
Abstract
Mitophagy is a degradative pathway that mediates the degradation of the entire mitochondria, and defects in this process are implicated in many diseases including cancer. In mammals, mitophagy is mediated by BNIP3L (also known as NIX) that is a dual regulator of mitochondrial turnover and programmed cell death pathways. Acute myeloid leukemia (AML) cells with deficiency of BNIP3L are more sensitive to mitochondria-targeting drugs. But small molecular inhibitors for BNIP3L are currently not available. Some immunomodulatory drugs (IMiDs) have been proved by FDA for hematologic malignancies, however, the underlining molecular mechanisms are still elusive, which hindered the applications of BNIP3L inhibition for AML treatment. In this study we carried out MS-based quantitative proteomics analysis to identify the potential neosubstrates of a novel thalidomide derivative CC-885 in A549 cells. In total, we quantified 5029 proteins with 36 downregulated in CRBN+/+ cell after CC-885 administration. Bioinformatic analysis showed that macromitophagy pathway was enriched in the negative pathway after CC-885 treatment. We further found that CC-885 caused both dose- and time-dependent degradation of BNIP3L in CRBN+/+, but not CRBN−/− cell. Thus, our data uncover a novel role of CC-885 in the regulation of mitophagy by targeting BNIP3L for CRL4CRBN E3 ligase-dependent ubiquitination and degradation, suggesting that CC-885 could be used as a selective BNIP3L degradator for the further investigation. Furthermore, we demonstrated that CC-885 could enhance AML cell sensitivity to the mitochondria-targeting drug rotenone, suggesting that combining CC-885 and mitochondria-targeting drugs may be a therapeutic strategy for AML patients.
Similar content being viewed by others


Introduction
Mitophagy is one type of macroautophagy and is necessary for the elimination of dysfunctional mitochondria and the maintenance of cellular homeostasis related to aging, degenerative diseases, and cancer [1,2,3]. In mammals, mitophagy is mediated by BNIP3L during red blood cell differentiation [4, 5]. BNIP3L, also known as Nix, is a dual regulator of mitochondrial turnover and programmed cell death pathways [6, 7]. Acute myeloid leukemia (AML) cells deficient in BNIP3L show increased sensitivity to mitochondria-targeting drugs [8], suggesting that BNIP3L could be a promising drug target for AML patients and a therapeutic strategy that combines BNIP3L inhibition and mitochondria-targeting drugs.
The thalidomide derivatives lenalidomide, pomalidomide, and CC-122 are classified as immunomodulatory drugs (IMiDs) and have been approved by the FDA for hematologic malignancies such as multiple myeloma [9,10,11]. A novel thalidomide derivative, CC-885, was also shown potent antitumor effects [12]. IMiDs confer substrate specificity by directly binding to the CRL4CRBN (CUL4–DDB1–RBX1–CRBN) E3 ubiquitin ligase [13], which leads to the ubiquitination and proteasome degradation of substrates. For example, all these drugs can induce the degradation of two key transcription factors, IKAROS (IKZF1) and AIOLOS (IKZF3), to exert clinical efficacy in multiple myeloma [14, 15]. Lenalidomide can also induce cytogenetic remission in more than half of the patients with myelodysplastic syndrome of deletion of chromosome 5q (del(5q) MDS) and apoptosis of del(5q) MDS cells via the selective degradation of CK1a [16]. In addition, a pulse-chase SILAC (stable-isotope labeling by amino acids in cell culture) mass spectrometry-based proteomics method identified ZFP91 as a substrate of IMiDs in both solid tumor and hematoma cells [17].
Of these thalidomide derivatives, CC-885 was particularly interesting because it shows potent anti-solid tumor activity. Its antitumor activity was reported to be mediated by the specific degradation of the translation termination factor GSPT1 [12]. However, whether there are additional substrates targeted by CC-885 for degradation remains unknown. Moreover, it is also unknown whether there is another mechanism of action of CC-885 other than apoptosis.
Here, through state-of-the-art mass spectrometry-based quantitative proteomic technology, we systematically screened the potential substrates degraded by CC-885. In total, we quantified 5029 proteins, and 36 were downregulated after CC-885 administration. Bioinformatic analysis showed that the macromitophagy pathway was enriched in the negative pathway after CC-885 treatment. Importantly, we demonstrated that CC-885 abrogated carbonyl cyanide m-chlorophenyl hydrazone (CCCP)-induced mitophagy by targeting BNIP3L for CRL4CRBN E3 ubiquitin ligase-dependent ubiquitination and degradation. In addition, our data indicated that CC-885 could enhance the sensitivity of AML cells to mitochondria-targeting drugs, suggesting that combining CC-885 and mitochondria-targeting drugs might be a therapeutic strategy for AML patients.
