 Tiina S. Salminen
Tiina S. Salminen Pedro F. Vale
Pedro F. Vale- 1School of Biological Sciences, Institute of Evolutionary Biology, The University of Edinburgh, Edinburgh, United Kingdom
- 2Faculty of Medicine and Health Technology, Tampere University, Tampere, Finland
Understanding why the response to infection varies between individuals remains one of the major challenges in immunology and infection biology. A substantial proportion of this heterogeneity can be explained by individual genetic differences which result in variable immune responses, and there are many examples of polymorphisms in nuclear-encoded genes that alter immunocompetence. However, how immunity is affected by genetic polymorphism in an additional genome, inherited maternally inside mitochondria (mtDNA), has been relatively understudied. Mitochondria are increasingly recognized as important mediators of innate immune responses, not only because they are the main source of energy required for costly immune responses, but also because by-products of mitochondrial metabolism, such as reactive oxygen species (ROS), may have direct microbicidal action. Yet, it is currently unclear how naturally occurring variation in mtDNA contributes to heterogeneity in infection outcomes. In this review article, we describe potential sources of variation in mitochondrial function that may arise due to mutations in vital nuclear and mitochondrial components of energy production or due to a disruption in mito-nuclear crosstalk. We then highlight how these changes in mitochondrial function can impact immune responses, focusing on their effects on ATP- and ROS-generating pathways, as well as immune signaling. Finally, we outline how being a powerful and genetically tractable model of infection, immunity and mitochondrial genetics makes the fruit fly Drosophila melanogaster ideally suited to dissect mitochondrial effects on innate immune responses to infection.
Introduction
Understanding why individuals vary in their response to infection is one of the major challenges in immunology (1, 2). This variation may arise from differences in host age (3), sex (4), nutrition or environmental stressors (5), and genetic variation present in immune related genes (2). Experimental immunology – mainly in model systems such as mice, zebra fish and fruit flies – has been successful in identifying the major immune pathways (Drosophila innate immunity summarized in Box 1) (6–8). Quantitative genetic and genomic approaches have identified polymorphisms in genes underlying these mechanisms, and these explain some of the variation in infection outcomes (9–12). While most of this work has focused on genetic variation in the nuclear genome, metazoan organisms have an additional genome, inherited maternally inside mitochondria (mtDNA). More than functioning as the powerhouses of the cell, a growing body of work in the last decade has shown that mitochondria play an important role in inflammation and immunity and contribute to the host response to infection (13–18).
BOX 1.
Drosophila innate immunity in a nutshell. Drosophila has been extensively utilized as a model system for innate immunity and it has led to many breakthrough in immunity field (6, 114, 115). Drosophila does not possess acquired/adaptive immunity and it relies on humoral and cell-mediated innate immunity for its defense against pathogens, such as bacteria, viruses, fungi, and parasites. Immune mechanisms against these invaders include activation of appropriate signal transduction pathways depending on the invading microbe, involving production of antimicrobial peptides (AMPs), phagocytosis of microbes, wound closure, and a melanization cascade involved in the encapsulation of foreign elements. Similar first-line innate immune defense mechanisms can be found from plants to humans.
Humoral Innate Immunity: In Drosophila, the humoral innate immune response to bacterial pathogens is characterized by the production and release of a cocktail of AMPs into the hemolymph. This response is driven by two evolutionarily conserved and largely independent pathways, Immune deficiency (IMD) and Toll pathways (116). The Toll pathway is induced by bacteria containing LYS-type peptidoglycan in their cell walls (mainly Gram-positive bacteria), while the IMD pathway is induced by DAP-type peptidoglycan (mainly Gram-negative) bacteria. These pathways culminate in the translocation of NF-κB dimers to the nucleus leading to infection-specific upregulation of AMPs targeted to clear the infection (117–119). The response to viral pathogens replicating within the host cells involves both the Janus kinase/signal transducers and activators of transcription (JAK-STAT) pathway (118, 120), RNA interference (RNAi) and antiviral effector molecules (121, 122). Viral infections involve cell-mediated responses like apoptosis and autophagy and humoral responses such as the expression of anti-viral genes, some of which overlap with genes induced upon bacterial and fungal infections, indicating the involvement of the NF-κB signaling upon viral infections. The response to fungal invaders includes both humoral and cellular arms of immunity and involves the expression of AMPs mainly via the Toll pathway.
Cell-Mediated Innate Immunity: In Drosophila, the cell-mediated innate immune system consists of hemocytes (blood cells) and is induced by epithelial damage and detection of foreign particles in the hemocoel. Hemocytes function in sealing of epithelial wounds, encapsulating and terminating parasites and engulfing apoptotic corpses [reviewed in (123)]. In Drosophila there are three major lineages of hemocytes: plasmatocytes (phagocytic), crystal cells (melanization) and lamellocytes (encapsulation). Plasmatocytes comprise the majority of the circulating hemocyte population and are responsible for the engulfment of small particles, participate in the encapsulation of foreign material and are able to trigger the systemic humoral immune response to secrete AMPs. Crystal cells usually make up less than 5% of the larval circulating hemocytes. Crystal cells contain prophenol oxidase which active form phenol oxidase is involved in the melanization cascade when the crystal cells rupture in response to immune activation (123). In uninfected larvae, the lamellocytes can be present in small numbers in the late third instar stage, otherwise healthy larvae do not contain them. Lamellocytes are produced upon invasion of parasitoid wasps and they form a multilayer capsule around the invading parasitic egg, with the help of plasmatocytes and crystal cells (124). Eventually the capsule is melanized and elevated levels of ROS terminate the intruder (125).
Here, we propose that the fruit fly (Drosophila melanogaster) offers an ideal model system to investigate the role of mitochondrial variation and mito-nuclear crosstalk in innate immunity. We start by discussing the sources of variation in mitochondrial function, using examples of mutations of nDNA and mtDNA encoded genes that have been shown to affect organismal phenotypes through changes in mitochondrial metabolism and signaling. This is followed by emphasizing the emerging role of mitochondria in immune responses through mitochondrial metabolites and by-products of mitochondrial metabolism, such as ROS. Finally, we describe methodology to investigate the role of mito-nuclear crosstalk and mtDNA variation in immunity in Drosophila. We emphasize how the use of cytoplasmic hybrid (cybrid) models allows to distinguish the effect of mtDNA variation from that arising from the nuclear genome. We conclude by highlighting the benefits of the cybrid model to further our understanding of mito-nuclear effects on heterogenous immune responses.
Sources of Mitochondrial Variation
Mitochondrial function depends on ∼1200 – 1500 proteins, the majority of which are encoded by the nuclear genome and transported to the mitochondria (19). Cellular energy production relies on mitochondria to produce ATP via oxidative phosphorylation (OXPHOS). OXPHOS requires the coordinated function of multiple protein subunits encoded by both the nuclear and mitochondrial genomes (nDNA and mtDNA, respectively – Box 2), and therefore both anterograde (from nucleus to mitochondria) and retrograde (from mitochondria to nucleus) signaling is required for optimal mitochondrial function. Mitochondrial variation arising from either nDNA or mtDNA can affect the transcription and translation of the mitochondrial proteins, signaling between the two genomes and through changes in the direct physical interactions among the OXPHOS components originating from the two genomes, ultimately affecting the function of mitochondria. Mitochondrial variation shows multiple mode of inheritance. When this variation originates from mtDNA it is maternally inherited and has a potential to become heteroplasmic even within mitochondria, and when originating from nuclear genome it can be X-linked, autosomal dominant, autosomal recessive or de novo. Here, we discuss potential sources of mitochondrial variation with examples of known nuclear and mitochondrial mutations that could also lead to variation in immune responses.
BOX 2.
