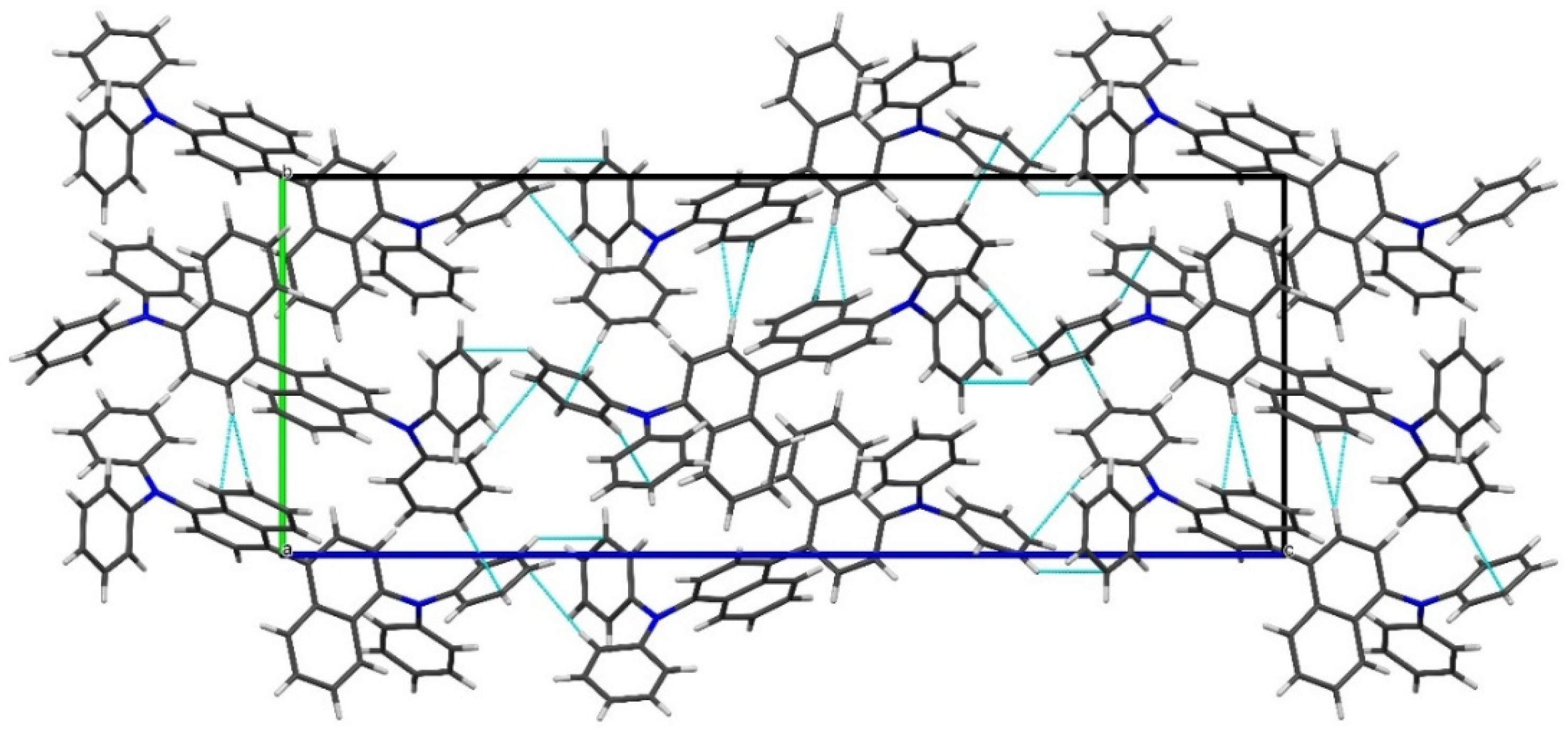
Figure 5.
Fragment of the crystal packing of N,N,N’,N’-tetraphenylnaphthidine (4) showing the unit cell and C–H···π interactions (view along the a-axis of the crystal).
Figure 5.
Fragment of the crystal packing of N,N,N’,N’-tetraphenylnaphthidine (4) showing the unit cell and C–H···π interactions (view along the a-axis of the crystal).

Figure 6.
Assignment of the 13C-NMR signals of the N,N,N’,N’-tetraarylnaphthidine (5) to the respective 1H-NMR signals by the HSQC spectrum.
Figure 6.
Assignment of the 13C-NMR signals of the N,N,N’,N’-tetraarylnaphthidine (5) to the respective 1H-NMR signals by the HSQC spectrum.