Experimental section
Materials
Mass spectrometry grade water and acetonitrile (ACN) were purchased from Thermo Fisher Scientific, Inc (USA). Formic acid (98%), potassium chloride (KCl), ammonium bicarbonate (NH4HCO3, 99%), trifluoroacetic acid (99%), and protease inhibitor mixture were purchased from Roche Diagnostics, Ltd. (Shanghai, China). SILAC medium and amine acid were purchased from Silantes (Munich, Germany). The BNIP3L antibody was purchased from Cell Signaling Technology (Danvers, USA), and the CRBN antibody was purchased from Atlas Antibody (Swenden). The compounds CC-885, thalidomide, and lenalidomide were purchased from MedChemExpress (Shanghai, China).
Cell culture and SILAC labeling
The human non-small cell lung cancer cell line A549, the human embryonic kidney cell line HEK293T and HeLa cells were cultured with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1× penicillin, and streptomycin. For SILAC labeling, the cells were cultured with DMEM medium containing 10% dialyzed FBS, 1× penicillin, and streptomycin, 1× GlutaMax and arginine (Arg0) and lysine (Lys0) (light) or lysine (13C6-Lys6) and arginine (13C615N4-Arg10) (heavy). The labeling efficiency was determined to be over 95% before proteomic analysis.
Sample preparation and basic reverse-phase fractionation
Cells were treated with 0.1% DMSO (light) or 200 nM CC-885 (heavy) for 12 h. Next, the cells were washed twice with cold PBS and then lysed in lysis buffer (8 M urea, 0.1 M NH4HCO3 and 1× protease inhibitor). Then, the sample was centrifuged at 20,000 × g for 20 min at 4 °C after sonication. The supernatant was transferred into another tube before the concentration was determined by a BCA assay. Protein lysates (200 μg) were mixed at a 1:1 ratio and reduced by 5 mM DTT (dithiothreitol) at 55 °C for 30 min and then incubated with 15 mM IAA (iodoacetamide) in the dark at room temperature for 30 min before quenching with 30 mM cysteine. The protein lysates were digested by lysyl endopeptidase (Lys-C) at a ratio of 1:100 (w/w) for 3 h at 37 °C, and then, trypsin was added to lysates for digestion again at a ratio of 1:50 (w/w) for 16 h at 37 °C. Finally, the peptides were desalted by a Sep-Pak C18 column.
To further reduce the complexity of the sample, tryptic peptides were resolved in buffer A (98% H2O, 2% ACN, pH 9.0) for preseparation by high-pH reversed-phase HPLC [18]. The sample was loaded onto a Waters XBridge Peptide BEH C18 column (100 Å, 5 µm, 3.5 × 100 mm) and eluted with a 57 min gradient from 2% to 95% of buffer B (98% ACN, 2% H2O) at a flow rate of 0.6 mL/min. Finally, the sample was combined into 15 or 20 fractions and dried by a SpeedVac for MS/MS analysis.
Nano-LC-MS/MS analysis and proteome database search
For the proteomic data of A549 cells, the samples were analyzed by an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). The peptides were separated from a reverse-phase C18 column (20 cm length, 75 μm ID, 3 μm particle size) with a 70 min gradient from 5% to 85% buffer B (90% ACN, 10% H2O, 0.1% formic acid) at a flow rate of 300 nL/min by an EASY-nLC 1000 system. The ions of m/z 350–1300 with two, three or four charges were analyzed at a resolution of 120,000 at m/z = 200 for full scans. The ion fragments obtained by high-energy collision dissociation (HCD) collision (normalized collision energy of 32%) were acquired and analyzed in top speed mode for MS/MS scans. The automatic gain control (AGC) was 1 E6 for a full scan and 7 E3 for a MS/MS scan, and dynamic exclusion was limited to 60 s. For the proteomic data of the 293 T CRBN−/− cells, the samples were analyzed by a Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). The peptides were separated with a reverse-phase C18 column (20 cm length, 75 μm ID, 1.9 μm particle size) with a 65 min gradient of 5% to 85% buffer B (90% ACN, 10% H2O, 0.1% formic acid) at a flow rate of 300 nL/min by an EASY-nLC 1200 system. The ions of m/z 350–1300 with two, three, or four charges were analyzed at a resolution of 60,000 at m/z = 200. The ion fragments obtained by high-energy collision dissociation (HCD) collision (normalized collision energy of 28%) were acquired and analyzed in the top 20 mode for the MS/MS scans. The automatic gain control (AGC) targets were 3 E6 for a full scan and 1 E5 for a MS/MS scan, and dynamic exclusion was limited to 60 s.