Mito-nuclear crosstalk is required for mitochondrial functions. Mitochondria are cellular organelles of eukaryotic cells that are thought to have originated by endosymbiotic phagocytosis of an oxygen-converting α-proteobacterium by archaebacterium (19, 126). The primary function of mitochondria is to produce ATP through oxidative phosphorylation (OXPHOS) complexes I-V, and mitochondrial matrix is also the site of tricarboxylic acid cycle. Mitochondria contains multiple copies of a circular mtDNA (mtDNA copy number) distinct from that of the nuclear genome (Figure 1). Majority of the mtDNA genes required for aerobic energy production through OXPHOS have been shifted to the nuclear chromosomes and the remaining mitochondrial genome in most metazoans encodes for 37 genes, all crucial in OXPHOS. From these, 13 are polypeptide subunits of four of the five OXPHOS complexes, with the majority of the polypeptides encoded by nDNA (Figure 1B). Mitochondria contains its own translational system and the mtDNA encodes two rRNA and 22 tRNA genes as the mt-aaRS genes are encoded by the nuclear genome. Beside the 84 nDNA genes functioning in OXPHOS, around 1200-1500 nDNA encoded polypeptides are imported to and assembled within mitochondria, required for the various mitochondrial functions (Figure 1A). Nucleus and the mitochondria maintain a bidirectional regulation where the nuclear genome can signal to the mitochondria (anterograde signaling) for example to increase mitochondrial respiration. Mitochondria can signal (retrograde signaling) for example to induce cell death by releasing cytochrome c, or by controlling mitochondrial fusion and fission by AMP-activated protein kinase (57).
Variation Arising From the Nuclear Genome
The vast majority of the proteins that are required for mitochondrial functions are encoded by the nuclear genome, translated in the cytosol and transported to mitochondria via mitochondrial targeting sequence which is removed upon entry into mitochondria. These proteins include the replication, transcription and translation machineries for mtDNA and the 84 polypeptide subunits needed for OXPHOS (summarized in Box 2). Mutations in nDNA directly affecting OXPHOS complex genes have been reviewed in (20). Maintenance genes of mitochondrial functions include regulatory genes of mitochondrial and cytosolic nucleotide pools to maintain balanced supply of mitochondrial dNTPs, involved with mtDNA nucleoid packaging, carrier proteins required for metabolite and cofactor transport across cellular and mitochondrial membranes, genes for mitochondrial lipid and membrane homeostasis, and mitochondrial fission/fusion and cristae organization [reviewed in (21)].
Mutations in mtDNA maintenance genes (replication and repair pathways and mtDNA nucleoid packaging) have been shown to cause mtDNA deletions, point mutations and even depletion (22). The most important mtDNA maintenance gene is the DNA polymerase gamma (POLG) which is responsible for the replication of the mtDNA. Almost 200 POLG mutations have been reported and these are the most common causes of mitochondrial disease. Mutations in POLG have been shown to cause large scale deletions and various other mutations to the mtDNA due to replication and/or repair machinery malfunctions and these have been connected to many mitochondrial diseases such as Alper’s syndrome, parkinsonism and multiple other neurodegenerative disorders (23). POLG was mutated in Drosophila to create a proofreading-deficient form resulting to drastically increased somatic mtDNA mutation frequency and mitochondrial dysfunction, which manifested as a shortened lifespan, a progressive locomotor deficit and a loss of dopaminergic neurons (24).
Regulation of OXPHOS gene transcription is tightly coordinated and must be able to establish efficient oxidative metabolism fulfilling the cell’s changing energy requirements. Components of the transcription machinery are encoded by the nuclear genome and have been reviewed in (25). Mossman et al., showed that in D. melanogaster cybrid lines the transcription of nuclear encoded mitochondrial genes were affected by mtDNA variation, indicating a retrograde signaling effect in transcription regulation (26). Hence, mutations in the transcription machinery can have a wide impact on the function of mitochondria, and variation in the mtDNA genes can affect the overall transcription efficiency of OXPHOS components, possibly also affecting their translation.
All proteins involved in mtDNA translation (27) are encoded by nuclear genes, involving ribosomal proteins, mt-aaRSs, tRNA modifying enzymes, and translation factors. Mutations in these genes have been shown to cause mitochondrial diseases due to dysfunction in the protein-synthesis machinery (27). mt-aaRSs are transported to mitochondria to catalyze an amino acid attachment to its complementary tRNA in aminoacylation reaction for translation of the thirteen mitochondrial proteins. All ribosomal RNAs and the transfer RNAs required for the translation of the mitochondrial proteins are encoded by the mtDNA (Figure 1). Hence, nuclear encoded components of the translation machinery need to be able to recognize mitochondrial counterparts for the production of mitochondrial proteins. mt-aaRS genes are central to cellular energy production and mutations in these can lead to variable disease phenotypes depending on the affected tissues and the energy demands of the cells in those tissue types (28). Mutations in both mt-tRNAs and mt-aaRSs can lead to disease and the clinical presentation has been shown to be highly specific to the affected mt-aaRS [reviewed in (29)]. However, diversity of pathologies is higher for mt-tRNA mutations than mt-aaRSs, possibly due to random distribution of heteroplasmic populations of mtDNA copies during mitotic segregation (28). In Drosophila simulans a variant of tyrosyl-tRNA synthetase interacts epistatically with a mitochondrially encoded tRNATyr variant, leading to decrease in the activities of OXPHOS complexes I, III, and IV (30). At the organismal level this manifests as developmental delay, compromised bristle formation and decreased fecundity (30).
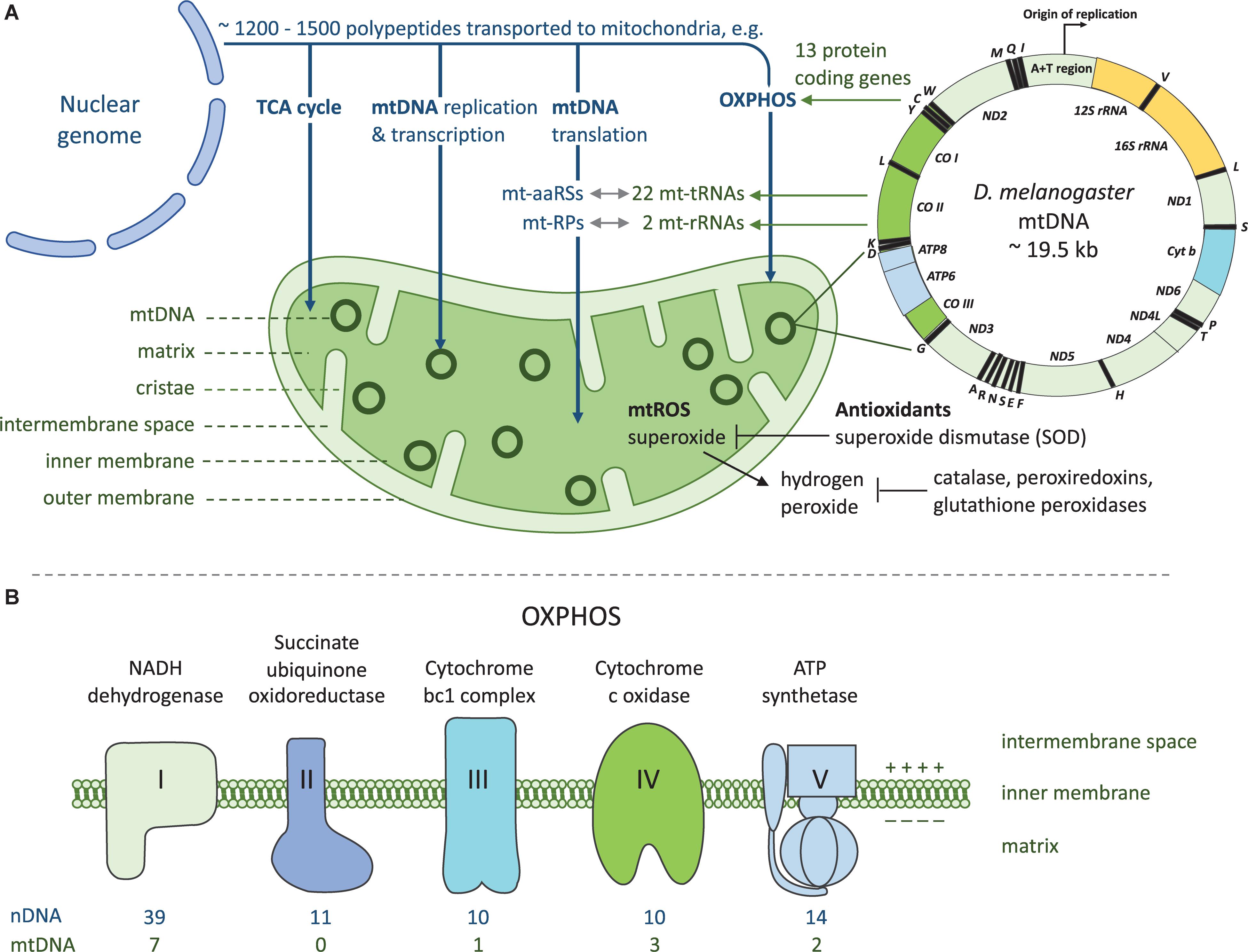
Figure 1. Mito-nuclear crosstalk in mitochondrial functions and energy production. (A) Mitochondria rely on coordinated functions of nuclear and mitochondrial genomes. Vast majority of the genes required for mitochondrial functions are encoded by the nuclear genome. These proteins are translated in the cytoplasmic compartment and transported into the mitochondrion post-translationally with a mitochondrial targeting signal. Products of both genomes are required for producing cellular energy in the form of ATP through OXPHOS. As a by-product of OXPHOS mitochondria produces reactive oxygen species (ROS) which are detoxified by antioxidants. mt-aaRSs, mitochondrial aminoacyl-tRNA synthetases; mt-RPs, mitochondrial ribosomal proteins. (B) OXPHOS takes place at the inner mitochondrial membrane and it comprises five enzyme complexes. Both nuclear and mitochondrial encoded proteins are required for OXPHOS complexes I and III-V, as complex II contains only nuclear encoded subunits. Complexes I-IV and two electron carriers form the respiratory chain which generates a proton gradient used by complex V to generate ATP.
