IRF7 Is Required for the Second Phase Interferon Induction during Influenza Virus Infection in Human Lung Epithelia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Preparation of Influenza Virus Stock
2.3. siRNA Transfection into BCi-NS1.1 and A549 Cells
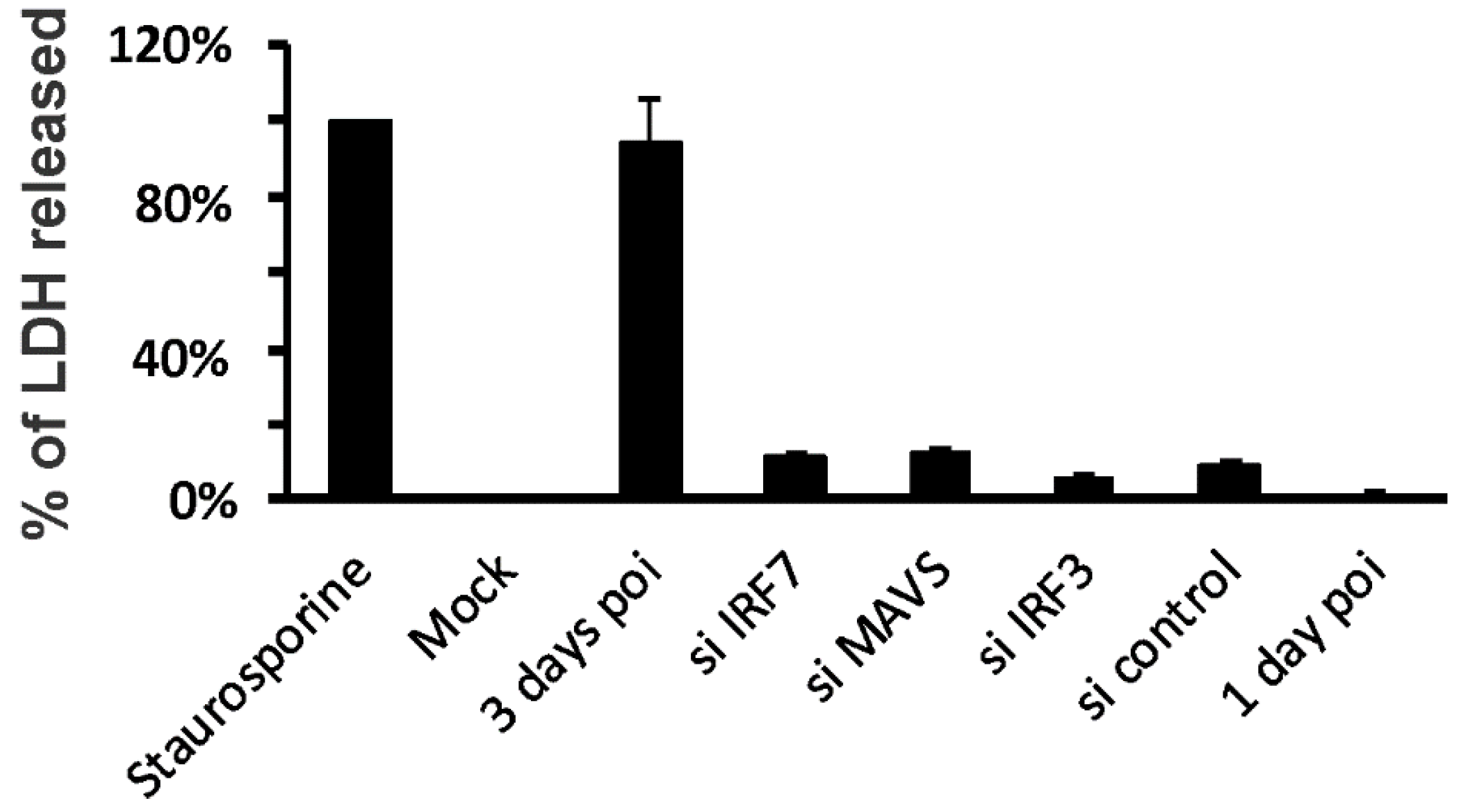
2.4. Lactate Dehydrogenase (LDH) Assay
2.5. Measurement of mRNA Expression by Quantitative Real-Time PCR (qRT-PCR)
2.6. RIG-I, TLR3 and IRF7 Protein Determination by Immunoblotting
2.7. Statistical Analysis
3. Results
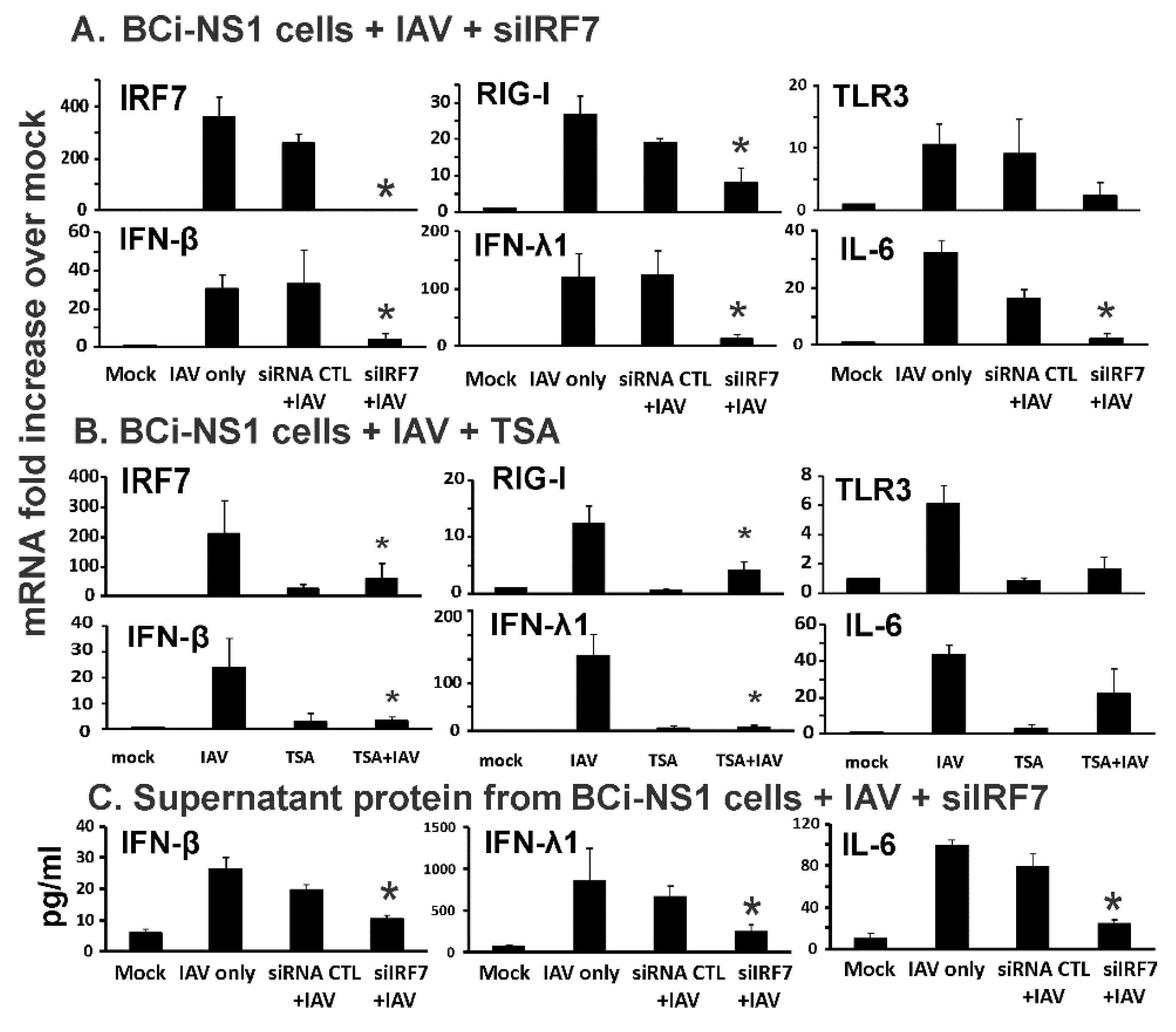
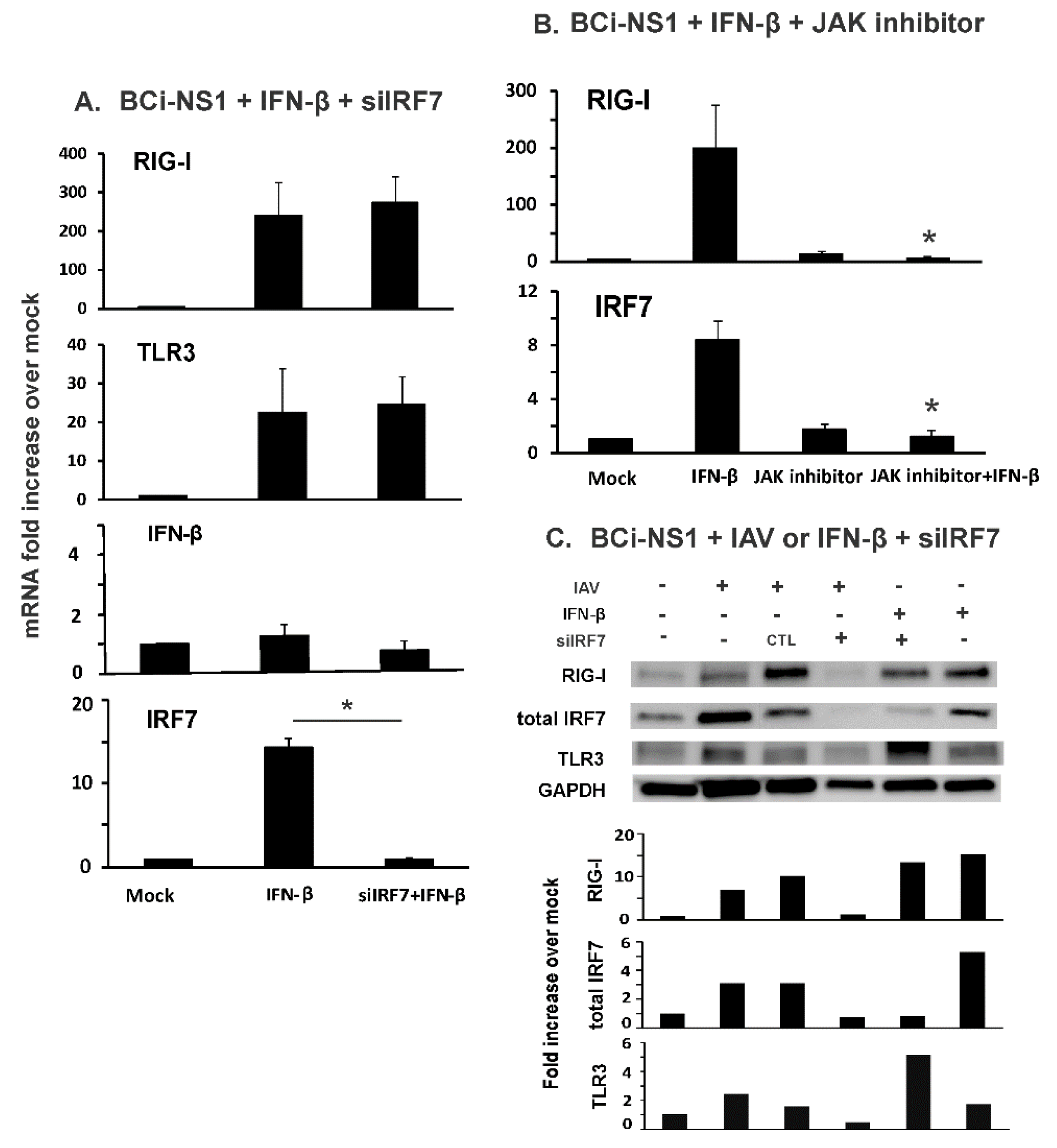
3.1. IRF7 Knockdown Inhibited Influenza-Initiated RIG-I and IFN Induction, but not IFN-β-Mediated RIG-I Induction in Human Bronchial BCi-NS1.1 Cells
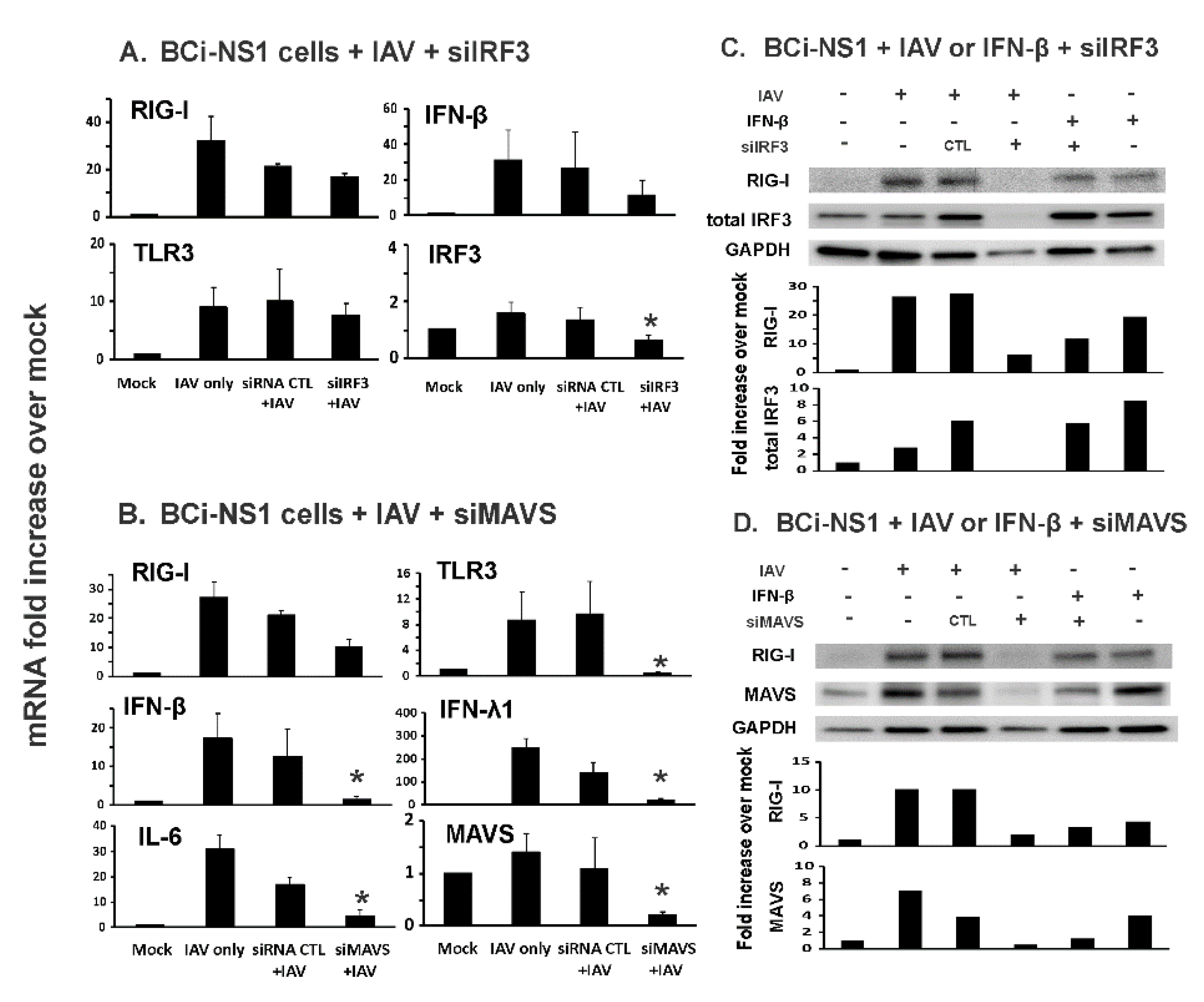
3.2. MAVS, but not IRF3, Knockdown Inhibited Influenza-Initiated RIG-I and IFN Induction in Human Bronchial BCi-NS1.1 Cells
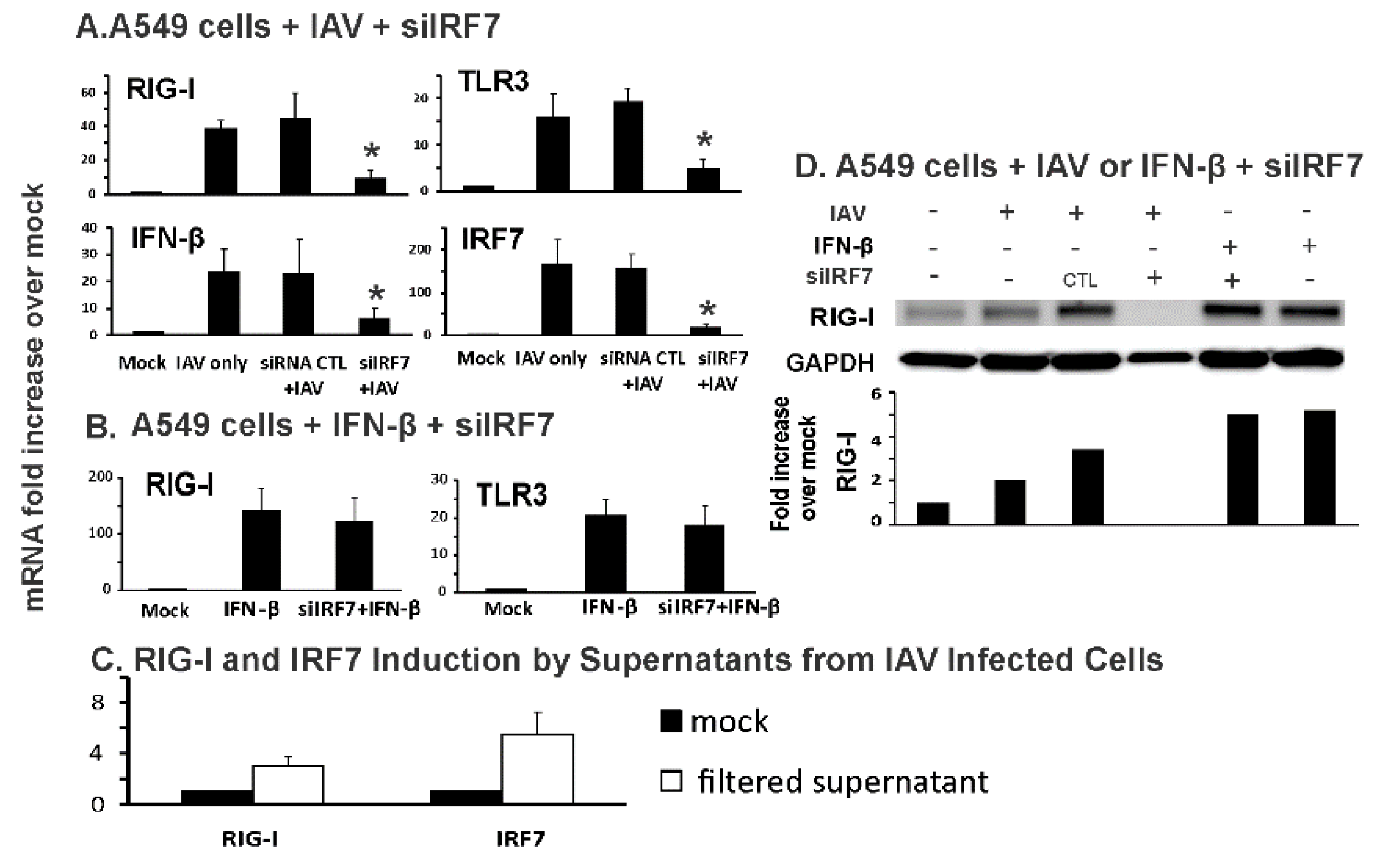