The MS/MS data were searched using MaxQuant software against the UniProt Human database (92,011 sequences, release date: September 2015) [19]. Trypsin/p was chosen as the specific enzyme, and the number of missed cleavages was limited to 2. 13C6-Lys and 13C615N4-Arg were set as “heavy”. Acetyl (protein N-term) and oxidation (M) were set as variable modifications, and carbamidomethyl (C) was the fixed modification. The protein and peptide FDRs were set to 1%. Statistical analysis was performed by two-tailed Student’s t test.
Bioinformatic analysis
For the proteomic data from the CRBN+/+ cells treated with or without CC-885, the protein intensity quantified by MaxQuant was used for enrichment analysis by Gene Set Enrichment Analysis (GSEA, version 3.0) with the Gene Ontology database [20]. Gene markers were ranked using Signal2Noise. The significance of the obtained terms was evaluated by using the FDR q-value. An FDR q value <0.25 was considered a criterion of statistically significant terms.
Cell viability assay
Cells were seeded and cultured in 96-well plates for 24 h. Then, the cells were treated with compounds at gradient concentrations for 48 h or 72 h. The absorbances were measured at wavelengths of 450 nm for CCK8 and 490 nm for MTT by an ELISA plate reader.
Immunoblotting analysis
Cells were washed twice with cold PBS and then boiled with loading buffer for 10 min. The sample was separated by 10% SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) and further transferred onto a NC membrane. After that, the membrane was blocked with 5% BSA for 1 h at room temperature before incubation with primary antibody overnight at 4 °C. The membrane was washed six times and incubated with secondary antibody for 1 h. Finally, the signals were determined by an ImageQuant LAS 4000 system.
CRISPR-Cas9
As described previously [21], CRISPR-Cas9-mediated CRBN KO 293 T cells were generated. Briefly, CRISPR-Cas9-related oligonucleotides were designed as follows: forward primer: 5′-CACCGCACCATACTGACTTCTTGA-3′; reverse primer: 5′-AAACTCAAGAAGTCAGTATGGTGC-3′. Guide RNA was cloned into PX459 plasmids, and then, the recombinant plasmids were validated by sequencing. Next, 293T cells were transfected with the PX495 guide RNA constructs. After 24 h, enrichment and isolation of the 293T cells transfected with the CRBN guide RNA were performed. Finally, the 293T CRBN−/− cells were confirmed by Western blotting and DNA sequencing.
Cellular immunoprecipitation
Cells stably expressing Flag-tagged BNIP3L were transfected with C-myc-tagged CRBN for 24 h and then treated with DMSO, 1 μM CC-885 or 10 μM CC-885 for 24 h in the presence of 2 μM MLN4924. Then, the cells were harvested and lysed with IP buffer (20 mM Tris (pH 8.0), 100 mM NaCl, 0.5% NP-40, 0.5 mM PMSF, 0.5 mM EDTA) and incubated on ice for half an hour. The lysates were centrifuged at 20,000 × g for 10 min at 4 °C after sonication. The supernatant was transferred to another tube. Anti-Flag beads were added to the lysates and incubated together at 4 °C overnight after washing three times with IP buffer. The anti-Flag beads were washed with IP buffer six times and resuspended in loading buffer. The bound proteins were eluted by boiling for 10 min and detected by Western blotting analysis.
In vivo ubiquitination
For analysis of the ubiquitination of BNIP3L, the 293T cells stably expressing Flag-tagged BNIP3L were treated with DMSO, 1 μM CC-885 or 10 μM CC-885 for 6 h in the presence of 10 μM MG132. Next, the cells were harvested and lysed with IP buffer containing 50 mM Tris-HCl, 0.15 M NaCl, 10% glycerol, 1% NP-40, 1 mM EDTA, 40 μM PR-619, and 10 μM MG132. Then, the ubiquitinated proteins were pulled down by TUBE2 Agarose (Boston Biochem) overnight at 4 °C. The ubiquitinated proteins were eluted by boiling with loading buffer for 5 min after washing four times with IP lysis buffer and finally detected by Western blotting analysis.