In D. melanogaster, a tko mutant (tko25t) carries a missense mutation in nuclear encoded mitoribosomal protein S12 causing a decrease in smaller ribosomal subunits. This causes decreased activity levels of all four OXPHOS complexes that contain mitochondrial encoded proteins (Figure 1), ultimately manifesting as developmental delay, temporary paralysis followed from vigorous shaking (bang sensitivity) and sensitivity to antibiotics and high sugar diet as well as impaired courtship behavior and hearing (31, 32). Similar defects have been found in fibroblasts of patients with antenatal encephalopathy caused by mutations in the MRPS22 gene coding mitochondrial ribosomal protein, which result in a reduction of 12s rRNA (33).
Variation Arising From the Mitochondrial Genome
The effect of the mitochondrial genome variation on innate immunity is intriguing as mtDNA does not follow the traditional Mendelian inheritance because it is inherited uniparentally through the maternal lineage. The mitochondrial genome contains 37 genes, from which 13 encode polypeptides, two rRNA genes and 22 tRNA genes (Box 2 and Figure 1A) and pathogenic mutations have been reported in all 37 mtDNA genes (34). mtDNA is subjected to much higher mutation rates than the nuclear genome and the mutation rate varies between species (35, 36). Besides mutations of the mtDNA replication and repair machinery, higher mutation rate of mtDNA is affected by the production of ROS within mitochondria as by-product of the OXPHOS. This can lead to a cycle in which ROS causes DNA damage, which in turn leads to dysregulation of respiration and accumulation of mutations.
Each cell contains numerous mitochondria and each mitochondrion may contain from a few to dozens of copies of mtDNA (the mtDNA copy number). Hence a given cell of a specific tissue type may contain thousands of copies of mtDNA and only one copy of nDNA. Unlike nDNA, mtDNA is not packed by histones but packed into protein-DNA complexes called nucleoids (37). Drosophila mtDNA contains one large A + T rich non-coding region (38), which in humans is called the D-loop. In both Drosophila and humans, the non-coding region is the starting point of mtDNA replication (39). Hence, variation in this region might cause changes in the mtDNA replication and have an effect on the mtDNA copy number content (40) and ultimately in energy production. mtDNA copy number is usually higher in tissues that have high energy demand such as brain, skeletal, and cardiac muscle tissues (41) and mtDNA mutations in these tissues can possibly lead to more pronounced phenotypes.
Besides heritable mtDNA mutations occurring in the germ line of the maternal lineage, the mtDNA of both females and males are subjected to spontaneous somatic mutations. mtDNA genetics is complicated due to its multi-copy nature. mtDNA mutations within a single cell (and even within a single mitochondria) can be either heteroplasmic due to a mix of mutated and wild type mtDNA, or homoplasmic where all mtDNAs contain either the mutated or the wild type form. In a heteroplasmic mutation the proportion of mutated mtDNA needs to exceed a certain threshold for the mutation to manifest. This can be due to the wild type mtDNA not being able to compensate the defect at that point. The threshold is also likely to be dependent on the mutation type and environmental effectors. Nuclear genotypes may also have variation in their ability to dampen or amplify the effects of specific deleterious mitochondrial mutations, which often demonstrate incomplete penetrance (42).
mtDNA mutations have been linked to various human diseases (43). In Leber hereditary optic neuropathy (LHON) the patient suffers from a loss of vision and the mtDNA mutations causing it are mostly considered homoplasmic (44). However, even though all the offspring of a homoplasmic mother inherits the LHON, only 50% of males and 10% of females develop the disease, showing that predicting the way the mtDNA mutation manifests, is difficult due to mito-nuclear crosstalk. Also, environmental factors can cause changes in the mtDNA mutation manifestation, as in the case of mtDNA encoded homoplasmic ribosomal RNA (RNR1) mutation that causes deafness early on in childhood. Specific antibiotics are associated with the manifestations of the clinical symptoms of the RNR1 mutation (45). Due to the complexity of the crosstalk of the two genomes and environmental factors, possible physiological compensations and the amplifying or dampening effects originating from nuclear genome variation and compatibility with the mtDNA, it is difficult to predict how the mtDNA mutations will eventually manifest themselves.
Disruption of Mito-Nuclear Crosstalk
The optimal functioning of mitochondria relies on the correct transcription and translation of genes involved in respiration, and as mentioned above, these genes are found on both the nuclear and mitochondrial genomes. Several signaling pathways between the nucleus and mitochondria have been uncovered recently (46). Mutations in either nDNA or mtDNA have the potential to disrupt the crosstalk between mitochondrial and nuclear proteins, and can therefore disrupt efficient gene transcription and translation (30), with consequences for metabolic rates (47), aging (48) and sperm competitiveness (49), which ultimately have detrimental effects on organismal fitness (50). Evidence for these detrimental effects is especially clear in hybrids between closely related species or between divergent populations within species, where long-term coevolution between the nuclear and mitochondrial genomes has been broken up, resulting in novel combinations of nuclear and mitochondrial genomes (51, 52). For example, hybrids of the marine copepod Tigriopus show reduced activity of OXPHOS Complex IV (cytochrome c oxidase) because of the breakup of coevolved nuclear and mitochondrial-encoded subunits of the Complex IV (51). This mismatch between nuclear and mitochondrial genes is thought to be a strong selection pressure for the fixation of compensatory mutations in nuclear-encoded OXPHOS subunits (53).
Mito-nuclear interactions can also have strong effects on the outcome of infection. In Drosophila, a mito-nuclear incompatibility resulted in energetically compromised flies that were more susceptible to infection by a bacterial pathogen (54). Salminen et al., identified an OXPHOS Complex III mutation D21N in D. melanogaster mitochondrial CYTB gene, that was shown to cause larval stage melanotic nodules in a healthy nuclear background (6%) and a significant increase (56%) of melanotic nodules when the mitotype was introgressed into a tko25t nuclear background (55). Formation of melanotic nodules is considered a sign of activated cell mediated innate immunity, and it usually involves the proliferation and aggregation of hemocytes, Drosophila blood cells (56). Furthermore, CYTB mutation bearing mitotype in a tko25t nuclear background caused 100% pupal lethality, which is a first report of synthetic lethality between nuclear-mitochondrial interaction within a metazoan species (55).