3.3. IRF7 Knockdown Inhibited Influenza-Initiated, but not IFN-β-Induced, RIG-I Induction in Human Alveolar Epithelial Cells
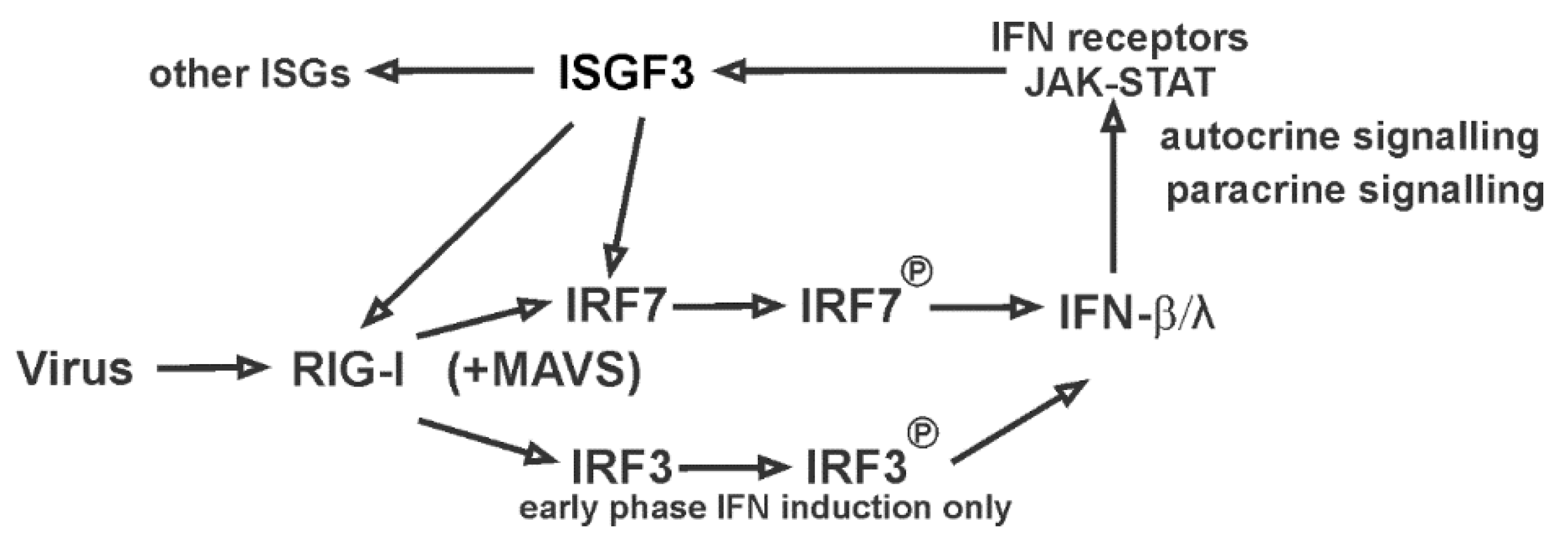
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Job, E.R.; Deng, Y.M.; Tate, M.D.; Bottazzi, B.; Crouch, E.C.; Dean, M.M.; Mantovani, A.; Brooks, A.G.; Reading, P.C. Pandemic H1N1 influenza A viruses are resistant to the antiviral activities of innate immune proteins of the collectin and pentraxin superfamilies. J. Immunol. 2010, 185, 4284–4291. [Google Scholar] [CrossRef] [PubMed]
- Secombes, C.J.; Zou, J. Evolution of Interferons and Interferon Receptors. Front. Immunol. 2017, 8, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Zhang, W.; Duggan, E.S.; Booth, J.L.; Zou, M.H.; Metcalf, J.P. RIG-I and TLR3 are both required for maximum interferon induction by influenza virus in human lung alveolar epithelial cells. Virology 2015, 482, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, H.