Figure 7.
Fluorescence of the compounds 3 (left), 4 (center), and 5 (right) in methanol (concentration c = 2 mg mL−1) by excitation with UV light (λex = 254 nm).
Figure 7.
Fluorescence of the compounds 3 (left), 4 (center), and 5 (right) in methanol (concentration c = 2 mg mL−1) by excitation with UV light (λex = 254 nm).

Figure 8.
Normalized fluorescence emission spectra of 1-(diphenylamino)naphthalene (3) (blue), N,N,N’,N’-tetraphenylnaphthidine (4) (orange), and naphthidine 5 (green) in methanol (excitation at λex = 291 nm).
Figure 8.
Normalized fluorescence emission spectra of 1-(diphenylamino)naphthalene (3) (blue), N,N,N’,N’-tetraphenylnaphthidine (4) (orange), and naphthidine 5 (green) in methanol (excitation at λex = 291 nm).

Figure 9.
Fluorescence of N,N,N’,N’-tetraphenylnaphthidine (4) by excitation with UV light (λex = 254 nm) in different solvents (concentration c = 2 mg mL−1 and from left to right, isohexane, dichloromethane, ethyl acetate, THF, and methanol).
Figure 9.
Fluorescence of N,N,N’,N’-tetraphenylnaphthidine (4) by excitation with UV light (λex = 254 nm) in different solvents (concentration c = 2 mg mL−1 and from left to right, isohexane, dichloromethane, ethyl acetate, THF, and methanol).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
UV absorption and fluorescence emission data of the compounds 3, 4, and 5 in methanol.
| Compound | λabs (nm) a | ε (M−1 cm−1) b | λem (nm) c | Δλ (nm) d |
|---|---|---|---|---|
| aminonaphthalene 3 | 290 | 15,700 | 439 | 149 |
| naphthidine 4 | 291 | 11,400 | 446 | 155 |
| naphthidine 5 | 291 | 6000 | 453 | 162 |
a UV absorption maximum; b molar extinction coefficient; c fluorescence emission maximum (excitation at λex = 291 nm); d Stokes shift, Δλ = λem − λabs.
Table 2.
Photophysical data of the naphthidines 4 and 5.
| Compound | λabs (nm) a | ε (M−1 cm−1) b | λem (nm) c | Φfl d,e | т (ns) f,e | CIE-coord. (x; y) g,e |
|---|---|---|---|---|---|---|
| naphthidine 4 | 294 | 32,000 | 449 | 0.68 | 4.2 | (0.153; 0.054) |
| naphthidine 5 | 294 | 25,600 | 453 | 0.65 | 3.6 | (0.152; 0.062) |
a UV absorption maximum in CH2Cl2; b molar extinction coefficient in CH2Cl2; c fluorescence emission maximum in CH2Cl2 (λex = 294 nm); d fluorescence quantum yield (λex = 310 nm); e in a 2% PMMA film; f fluorescence lifetime (λex = 360 nm); g Commission internationale de l’éclairage (CIE) (λex = 310 nm).
Table 3.
UV absorption and fluorescence emission data of the naphthidine 4 in different solvents.
| Isohexane | CH2Cl2 | Ethyl Acetate | THF | Methanol | |
|---|---|---|---|---|---|
| λabs (nm) a | 291 | 294 | 291 | 292 | 291 |
| λem (nm) b | 428 | 449 | 439 | 445 | 446 |
a UV absorption maximum and b fluorescence emission maximum (for each solvent λex = λabs).
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Purtsas, A.; Stipurin, S.; Kataeva, O.; Knölker, H.-J. Iron-Catalyzed Synthesis, Structure, and Photophysical Properties of Tetraarylnaphthidines. Molecules 2020, 25, 1608. https://doi.org/10.3390/molecules25071608
AMA Style
Purtsas A, Stipurin S, Kataeva O, Knölker H-J. Iron-Catalyzed Synthesis, Structure, and Photophysical Properties of Tetraarylnaphthidines. Molecules. 2020; 25(7):1608. https://doi.org/10.3390/molecules25071608
Chicago/Turabian StylePurtsas, Alexander, Sergej Stipurin, Olga Kataeva, and Hans-Joachim Knölker. 2020. "Iron-Catalyzed Synthesis, Structure, and Photophysical Properties of Tetraarylnaphthidines" Molecules 25, no. 7: 1608. https://doi.org/10.3390/molecules25071608