Mitochondrial Variation Can Affect Infection Outcome
The role of mitochondria in the response to infection is central, impacting multiple functions. First, intermediates of the mitochondrial tricarboxylic acid (TCA) cycle have a signaling function in innate immune responses. Second, mitochondria generate energy by producing ATP during OXPHOS (Box 2 and Figure 1B), and given the elevated energetic requirements of immunity, we may expect variation in mitochondrial functions to result in changes in ATP production, thereby generating heterogeneity in the response to infection. Third, mitochondrial metabolism may further promote protection against pathogens by producing ROS, with direct antimicrobial action. Finally, in mammalian models of immunity, damaged mtDNA has been shown to act as DAMP triggering inflammatory responses akin to those seen during infection. There is therefore increasing evidence that mitochondrial functions contribute to the host response to infection.
Mitochondrial TCA Cycle Metabolites With Immune Signaling Functions
Mitochondrial tricarboxylic acid cycle (TCA, also called Krebs cycle and citric acid cycle) consists of a series of reactions where substrates originating from carbohydrates, fats and proteins have been fed into it and the metabolites from the cycle are transported into cytosol as building blocks for macromolecules or energy is released through the oxidation of acetyl-CoA. However, metabolites in the TCA cycle have also been shown to be involved in regulation of chromatin modifications, DNA methylation and post-translational modifications of proteins [reviewed in (57)]. Intermediates and derivatives of the TCA cycle have been shown to have non-metabolic signaling functions, in addition to their more conventional role as metabolites associated with bioenergetics (58). Non-metabolic functions of the TCA cycle intermediates succinate, itaconate, fumarate, 2-hydroxyglutarate and acetyl-CoA play a role in inflammation, and immune cell activation (58). For example, succinate is a pro-inflammatory metabolite as its production is enhanced during inflammation (59) and it acts as a signal from mitochondria to the cytosol to induce the expression of pro-inflammatory genes and increases the levels of antioxidant superoxide as a proinflammatory redox signal (60). Succinate has also been shown to accumulate in lipopolysaccharide treated macrophages (59). Itaconate on the other hand is endogenous protective and anti-inflammatory molecule that negatively regulates the inflammatory response and cytokine production (61–63) and also has direct antibacterial effects (64). TCA cycle intermediates have also been connected to epigenetic signaling (58). For example, fumarate has a role as an epigenetic inflammatory signal. Arts et al., showed that the accumulation of fumarate in immune activated monocytes was needed for trained immunity by enhancing cytokine production upon re-activation with lipopolysaccharide (65). Further, Acetyl-CoA has been shown to drive histone acetylation which can have profound impact on immune cell function (66). To summarize, mitochondrial variation may impact infection outcomes via their effect on TCA cycle products that have immune signaling functions.
Changes in ATP Production
Mitochondria generate energy by producing ATP during OXPHOS (Box 2), and given the elevated energetic requirements of immunity, we may expect variation in mitochondrial function to result in changes in ATP production, thereby generating heterogeneity in the response to infection (54). Mutations in any of the nuclear or mitochondrial encoded OXPHOS complexes or in nuclear genes affecting replication, transcription or translation of mtDNA can affect the total electron transfer chain outcome, potentially causing a decrease in the total production of ATP. Severely decreased ATP synthesis is an obvious problem for cells with constant high energy demands such as cardiomyocytes and neurons (67) and decreased ATP synthesis can also increase the AMP/ATP ratio that can lead to activation of AMP-activated protein kinase and multiple signaling pathways (46). We might therefore expect mutations in nuclear or mitochondrial encoded components that cause a reduction in ATP to result in a decrease of immune cell function.
Role of ROS in Immune Responses
Reactive oxygen species are a group of reactive molecules and free radicals derived from molecular oxygen which are now known to have a role in cellular homeostasis (68). Elevated levels of ROS can cause oxidative stress, cellular-, and DNA damage in eukaryotic cells. Mutations in nuclear or mitochondrial genes encoding the protein subunits of OXPHOS complexes I and III can cause a decrease or an increase in ROS production, depending on the mutation. One of the most evident roles of mitochondrial functions in innate immunity is the production of ROS by leakage from mitochondrial ETC. The majority of ROS are produced during mitochondrial ETC (mtROS), and some by oxidoreductase enzymes such as NADPH oxidase, a multicomponent membrane bound enzyme complex. Common ROS include superoxide (O–2), hydrogen peroxide (H202), hydroxyl radical (OH), hydroxide ion (OH–) and nitric oxide (NO). Prolonged oxidative stress is harmful and so detoxification of ROS via scavenging enzymes and antioxidants is vital. Therefore, mutations in the nuclear encoded antioxidants that are targeted to detoxify ROS, can also have an impact on immune response. Antioxidant superoxide dismutase (SOD) is transported to mitochondria where it converts superoxide to hydrogen peroxide. Hydrogen peroxide outside mitochondria is converted to water and oxygen with the help of catalase, peroxiredoxins and glutathione peroxidases (Figure 1).
mtROS is produced in all cell types that contain mitochondria and it has been connected to regulation of signaling pathways (69), apoptosis (70), inflammation (71), cellular adaptation to hypoxia (72), cellular differentiation (73), and autophagy (74). In addition to the regulative role of mtROS, a growing body of evidence has highlighted the role of ROS as a target of regulation of immune signaling pathways (14, 75). mtROS serves several roles within both the humoral and cell-mediated arms of innate immunity, including direct elimination of pathogens through its microbicidal effects. However, it is still unclear what is the exact mechanism of the bactericidal effect of mtROS upon bacterial infection as the effect seem to be the type of ROS and pathogen specific [reviewed in (68)] and aligns with the increased heterogeneity of the innate immune response.
Alongside its role in promoting bacterial clearance, mtROS also functions in signaling for hemocyte proliferation and differentiation in Drosophila and has been identified as an essential signaling molecule in Drosophila’s cellular immune response to parasitoid infection (76). ROS plays a likely role as a key signaling molecule within the Drosophila lymph gland as it has been suggested to prime the quiescent hemocyte progenitors within the lymph gland for differentiation (77). It has been shown that reduction of ROS significantly retards progenitor differentiation whereas upregulation of ROS via OXPHOS Complex I disruption produces a phenotype with a significantly higher hemocyte population (77).
Transmitochondrial cell lines can be created by combining enucleated cells that contain the mtDNA of interest with cells that lack their mtDNA (78). Data obtained from transmitochondrial cell lines suggest that mtDNA variants on a controlled nuclear background can alter ROS levels of cells (79). Organismal cybrids have also been shown to differ in OXPHOS parameters (34) and effect of mtDNA variation on altered ROS production has been studied in Drosophila in vivo, showing that specific mtDNA variants can elevate ROS production (80).
Cytosolic mtDNA as a Danger Signal
During infection, elevated levels of ROS can cause mitochondrial and cellular damage resulting mtDNA leakage. Due to the evolutionary origin of mitochondria it harbors resemblance to bacterial DNA making it appear non-self. mtDNA is surrounded by a double-membrane structure and membrane damage could lead to leakage of mtDNA outside mitochondria and elicit self-derived immune activation. When mtDNA is located outside mitochondria in the cytoplasm of the cell or in extracellular space, it can trigger immune responses by directly engaging the host’s innate immune pattern-recognition receptors (PRRs) [reviewed in (81)]. PRRs are conserved receptors that recognize viral, bacterial and fungal particles as well as molecules released from injured cells. The release of mtDNA outside of mitochondria can occur from dying cells, during injury, cellular stress or infection, and mtDNA outside of mitochondria engages PRRs and functions as damage-associated molecular pattern (DAMP) leading to enhancement of pro-inflammatory responses (71). In mammalian models of immunity, inflammasomes are innate immune related signaling complexes that monitor the cytosolic compartment of the cell and are involved in the secretion of cytokines upon infection and recognition of DAMPs (81). Altered mitochondrial dynamics, production of mROS and release of mtDNA outside mitochondria have all been linked to inflammasome activation (82). In mammalian system extracellular circulatory mtDNA has been shown to act as an endogenous Toll-like receptor TLR9 agonist and been connected to many TLR9- dependent inflammatory diseases (81). Cytosolic mtDNA (83) as well as cytosolic double-stranded RNA created during bidirectional transcription of mtDNA (84) has been shown to trigger antiviral responses in human.