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Uematsu, S.; Matsui, K.; Tsujimura, T.; Takeda, K.; Fujita, T.; Takeuchi, O.; et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity 2005, 23, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Guillot, L.; Le Goffic, R.; Bloch, S.; Escriou, N.; Akira, S.; Chignard, M.; Si-Tahar, M. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J. Biol. Chem. 2005, 280, 5571–5580. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef]
- Wu, W.; Booth, J.L.; Duggan, E.S.; Wu, S.; Patel, K.B.; Coggeshall, K.M.; Metcalf, J.P. Innate immune response to H3N2 and H1N1 influenza virus infection in a human lung organ culture model. Virology 2010, 396, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Teijaro, J.R. Type I interferons in viral control and immune regulation. Curr. Opin Virol. 2016, 16, 31–40. [Google Scholar] [CrossRef]
- Schindler, C.; Darnell, J.E., Jr. Transcriptional responses to polypeptide ligands: The JAK-STAT pathway. Annu. Rev. Biochem. 1995, 64, 621–651. [Google Scholar] [CrossRef]
- Zhou, A.; Paranjape, J.M.; Der, S.D.; Williams, B.R.; Silverman, R.H. Interferon action in triply deficient mice reveals the existence of alternative antiviral pathways. Virology 1999, 258, 435–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.P.; Lee, S.M.; Cheung, C.Y.; Mao, H.; Lai, A.K.; Chan, R.W.; Chan, M.C.; Tu, W.; Guan, Y.; Lau, Y.L.; et al. H5N1 influenza virus-induced mediators upregulate RIG-I in uninfected cells by paracrine effects contributing to amplified cytokine cascades. J. Infect. Dis. 2011, 204, 1866–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef]
- Hatesuer, B.; Hoang, H.T.; Riese, P.; Trittel, S.; Gerhauser, I.; Elbahesh, H.; Geffers, R.; Wilk, E.; Schughart, K. Deletion of Irf3 and Irf7 Genes in Mice Results in Altered Interferon Pathway Activation and Granulocyte-Dominated Inflammatory Responses to Influenza A Infection. J. Innate Immun. 2017, 9, 145–161. [Google Scholar] [CrossRef]