Real-time Q-PCR
Total RNA was extracted from A549 cells treated with or without CC-885 using TRIzol reagents. The purified RNA was reverse-transcribed by PrimeScript RT Master Mix. The cDNA was quantified with a SYBR® Premix ExTaq II kit. The data were analyzed using actin as the control. The BNIP3L primer sequences used in the above assays were as follows: forward primer: 5′-CATCCTCATCCTCCATCCAC-3′; reverse primer: 5′-GATCTGCCCATCTTCTTGTG-3′.
Assessment of mitophagy
HeLa cells or HCT116 cells (300,000 cells) were transfected with GFP-tagged LC3. After 24 h, 20,000 HeLa cells or HCT116 cells expressing GFP-LC3 were seeded onto a 24-well dish. Next, the cells were treated with 10 μM CCCP for 4 h after treatment with DMSO or 1 μM CC-885 for 24 h. The cells were then incubated with CellLight Mitochondria-RFP BacMan 2.0 (Invitrogen) for 16 h to label the mitochondria before the cells were harvested according to the manufacturer’s instructions. Then, the cells were imaged by a Leica TCS SP5 II confocal microscope (Leica Microsystems). The relative quantification was performed by using ImageJ software. Statistical differences were determined by Student’s t test.
Results
Identification of potential CRBN-dependent neosubstrates of CC-885 by quantitative proteomics
Our preliminary experiment showed that the novel thalidomide derivative CC-885 had potent antitumor activity, as reported in Supplementary Fig. S1. To provide a global view of the CC-885-regulated proteins, we carried out a mass spectrometry (MS)-based quantitative proteomics analysis based on stable-isotope labeling with amino acids in cell culture (SILAC) [22] in A549 cells with or without CC-885 (Fig. 1a). In total, 7353 proteins were identified in two biological replicates. Of these identified proteins, 5029 proteins were quantified (Fig. 1b). Correlation analysis found that the two replicates showed a high correlation (R2 = 0.71), which demonstrates good reproducibility between the two replicates (Fig. 1c).

Analysis of SILAC-based quantitative proteomics. a Workflow of the SILAC-based quantified proteomics. b Overlap of the quantified proteins in two replicates. c The correlation of the quantified proteins in two replicates. The x-axis indicates the log2 ratios of protein abundance in one replicate, and the y-axis indicates another replicate
A P value < 0.01 and fold change > 2 were set as a cutoff point to identify the high-confidence candidate degradation substrates between the DMSO- and CC-885-treated groups (Supplementary Fig. S2 and Supplementary Table S1). Of these quantified proteins, 36 proteins were downregulated, which may contain the potential neosubstrates of CC-885 (Fig. 2a). Consistent with a recent study, the only known CC-885 substrate, GSPT1, was successfully identified in our protein list. To gain insights into the biological function of CC-885, we conducted a bioinformatic analysis of the proteome data by gene set enrichment analysis (GSEA) with the Gene Ontology (GO) database [20]. Interestingly, we found that the macromitophagy pathway, regulation of the extent of cell growth and positive regulation of developmental growth was significantly enriched in the negative pathway after CC-885 treatment (Fig. 2b and Supplementary Table S2). As CC-885-induced protein degradation was dependent on cereblon (CRBN) [12], we then attempted to identify the degraded proteins in our list that were regulated by CRBN. Toward this goal, we generated CRBN−/− 293T cells by the CRISPR/Cas9 system (Supplementary Fig. S3a). Then, we used this cell line to perform proteome-wide protein quantification experiments via MS-based quantitative proteomics with or without CC-885 treatment (Supplementary Table S3). We found that 19 proteins, including the known substrate GSPT1, were unchanged in the CRBN−/− cells following CC-885 treatment (Fig. 2c and Supplementary Table S4). Thus, our results suggested that these proteins represented potential CRBN-dependent neosubstrates of CC-885.

Identification of the CC-885-induced neosubstrate BNIP3L. a Log2 ratios of protein abundance of the CC-885- vs. DMSO-treated A549 cells. Each plot indicates a distinct protein group. Data are representative of two replicates. b Bioinformatic analysis of the protein levels in the A549 cells treated with CC-885. c Venn diagram representing the overlap between the downregulated proteins in the CRBN+/+ cells and the stably expressed proteins in the CRBN−/− cells after treatment with CC-885. d The MS/MS spectrum of a unique peptide of the BNIP3L protein
CC-885 induces the proteasome-dependent degradation of BNIP3L
As macromitophagy could be regulated by CC-885 treatment, we focused on proteins with regulatory roles in macromitophagy. Interestingly, we found that the macromitophagy master protein BNIP3L was downregulated in the CRBN+/+ cells after treatment with CC-885 but not in the CRBN−/− cells. Thus, we hypothesized that BNIP3L could be a critical neosubstrate of CRBN induced by CC-885 in the macromitophagy pathway.