Using Drosophila to Study Mitochondrial Variation and Mito-Nuclear Interactions in Immunity
Mitochondrial function and the content of the mitochondrial genome are highly conserved among metazoans e.g., between humans and the fruit fly D. melanogaster and the latter has been widely used to model human mitochondrial diseases [reviewed in (85)]. Besides studying the naturally occurring variation of nuclear genes affecting mitochondrial function, i.e., mtDNA replication, transcription and translation as well as polypeptides needed for TCA and OXPHOS, it is possible to exploit the highly sophisticated genetic toolbox that exists for D. melanogaster. With the binary gene expression systems, such as GAL4-UAS, one can modify the expression of a desired gene within a specific tissue within a specific time, providing a route to investigate the effect of specific genes in chosen tissues on a given phenotype (86). For example, overexpression and gene knock-down methods could allow the modification of the gene expression of nuclear encoded genes that are transported to mitochondria, hence altering the mitochondrial function, assuming that the mitochondrial import stage does not dampen the effect of genetic modification.
Another approach to study mitochondrial variation is to focus on naturally occurring variation in mtDNA. Investigating the effect of mtDNA mutations is complicated by cross-talk between the mitochondria and the nucleus of a cell, with nuclear genes generally responsible for controlling mitochondrial activity. With the cytoplasmic hybrid, aka. cybrid model, specific mtDNAs can be introgressed onto controlled nuclear backgrounds, making it possible to focus on the effects arising from the mitochondrial genome (Figure 2). It is presently not possible to genetically target and modify the gene expression of specific mtDNA genes, and so the cybrid model relies on using natural mtDNA variants found through different genetic screens. However, there are methods for creating random mutations to mtDNA genome such as using POLG mutants (87, 88) or to more specific regions with targeted restriction enzymes (89).
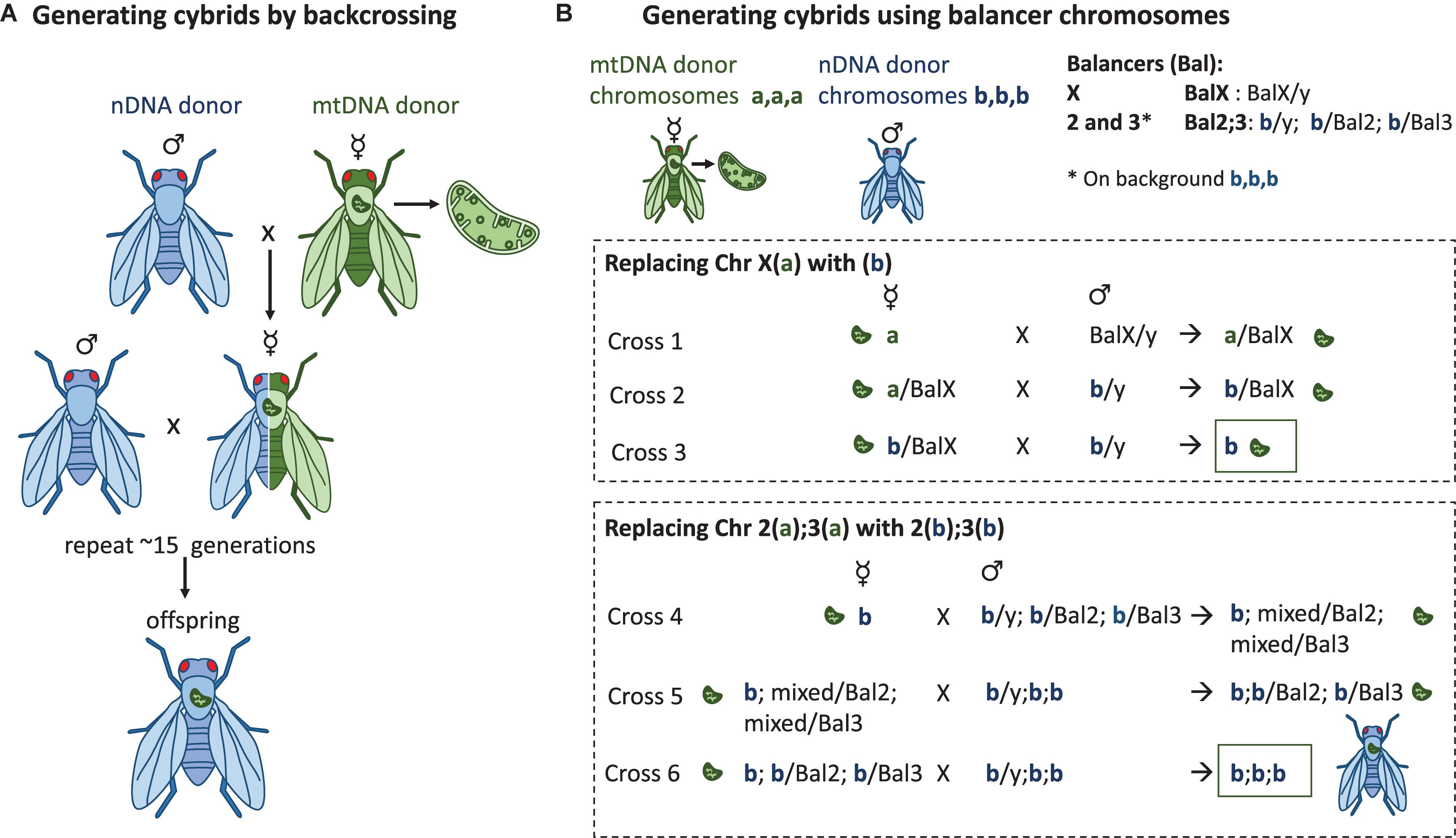
Figure 2. Methods to generate Drosophila in vivo cybrid lines. (A) Repeated backcrossing. The chosen mtDNA variant is added during the first cross where the virgin females from the mtDNA donor strain are crossed with the males of the nuclear donor strain. Virgin females of the following progeny are crossed again with the males of the nuclear donor strain. This will be repeated for >15 generations, after which the cybrid progeny contain the wanted mtDNA variant on a nuclear background that in theory is the same as the one in the nuclear donor strain. This method also has the potential to reveal possible mito-nuclear epistasis during the course of the backcrossing. (B) Balancer chromosome method. Drosophila males do not go through recombination, whereas in females the recombination can be controlled with the balancer chromosomes which do not recombine with the normal chromosome homologs during meiotic prophase. The presence of balancer chromosomes in the progeny can be recognized by the dominant marker mutations that the balancers carry, e.g., mutations affecting eye shape, body color, or wing morphology. If the progeny do not have the balancer, it has the normal homolog of the wanted chromosome. Three (labeled here a,a,a or b,b,b) of the four D. melanogaster chromosomes can be replaced to contain the genetic content of the wanted nuclear donor strain by using the balancer chromosome method. The mtDNA from the maternal donor strain is introgressed to the strain during the first cross and the chromosome content is replaced chromosome at a time by using the correct progeny of the previous cross. For clarity, chromosomes 2 and 3 have not been marked to the cross where the X(a) is replaced with the X(b).
By generating transmitochondrial cybrid cells, it is possible to study the effects of mtDNA variation at the cellular level (78). This in vitro approach has been used previously to investigate the cellular effects of mtDNA polymorphism associated with an aggressive form of breast cancer (90). Similarly, Bellizzi et al., hypothesized that the transcription of stress-responder nuclear genes can be modulated according to the mtDNA variability. They showed that osteosarcoma cells depleted of their own mitochondria and repopulated with different ones, modulated the expression of cytokines and cytokine receptors due to the variability of the mtDNA (91).