- Ciancanelli, M.J.; Huang, S.X.; Luthra, P.; Garner, H.; Itan, Y.; Volpi, S.; Lafaille, F.G.; Trouillet, C.; Schmolke, M.; Albrecht, R.A.; et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 2015, 348, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Pulendran, B.; Maddur, M.S. Innate immune sensing and response to influenza. Curr. Top. Microbiol. Immunol. 2015, 386, 23–71. [Google Scholar] [CrossRef]
- Manicassamy, B.; Manicassamy, S.; Belicha-Villanueva, A.; Pisanelli, G.; Pulendran, B.; Garcia-Sastre, A. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc. Natl. Acad. Sci. USA 2010, 107, 11531–11536. [Google Scholar] [CrossRef] [Green Version]
- Walters, M.S.; Gomi, K.; Ashbridge, B.; Moore, M.A.; Arbelaez, V.; Heldrich, J.; Ding, B.S.; Rafii, S.; Staudt, M.R.; Crystal, R.G. Generation of a human airway epithelium derived basal cell line with multipotent differentiation capacity. Respir. Res. 2013, 14, 135. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Harrison, C.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bals, R.; Hiemstra, P.S. Innate immunity in the lung: How epithelial cells fight against respiratory pathogens. Eur. Respir. J. 2004, 23, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Jakiela, B.; Brockman-Schneider, R.; Amineva, S.; Lee, W.M.; Gern, J.E. Basal cells of differentiated bronchial epithelium are more susceptible to rhinovirus infection. Am. J. Respir. Cell Mol. Biol. 2008, 38, 517–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, R.R.; Russell, M.L.; Roggli, V.L.; Crapo, J.D. Cell number and distribution in human and rat airways. Am. J. Respir. Cell Mol. Biol. 1994, 10, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef]
- Sato, M.; Hata, N.; Asagiri, M.; Nakaya, T.; Taniguchi, T.; Tanaka, N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 1998, 441, 106–110. [Google Scholar] [CrossRef] [Green Version]
- Killip, M.J.; Smith, M.; Jackson, D.; Randall, R.E. Activation of the interferon induction cascade by influenza a viruses requires viral RNA synthesis and nuclear export. J. Virol. 2014, 88, 3942–3952. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, W.; Zhang, W.; Tian, L.; Brown, B.R.; Walters, M.S.; Metcalf, J.P. IRF7 Is Required for the Second Phase Interferon Induction during Influenza Virus Infection in Human Lung Epithelia. Viruses 2020, 12, 377. https://doi.org/10.3390/v12040377
Wu W, Zhang W, Tian L, Brown BR, Walters MS, Metcalf JP. IRF7 Is Required for the Second Phase Interferon Induction during Influenza Virus Infection in Human Lung Epithelia. Viruses. 2020; 12(4):377. https://doi.org/10.3390/v12040377
Chicago/Turabian StyleWu, Wenxin, Wei Zhang, Lili Tian, Brent R. Brown, Matthew S. Walters, and Jordan P. Metcalf. 2020. "IRF7 Is Required for the Second Phase Interferon Induction during Influenza Virus Infection in Human Lung Epithelia" Viruses 12, no. 4: 377. https://doi.org/10.3390/v12040377