To validate our hypothesis, we initially verified the reliability of the identified BNIP3L protein by manually assessing the MS/MS spectrum of the unique peptides of the BNIP3L protein (Fig. 2d). Subsequently, we monitored the changes in the BNIP3L protein levels after CC-885 treatment at different concentrations for 24 h by Western blotting analysis. We found that CC-885 decreased the BNIP3L protein levels in a dose-dependent manner (Fig. 3a). However, the mRNA levels of BNIP3L were unaltered after CC-885 treatment (Fig. 3b), suggesting that protein downregulation induced by CC-885 is a post-transcriptional mechanism. To further rule out the possibility of transcriptional regulation, we generated 293T cells stably expressing Flag-tagged BNIP3L. Similarly, CC-885 treatment time-dependently decreased the Flag-BNIP3L protein levels, which occurred at 6 h after CC-885 treatment (Fig. 3c and Supplementary Fig. S3b). Consistent with these observations, the half-life of the Flag-BNIP3L protein was significantly decreased in the presence of CC-885 (Fig. 3d). Interestingly, other IMiDs, such as lenalidomide and thalidomide, which have minimal anti-solid tumor activity, failed to degrade the BNIP3L protein (Fig. 3e), suggesting that BNIP3L could be specifically induced for degradation by CC-885.

Effect of CC-885 on the BNIP3L protein levels. a Western blotting analysis of the endogenous BNIP3L level in the A549 cells treated with CC-885 at different concentrations for 24 h. b The real-time qPCR analysis of BNIP3L in the A549 cells treated with DMSO or CC-885 for 12 h. Data are representative of two replicates. Data are the mean ± SEM . c Western blotting analysis of the 293T cells stably transfected with Flag-tagged BNIP3L after treatment with 1 μM CC-885 for the indicated times. d Western blotting analysis of the 293T cells transfected with Flag-tagged BNIP3L treated with cycloheximide (CHX) and CC-885 for the indicated times. Cells were pretreated with CC-885 for 2 h (top). Statistical results of Western blotting analyses were obtained by ImageJ and normalized to actin intensities (ratio of Flag-BNIP3L to actin). Data are the mean ± SEM, n = 3 technical replicates (bottom). e Western blotting analysis of the 293T cells stably transfected with Flag-tagged BNIP3L after treatment with CC-885, thalidomide or lenalidomide for 24 h
To further test whether the CC-885-induced BNIP3L protein reduction is mediated by the proteasome pathway, we assessed the effect of the proteasome inhibitor MG132 and NEDD8-activating enzyme (NAE) inhibitor MLN4924 [23] after CC-885 treatment. We found that MG132 completely blocked the CC-885-induced reduction of BNIP3L, as did MLN4924, suggesting that CC-885-induced proteasomal-dependent degradation of BNIP3L is mediated by a Cullin-based ubiquitination E3 ligase (Fig. 4a). It has recently been shown that valosin-containing protein (VCP)/p97 is required for IMiD-induced degradation of CRBN neosubstrates [24]. We found that the p97 inhibitor CB-5083 could also block the CC-885-induced BNIP3L degradation, further suggesting the involvement of CRBN in this process (Fig. 4a). By using a TUBE2 pulldown assay, we further found that CC-885 induced the ubiquitination of BNIP3L (Fig. 4b). Together, these data suggested that CC-885 could specifically induce the proteasomal-dependent degradation of BNIP3L via a Cullin-based E3 ligase.