The effects of mtDNA variation at the organismal level may vary significantly from what can be inferred from cellular level. The transmitochondrial in vitro method is limited in investigating the impact of mtDNA variation affecting an entire organism, for example upon environmental stress, limited diet or when fighting infection. However, D. melanogaster offers a feasible way of creating and utilizing cybrid strains for in vivo experiments of the effects of mtDNA variation at the organismal level (26, 30, 50), as well as quantifying the effects of mtDNA, nDNA and their interaction. Given the ease with which flies can be sampled from natural populations, options of screening the mtDNA variation by sequencing and crossing and breeding the lines in constant laboratory conditions, Drosophila cybrid lines offer a unique opportunity to disentangle variation in immunity arising from mtDNA polymorphism from the more commonly investigated variation in nuclear-encoded genes associated with canonical immune pathways.
There are three ways of creating cybrids and two of them, repeated backcrossing and using balancer chromosomes, are common methods in Drosophila, explained in detail below and in Figure 2. In the third method, Mitochondrial Replacement Therapy (MRT), mtDNA is introduced directly to a novel nuclear environment. In MRT the nucleus of a mtDNA mutation bearing female is transferred to an enucleated egg of a mitochondrially healthy donor (92). To date, MRT has been performed in two other mammalian species beside humans, in macaques (93) and mice (94, 95).
Generating Drosophila Cybrids by Repeated Backcrossing
Because mtDNA is inherited uniparentally through the mother, the chosen donor mtDNA can be introgressed onto wanted nuclear background by multiple generations of backcrossing (Figure 2A). In the first cross the virgin females containing the donor mtDNA are crossed with males containing the wanted nuclear background, resulting in progeny with 100% maternal mtDNA, 50% maternal nDNA and 50% paternal nDNA. This is followed by a series of crosses where the virgin females of each generation are crossed with males of the nuclear donor strain. With each cross, the proportion of paternal nDNA increases by 50% from the previous generation, meaning that in theory, after 10 generations of backcrossing, less than 0.1% of the maternal nDNA should be present. An important caveat to introgression by repeated backcrossing is that although in theory only 10 generations should be required to obtain a line over 99.9% of the paternal nuclear genome, in practice this value will be lower due to strong linkage disequilibrium between loci that have a close genetic distance (called linkage drag) (96). From a mapping perspective this linkage can be advantageous, but the backcrossing approach has the potential to select for compatible nuclear partners, masking true incompatibilities (also lethal combinations). The number of generations of backcrossing required to break linkage between two loci will therefore depend on the genetic distance such that the number of backcrosses N = Log(1-r)(1-P) where r is the recombination frequency, and P is the probability of separation (97). For example, for two loci separated by a 20 cM interval, at least 17 generations of backcrossing are required to have a 95% certainty of a crossover event (97). However, even after multiple generations of backcrossing, some loci are likely to never recombine if they result in strongly deleterious or even lethal phenotypes, and such loci will remain a source of residual heterozygosity regardless of the number of generations of backcrossing.
This method has been used in D. melanogaster experiments to study the effect of mito-nuclear interactions on mtDNA copy number, respiration, development time and weight (40) as well as in experiments where the effect of mtDNA variants on mitochondrial diseases has been assayed in combination with the balancer chromosome method (55, 98). The method has been also employed in other insect species where the use of balancer chromosomes is not possible, where the effect of mito-nuclear crosstalk has been studied on traits such as metabolism and aging in D. simulans (99), metabolic rate in Drosophila subobscura (100), and personality (101), bioenergetics, aging, life history traits (102), and male mating costs (47) in seed beetles.
Utilizing Drosophila Balancer Chromosomes
Drosophila has four chromosomes that include the X/Y sex chromosome pair and autosomal chromosomes 2, 3, and 4. The fourth chromosome is very small, contains only around 80 genes (103) and does not recombine. The genetic tool box developed for D. melanogaster allows the replacement of the entire chromosomes with the wanted content of specific strains or mutation. This is done with the help of balancer chromosomes. Balancer chromosomes (104) are multiply inverted and scrambled chromosomes that are not able to undergo crossover with their normal chromosome homologs. They also contain genetic markers that enable the recognition of their segregation. As the small chromosome 4 does not go through crossing over there are no balancers designed for this chromosome. Nuclear genomes can be constructed by replacing the chromosomes in multiple crosses in a properly planned crossing scheme, and eventually controlling the nuclear genome (Figure 2B). For creating Drosophila lines that contain the wanted mtDNA variant on a controlled nuclear background, the first cross includes the mtDNA donor females after which the wild-type chromosomes will be replaced one by one with the wanted isogenic chromosomes [crossing scheme explained e.g., in (30, 105, 106)]. This method has been utilized to study the effect of mtDNA variation and mito-nuclear interaction on aging (105), sex differences in aging, respiration and fertility (107, 108), starvation resistance, lipid proportion and physical activity (106) as well as ROS production and mtDNA copy number (109).
Conclusion and Future Perspectives
Drosophila has contributed significantly for our understanding of mitochondrial variation in both mitochondrial diseases and in immunity. Here we have highlighted strengths of this experimental powerhouse and described approaches that will link these two important fields to address the question how mitochondrial variation and specifically mtDNA variation affect innate immune functions. This is significant because a number of mitochondrial mutations have been associated with increased susceptibility to infection in humans (110, 111) and recent genetic screens have revealed vast variation in Drosophila mtDNA (112). The Drosophila model will also be relevant in the context of mitochondrial replacement therapy as a tool to test for potential incompatibilities that may result from specific mito-nuclear combinations (113). Given the homology between vertebrate and invertebrate innate immunity (114), the Drosophila model has the translational potential to generate novel candidate genes originating from mitochondrial sources of disease susceptibility and resistance, and for development of new therapeutic targets.
Author Contributions
TS and PV wrote the manuscript.
Funding
TS was supported by an Academy of Finland Fellowship (grants 322732 and 32879). PV was supported by a Leverhulme Trust Research Project Grant RPG-2018-369, a Branco Weiss Fellowship (https://brancoweissfellowship.org/), and a Chancellor’s Fellowship (School of Biological Sciences, University of Edinburgh).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AMP, antimicrobial peptide; ATP, adenosine triphosphate; DAMP, danger-associated molecular pattern; ETC, electron transport chain; IMD, immune deficiency; mt-aaRS, mitochondrial aminoacyl-tRNA synthetase; mtDNA, mitochondrial DNA; mtROS, mitochondrial reactive oxygen species; nDNA, nuclear DNA; NF- κ B, nuclear factor kappa-light-chain-enhancer of activated B cells; OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species.