CC-885 induces the degradation of BNIP3L by the Cullin-RING CRBN E3 ligase. a Western blotting analysis of the Flag-tagged BNIP3L protein levels in the 293T cells treated with CC-885 or in the presence of 2 μM MLN4924, 2 μM MG132 or 2 μM CB-5083 for 24 h. b Ubiquitination of the Flag-tagged BNIP3L in the 293T cells after treatment with DMSO, 1 μM or 10 μM CC-885 in the presence of 2 μM MG132. Ubiquitinated proteins were analyzed by Western blotting with an anti-Flag antibody. c Immunoblotting analysis of the 293T parental or CRBN−/− cells transfected with the Flag-tagged BNIP3L after treatment with DMSO or CC-885 for 24 h. d Immunoprecipitation of the Flag-tagged BNIP3L in the 293T cells transfected with Myc-tagged CRBN after treatment with DMSO, 1 μM or 10 μM CC-885 in the presence of 2 μM MLN4924. IP Immunoprecipitation
BNIP3L is a CRL4CRBN-dependent neosubstrate of CC-885
The abovementioned data suggested that the CC-885-dependent BNIP3L degradation could be mediated by the CRL4CRBN E3 ligase. Indeed, CC-885 treatment failed to decrease the endogenous BNIP3L protein level in the CRBN−/− cells, suggesting that CRBN is required for the CC-885-induced BNIP3L degradation. Interestingly, the BNIP3L protein levels were not changed in the CRBN−/− cells, suggesting that BNIP3L is not a native substrate of CRBN in cell lines cultured in vitro (Fig. 4c). Importantly, BNIP3L was not associated with CRBN in the cells that were not treated with CC-885. However, in the presence of CC-885, the interaction between BNIP3L and CRBN was readily detected, suggesting that CC-885 could bridge their interaction (Fig. 4d). Thus, these data indicated that BNIP3L is a CRL4CRBN-dependent neosubstrate of CC-885.
CC-885 abrogates the BNIP3L-dependent mitophagy
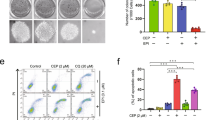
BNIP3L is partially located in the outer mitochondrial membrane and is a master regulator of mitophagy [25]. As BNIP3L downregulation caused inhibition of mitophagy and CC-885 could induce rapid ubiquitination and degradation of BNIP3L, we next examined whether BNIP3L-mediated mitophagy could be abrogated by CC-885 treatment. We detected the level of mitophagy using colocalization between mitochondria (Red Fluorescent Protein (RFP-mito)) and autophagosomes (Green Fluorescent Protein-tagged microtubule-associated protein light chain (GFP-LC3)) by confocal microscopy in the HCT116 cells after sequential treatment with CC-885 and CCCP (carbonyl cyanide m-chlorophenyl hydrazone), the latter of which is a classical mitochondrial fragmentation and mitophagy inducer. CCCP treatment induced colocalization between GFP-LC3 and RFP-mitochondria in the HCT116 cells, which was significantly prevented in the presence of CC-885 (Fig. 5a, b and Supplementary Fig. S4). Together, these results demonstrated that CC-885 can abrogate mitophagy by targeting BNIP3L for destruction (Fig. 5c).

CC-885 abrogates the BNIP3L-mediated mitophagy and enhances the sensitivity of AML cells to mitochondria-targeting drugs. a Live cell imaging of RFP-Mito (red fluorescence in the left position) and GFP-LC3 (green fluorescence in the middle position) in the HCT116 cells. Merge indicates the colocalization of RFP-Mito and GFP-LC3 (yellow signals in the right position) under different treatments in the HCT116 cells. b Statistical results of the confocal assays were obtained by ImageJ. Data are the mean ± SEM, n = 3 technical replicates. ***P < 0.01. c Diagram of the BNIP3L-mediated mitophagy abrogated by CC-885. d, e The results of the cell viability assays. Cells were treated with CC-885 and rotenone, alone or in combination for 48 h. Data are shown as the mean ± SEM, n = 2 replicates
CC-885 enhances the sensitivity of AML cells to mitochondria-targeting drugs
A recent study demonstrated that AML cells deficient in BNIP3L show increased sensitivity to mitochondria-targeting drugs [8]. Our findings confirmed that CC-885 could induce the ubiquitination and degradation of BNIP3L by targeting the CRL4CRBN E3 ligase. Next, we explored whether CC-885 treatment could increase the sensitivity of AML cells to mitochondria-targeting drugs. We treated NB4 cells with the mitochondria-targeting compound rotenone alone or in the presence of CC-885. The results indicated that the NB4 cells were more sensitive to rotenone after cotreatment with CC-885 and that rotenone could also enhance the cytotoxicity of CC-885 (Fig. 5d, e), which suggested that CC-885 could enhance the sensitivity of AML cells to mitochondria-targeting drugs.
Discussion
Targeting the degradation of specific proteins via the ubiquitin–proteasome pathway opens a new area for cancer therapy. Recently, immunomodulatory drugs (IMiDs), such as thalidomide, lenalidomide, and pomalidomide, have been demonstrated to have clinical efficacy in hematologic malignancies by the ubiquitination and degradation of specific proteins via the CRL4CRBN E3 ligase. CC-885, a novel thalidomide derivative, exhibited potent anti-solid tumor efficacy via the specific degradation of the translation termination factor GSPT1 [12]. However, it is unknown whether CC-885 targets additional substrates for degradation.