References
1. Lloyd-Smith JO, Schreiber SJ, Kopp PE, Getz WM. Superspreading and the effect of individual variation on disease emergence. Nature. (2005) 438:355–9. doi: 10.1038/nature04153
2. Brodin P, Davis MM. Human immune system variation. Nat Rev Immunol. (2017) 17:21–9. doi: 10.1038/nri.2016.125
3. Pawelec G. Age and immunity: what is “immunosenescence”? Exp Gerontol. (2018) 105:4–9. doi: 10.1016/j.exger.2017.10.024
4. Lotter H, Altfeld M. Sex differences in immunity. Semin Immunopathol. (2019) 41:133–5. doi: 10.1007/s00281-018-00728-x
5. Martin LB. Stress and immunity in wild vertebrates: timing is everything. Gen Comp Endocrinol. (2009) 163:70–6. doi: 10.1016/j.ygcen.2009.03.008
6. Lemaitre B, Hoffmann J. The host defense of Drosophila melanogaster. Annu Rev Immunol. (2007) 25:697–743. doi: 10.1146/annurev.immunol.25.022106.141615
7. Tao L, Reese TA. Making mouse models that reflect human immune responses. Trends Immunol. (2017) 38:181–93. doi: 10.1016/j.it.2016.12.007
8. Renshaw SA, Trede NS. A model 450 million years in the making: zebrafish and vertebrate immunity. Dis Model Mech. (2012) 5:38–47. doi: 10.1242/dmm.007138
9. Barreiro LB, Quintana-Murci L. From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat Rev Genet. (2010) 11:17–30. doi: 10.1038/nrg2698
10. Timmann C, Thye T, Vens M, Evans J, May J, Ehmen C, et al. Genome-wide association study indicates two novel resistance loci for severe malaria. Nature. (2012) 489:443–6. doi: 10.1038/nature11334
11. McLaren PJ, Coulonges C, Bartha I, Lenz TL, Deutsch AJ, Bashirova A, et al. Polymorphisms of large effect explain the majority of the host genetic contribution to variation of HIV-1 virus load. Proc Natl Acad Sci USA. (2015) 112:14658–63. doi: 10.1073/pnas.1514867112
12. Tian C, Hromatka BS, Kiefer AK, Eriksson N, Noble SM, Tung JY, et al. Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. Nat Commun. (2017) 8:599. doi: 10.1038/s41467-017-00257-5
13. Arnoult D, Carneiro L, Tattoli I, Girardin SE. The role of mitochondria in cellular defense against microbial infection. Semin Immunol. (2009) 21:223–32. doi: 10.1016/j.smim.2009.05.009
14. West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. (2011) 11:389–402. doi: 10.1038/nri2975
15. Cloonan SM, Choi AM. Mitochondria: commanders of innate immunity and disease? Curr Opin Immunol. (2012) 24:32–40. doi: 10.1016/j.coi.2011.11.001
16. Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. (2015) 42:406–17. doi: 10.1016/j.immuni.2015.02.002
17. Fang C, Wei X, Wei Y. Mitochondrial DNA in the regulation of innate immune responses. Protein Cell. (2016) 7:11–6. doi: 10.1007/s13238-015-0222-9
18. Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol. (2017) 18:488–98. doi: 10.1038/ni.3704
20. Zeviani M, Spinazzola A, Carelli V. Nuclear genes in mitochondrial disorders. Curr Opin Genet Dev. (2003) 13:262–70. doi: 10.1016/S0959-437X(03)00052-2
21. Frazier AE, Thorburn DR, Compton AG. Mitochondrial energy generation disorders: genes, mechanisms, and clues to pathology. J Biol Chem. (2019) 294:5386–95. doi: 10.1074/jbc.R117.809194
22. El-Hattab AW, Craigen WJ, Scaglia F. Mitochondrial DNA maintenance defects. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1539–55. doi: 10.1016/j.bbadis.2017.02.017
23. Cohen BH, Chinnery PF, Copeland WC. POLG-related disorders. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, et al. editors. GeneReviews. Seattle, WA: University of Washington. (2019).
24. Samstag CL, Hoekstra JG, Huang C-H, Chaisson MJ, Youle RJ, Kennedy SR, et al. Deleterious mitochondrial DNA point mutations are overrepresented in Drosophila expressing a proofreading-defective DNA polymerase γ. PLoS Genet. (2018) 14:e1007805. doi: 10.1371/journal.pgen.1007805
25. Rebelo AP, Dillon LM, Moraes CT. Mitochondrial DNA transcription regulation and nucleoid organization. J Inherit Metab Dis. (2011) 34:941–51. doi: 10.1007/s10545-011-9330-8
26. Mossman JA, Biancani LM, Zhu C, Rand DM. Mitonuclear epistasis for development time and its modification by diet in Drosophila. Genetics. (2016) 203:463–84. doi: 10.1534/genetics.116.187286
27. Jacobs HT, Turnbull DM. Nuclear genes and mitochondrial translation: a new class of genetic disease. Trends Genet. (2005) 21:312–4. doi: 10.1016/j.tig.2005.04.003
28. Sissler M, González-Serrano LE, Westhof E. Recent advances in mitochondrial aminoacyl-tRNA synthetases and disease. Trends Mol Med. (2017) 23:693–708. doi: 10.1016/j.molmed.2017.06.002
29. Konovalova S, Tyynismaa H. Mitochondrial aminoacyl-tRNA synthetases in human disease. Mol Genet Metab. (2013) 108:206–11. doi: 10.1016/j.ymgme.2013.01.010
30. Meiklejohn CD, Holmbeck MA, Siddiq MA, Abt DN, Rand DM, Montooth KL. An incompatibility between a mitochondrial tRNA and its nuclear-encoded tRNA synthetase compromises development and fitness in Drosophila. PLoS Genet. (2013) 9:e1003238. doi: 10.1371/journal.pgen.1003238
31. Toivonen JM, O’Dell KMC, Petit N, Irvine SC, Knight GK, Lehtonen M, et al. Technical knockout, a Drosophila model of mitochondrial deafness. Genetics. (2001) 159:241–54.
32. Kemppainen E, George J, Garipler G, Tuomela T, Kiviranta E, Soga T, et al. Mitochondrial dysfunction plus high-sugar diet provokes a metabolic crisis that inhibits growth. PLoS One. (2016) 11:e0145836. doi: 10.1371/journal.pone.0145836
33. Saada A, Shaag A, Arnon S, Dolfin T, Miller C, Fuchs-Telem D, et al. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J Med Genet. (2007) 44:784–6. doi: 10.1136/jmg.2007.053116
34. Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW. The genetics and pathology of mitochondrial disease. J Pathol. (2017) 241:236–50. doi: 10.1002/path.4809
35. Montooth KL, Rand DM. The spectrum of mitochondrial mutation differs across species. PLoS Biol. (2008) 6:e213. doi: 10.1371/journal.pbio.0060213
36. Nabholz B, Glémin S, Galtier N. Strong variations of mitochondrial mutation rate across mammals–the longevity hypothesis. Mol Biol Evol. (2008) 25:120–30. doi: 10.1093/molbev/msm248
37. Spelbrink JN. Functional organization of mammalian mitochondrial DNA in nucleoids: history, recent developments, and future challenges. IUBMB Life. (2010) 62:19–32. doi: 10.1002/iub.282
38. Lewis DL, Farr CL, Farquhar AL, Kaguni LS. Sequence, organization, and evolution of the A+T region of Drosophila melanogaster mitochondrial DNA. Mol Biol Evol. (1994) 11:523–38.
39. Falkenberg M. Mitochondrial DNA replication in mammalian cells: overview of the pathway. Essays Biochem. (2018) 62:287–96. doi: 10.1042/EBC20170100
40. Salminen TS, Oliveira MT, Cannino G, Lillsunde P, Jacobs HT, Kaguni LS. Mitochondrial genotype modulates mtDNA copy number and organismal phenotype in Drosophila. Mitochondrion. (2017) 34:75–83. doi: 10.1016/j.mito.2017.02.001
41. D’Erchia AM, Atlante A, Gadaleta G, Pavesi G, Chiara M, De Virgilio C, et al. Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion. (2015) 20:13–21. doi: 10.1016/j.mito.2014.10.005
42. Giordano C, Iommarini L, Giordano L, Maresca A, Pisano A, Valentino ML, et al. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy. Brain J Neurol. (2014) 137:335–53. doi: 10.1093/brain/awt343
43. Tuppen HAL, Blakely EL, Turnbull DM, Taylor RW. Mitochondrial DNA mutations and human disease. Biochim Biophys Acta Bioenerg. (2010) 1797:113–28. doi: 10.1016/j.bbabio.2009.09.005
44. Man PYW, Griffiths PG, Brown DT, Howell N, Turnbull DM, Chinnery PF. The epidemiology of leber hereditary optic neuropathy in the North East of England. Am J Hum Genet. (2003) 72:333–9. doi: 10.1086/346066
45. Prezant TR, Agapian JV, Bohlman MC, Bu X, Oztas S, Qiu W, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet. (1993) 4:289–94.