In the present study, by using mass spectrometry-based quantitative proteomic technology, we systematically screened the neosubstrates of the thalidomide derivative CC-885, which has potent antitumor activity [12]. Thirty-six proteins were downregulated upon CC-885 treatment. These data along with bioinformatic analysis showed that the pathway related to macromitophagy was significantly enriched in the negative pathway after CC-885 treatment. To identify the CRBN-dependent neosubstrates, we generated CRBN−/− cells by CRISPR/Cas9 methods. These cells were SILAC labeled, treated with DMSO or CC-885 and subjected to MS spectrum analysis. Our proteomic data suggested that 19 proteins, such as BNIP3L, UBE2S, and UBE2C, were downregulated in the CRBN+/+ cells but not in the CRBN−/− cells after treatment with CC-885. Western blotting data also confirmed that CC-885 treatment could decrease these exogenously expressed proteins in the CRBN+/+ cells (Supplementary Fig. S5), which represented the high-confidence neosubstrates of CC-885. Given the critical role of BNIP3L in the regulation of mitophagy, we then selected BNIP3L for further investigation and proved that BNIP3L is a bona fide neosubstrate of CC-885. First, consistent with the MS data, CC-885 induced rapid BNIP3L destruction in both a time- and dose-dependent manner. Second, CC-885 failed to decrease the BNIP3L protein level when CRBN was depleted, indicating that CRBN is required for CC-885-induced BNIP3L degradation. Third, CC-885 bridges the interaction between BNIP3L and CRBN.
Mitophagy is a process that eliminates damaged and redundant mitochondria. BNIP3L is located in the mitochondrial membrane and is a crucial protein during mitophagy [25]. The rapid degradation of BNIP3L by CC-885 drove us to ask whether CC-885 could repress BNIP3L-dependent mitophagy. Indeed, CC-885 treatment significantly prevented CCCP-induced mitophagy, as shown by confocal microscopy assays. Recently, targeting protein degradation has been shown to be a novel strategy for drug development [26, 27], and a previous study demonstrated that knockdown of BNIP3L would increase the sensitivity to mitochondrial drugs in AML patients [8]. Our above data suggest that CC-885 might be used to treat AML patients combined with mitochondria-targeting drugs by targeting BNIP3L degradation. Thus, further work is warranted to expend the clinical use of this potent antitumor thalidomide derivative.
A recent structural study revealed that substrates could directly bind to CRBN and thalidomide derivative complexes by two common domains, the Cys2-His2 (C2H2) zinc finger “degrome” or a beta-sheet hairpin including a key glycine, which leads to several substrates for ubiquitination and degradation, such as IKZF1, IKZF3, ZFP91, SALL4, CK1a, and GSPT1 [12, 14, 16, 17, 28,29,30]. Therefore, we analyzed the potential substrates we identified by sequence BLAST analysis, but unfortunately, none of these proteins, including BNIP3L had a common domain. Nevertheless, our biochemical results demonstrated that BNIP3L could still directly bind to CRBN in the presence of CC-885. In addition, a molecular docking model showed that hydrophobic interactions might exist between F181/F185/F189 in the BNIP3L protein and the CRBN-CC-885 complex, which suggested that F181/F185/F189 in the BNIP3L protein might directly bind to the CRBN-CC-885 complex (Supplementary Fig. S6). Thus, further works are important to identify the unknown “degron” of BNIP3L recognized by CRBN.
Together, our results demonstrated that the thalidomide analog CC-885 could abrogate mitophagy by inducing the ubiquitination and degradation of BNIP3L, representing a promising strategy to treat AML patients by combining CC-885 with mitochondria-targeting drugs.
Data availability
The proteomics data in this study have been uploaded to the iProx database (project ID: IPX0001589000).
References
O’Sullivan TE, Johnson LR, Kang HH, Sun JC. BNIP3- and BNIP3L-mediated mitophagy promotes the generation of natural killer cell memory. Immunity. 2015;43:331–42.
Lazarini M, Machado-Neto JA, Duarte AD, Pericole FV, Vieira KP, Niemann FS, et al. BNIP3L in myelodysplastic syndromes and acute myeloid leukemia: impact on disease outcome and cellular response to decitabine. Haematologica. 2016;101:e445–8.
Gustafsson AB, Dorn GW 2nd. Evolving and expanding the roles of mitophagy as a homeostatic and pathogenic process. Physiol Rev. 2019;99:853–92.
Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104:19500–5.
Diwan A, Koesters AG, Odley AM, Pushkaran S, Baines CP, Spike BT, et al. Unrestrained erythroblast development in Nix-/- mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc Natl Acad Sci U S A. 2007;104:6794–9.
Gustafsson AB. Bnip3 as a dual regulator of mitochondrial turnover and cell death in the myocardium. Pediatr Cardiol. 2011;32:267–74.
Chen Y, Lewis W, Diwan A, Cheng EH, Matkovich SJ, Dorn GW 2nd. Dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc Natl Acad Sci U S A. 2010;107:9035–42.
Rodrigo R, Mendis N, Ibrahim M, Ma C, Kreinin E, Roma A, et al. Knockdown of BNIP3L or SQSTM1 alters cellular response to mitochondria target drugs. Autophagy. 2018;15:900–7.
Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddlemon P, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N. Engl J Med. 1999;341:1565–71.
Rasco DW, Papadopoulos KP, Pourdehnad M, Gandhi AK, Hagner PR, Li Y, et al. A first-in-human study of novel cereblon modulator avadomide (CC-122) in advanced malignancies. Clin Cancer Res. 2019;25:90–8.
Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343:305–9.
Matyskiela ME, Lu G, Ito T, Pagarigan B, Lu CC, Miller K, et al. A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature. 2016;535:252–7.
Zhu YX, Braggio E, Shi CX, Bruins LA, Schmidt JE, Van Wier S, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood. 2011;118:4771–9.
Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301–5.
Fischer ES, Bohm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512:49–53.
Kronke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, et al. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature. 2015;523:183–8.
An J, Ponthier CM, Sack R, Seebacher J, Stadler MB, Donovan KA, et al. pSILAC mass spectrometry reveals ZFP91 as IMiD-dependent substrate of the CRL4(CRBN) ubiquitin ligase. Nat Commun. 2017;8:15398.
Wang Y, Yang F, Gritsenko MA, Wang Y, Clauss T, Liu T, et al. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics. 2011;11:2019–26.
Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–72.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50.
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–308.
Ong S-E, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteom. 2002;1:376–86.
Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–6.
Nguyen TV, Li J, Lu CJ, Mamrosh JL, Lu G, Cathers BE, et al. p97/VCP promotes degradation of CRBN substrate glutamine synthetase and neosubstrates. Proc Natl Acad Sci U S A. 2017;114:3565–71.
Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51.
Deshaies RJ. Protein degradation: prime time for PROTACs. Nat Chem Biol. 2015;11:634–5.
Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, et al. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–81.
Sievers QL, Petzold G, Bunker RD, Renneville A, Slabicki M, Liddicoat BJ, et al. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science. 2018;362:eaat0572.
Matyskiela ME, Couto S, Zheng X, Lu G, Hui J, Stamp K, et al. SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat Chem Biol. 2018;14:981–7.
Donovan KA, An J, Nowak RP, Yuan JC, Fink EC, Berry BC, et al. Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. Elife. 2018;7:e38430.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81872888, No. 91753203, No. 81773018), the Natural Science Foundation of China for Innovation Research Group (No. 81821005), and the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program” (No. 2018ZX09711002-004).
Author information
Authors and Affiliations
Contributions
MJT and BL conceived the project and designed the experiments. BBH performed the proteomic experiments and data analysis with the help of MJT, GFX, and LHZ. BBH, XJL, XLJ, YXW, DZL, JL, DZ, YLC, YHX, and SKL carried out other experiments with the help of XHC, BL and YJD. BL and BBH wrote the manuscript, and MJT revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Hao, Bb., Li, Xj., Jia, Xl. et al. The novel cereblon modulator CC-885 inhibits mitophagy via selective degradation of BNIP3L. Acta Pharmacol Sin 41, 1246–1254 (2020). https://doi.org/10.1038/s41401-020-0367-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-0367-9
Keywords
This article is cited by
-
Leukemia and mitophagy: a novel perspective for understanding oncogenesis and resistance
Annals of Hematology (2024)
-
Proteomic characterization of post-translational modifications in drug discovery
Acta Pharmacologica Sinica (2022)
-
Targeting CRL4 suppresses chemoresistant ovarian cancer growth by inducing mitophagy
Signal Transduction and Targeted Therapy (2022)
-
Key regulators of sensitivity to immunomodulatory drugs in cancer treatment
Biomarker Research (2021)
-
Neddylation regulation of mitochondrial structure and functions
Cell & Bioscience (2021)