46. Quirós PM, Mottis A, Auwerx J. Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol. (2016) 17:213–26. doi: 10.1038/nrm.2016.23
47. Immonen E, Rönn J, Watson C, Berger D, Arnqvist G. Complex mitonuclear interactions and metabolic costs of mating in male seed beetles. J Evol Biol. (2016) 29:360–70. doi: 10.1111/jeb.12789
48. Immonen E, Collet M, Goenaga J, Arnqvist G. Direct and indirect genetic effects of sex-specific mitonuclear epistasis on reproductive ageing. Heredity. (2016) 116:338–47. doi: 10.1038/hdy.2015.112
49. Dowling DK, Friberg U, Arnqvist G. A comparison of nuclear and cytoplasmic genetic effects on sperm competitiveness and female remating in a seed beetle. J Evol Biol. (2007) 20:2113–25. doi: 10.1111/j.1420-9101.2007.01433.x
50. Montooth KL, Meiklejohn CD, Abt DN, Rand DM. Mitochondrial–nuclear epistasis affects fitness within species but does not contribute to fixed incompatibilities between species of Drosophila. Evolution. (2010) 64:3364–79. doi: 10.1111/j.1558-5646.2010.01077.x
51. Ellison CK, Burton RS. Disruption of mitochondrial function in interpopulation hybrids of Tigriopus californicus. Evolution. (2006) 60:1382–91. doi: 10.1111/j.0014-3820.2006.tb01217.x
52. Lee-Yaw JA, Jacobs CGC, Irwin DE. Individual performance in relation to cytonuclear discordance in a northern contact zone between long-toed salamander (Ambystoma macrodactylum) lineages. Mol Ecol. (2014) 23:4590–602. doi: 10.1111/mec.12878
53. Osada N, Akashi H. Mitochondrial-nuclear interactions and accelerated compensatory evolution: evidence from the primate cytochrome C oxidase complex. Mol Biol Evol. (2012) 29:337–46. doi: 10.1093/molbev/msr211
54. Buchanan JL, Meiklejohn CD, Montooth KL. Mitochondrial dysfunction and infection generate immunity–fecundity tradeoffs in Drosophila. Integr Comp Biol. (2018) 58:591–603. doi: 10.1093/icb/icy078
55. Salminen TS, Cannino G, Oliveira MT, Lillsunde P, Jacobs HT, Kaguni LS. Lethal interaction of nuclear and mitochondrial genotypes in Drosophila melanogaster. G3 (Bethesda). (2019) 9:2225–34. doi: 10.1534/g3.119.400315
56. Minakhina S, Steward R. Melanotic mutants in Drosophila: pathways and phenotypes. Genetics. (2006) 174:253–63. doi: 10.1534/genetics.106.061978
57. Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. (2020) 11:102. doi: 10.1038/s41467-019-13668-3
58. Ryan DG, Murphy MP, Frezza C, Prag HA, Chouchani ET, O’Neill LA, et al. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat Metab. (2019) 1:16–33. doi: 10.1038/s42255-018-0014-7
59. Tannahill GM, Curtis AM, Adamik J, Palsson-Mcdermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. (2013) 496:238–42. doi: 10.1038/nature11986
60. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. (2016) 167:457–70.e13. doi: 10.1016/j.cell.2016.08.064
61. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. (2016) 24:158–66. doi: 10.1016/j.cmet.2016.06.004
62. Bambouskova M, Gorvel L, Lampropoulou V, Sergushichev A, Loginicheva E, Johnson K, et al. Electrophilic properties of itaconate and derivatives regulate the IκBζ-ATF3 inflammatory axis. Nature. (2018) 556:501–4. doi: 10.1038/s41586-018-0052-z
63. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. (2018) 556:113–7. doi: 10.1038/nature25986
64. McFadden BA, Purohit S. Itaconate, an isocitrate lyase directed inhibitor in Pseudomonas indigofera. J Bacteriol. (1977) 131:136–44.
65. Arts RJW, Novakovic B, ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. (2016) 24:807–19. doi: 10.1016/j.cmet.2016.10.008
66. Kurdistani SK, Grunstein M. Histone acetylation and deacetylation in yeast. Nat Rev Mol Cell Biol. (2003) 4:276–84. doi: 10.1038/nrm1075
67. Smeitink JA, Zeviani M, Turnbull DM, Jacobs HT. Mitochondrial medicine: a metabolic perspective on the pathology of oxidative phosphorylation disorders. Cell Metab. (2006) 3:9–13. doi: 10.1016/j.cmet.2005.12.001
68. Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol. (2015) 6:472–85. doi: 10.1016/j.redox.2015.09.005
69. Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. (2015) 163:560–9. doi: 10.1016/j.cell.2015.10.001
70. Bender T, Martinou JC. Where killers meet-permeabilization of the outer mitochondrial membrane during apoptosis. Cold Spring Harb Perspect Biol. (2013) 5:a011106. doi: 10.1101/cshperspect.a011106
71. Blaser H, Dostert C, Mak TW, Brenner D. TNF and ROS crosstalk in inflammation. Trends Cell Biol. (2016) 26:249–61. doi: 10.1016/j.tcb.2015.12.002
72. Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol. (2006) 91:807–19. doi: 10.1113/expphysiol.2006.033506
73. Mandal S, Lindgren AG, Srivastava AS, Clark AT, Banerjee U. Mitochondrial function controls proliferation and early differentiation potential of embryonic stem cells. Stem Cells. (2011) 29:486–95. doi: 10.1002/stem.590
74. Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. (2009) 16:1040–52. doi: 10.1038/cdd.2009.49
75. Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY, et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J Exp Med. (2011) 208:519–33. doi: 10.1084/jem.20102049
76. Meister M, Ferrandon D. Immune cell transdifferentiation: a complex crosstalk between circulating immune cells and the haematopoietic niche. EMBO Rep. (2012) 13:3–4. doi: 10.1038/embor.2011.238
77. Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature. (2009) 461:537–41. doi: 10.1038/nature08313
78. Vithayathil SA, Ma Y, Kaipparettu BA. Transmitochondrial cybrids: tools for functional studies of mutant mitochondria. Methods Mol Biol. (2012) 837:219–30. doi: 10.1007/978-1-61779-504-6_15
79. Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. (2008) 320:661–4. doi: 10.1126/science.1156906
80. Matoo OB, Julick CR, Montooth KL. Genetic variation for ontogenetic shifts in metabolism underlies physiological homeostasis in Drosophila. Genetics. (2019) 212:537–52. doi: 10.1534/genetics.119.302052
81. West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. (2017) 17:363–75. doi: 10.1038/nri.2017.21
82. Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev. (2015) 265:35–52. doi: 10.1111/imr.12286
83. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. (2015) 520:553–7. doi: 10.1038/nature14156
84. Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rötig A, et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature. (2018) 560:238–42. doi: 10.1038/s41586-018-0363-0
85. Sánchez-Martínez A, Luo N, Clemente P, Adán C, Hernández-Sierra R, Ochoa P, et al. Modeling human mitochondrial diseases in flies. Biochim Biophys Acta. (2006) 1757:1190–8. doi: 10.1016/j.bbabio.2006.05.008
86. Caygill EE, Brand AH. The GAL4 system: a versatile system for the manipulation and analysis of gene expression. Methods Mol Biol. (2016) 1478:33–52. doi: 10.1007/978-1-4939-6371-3_2
87. Spelbrink JN, Toivonen JM, Hakkaart GAJ, Kurkela JM, Cooper HM, Lehtinen SK, et al. In Vivo functional analysis of the human mitochondrial DNA polymerase POLG expressed in cultured human cells. J Biol Chem. (2000) 275:24818–28. doi: 10.1074/jbc.M000559200
88. Ahlqvist KJ, Hämäläinen RH, Yatsuga S, Uutela M, Terzioglu M, Götz A, et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in polg mutator mice. Cell Metab. (2012) 15:100–9. doi: 10.1016/j.cmet.2011.11.012
89. Xu H, DeLuca SZ, O’Farrell PH. Manipulating the metazoan mitochondrial genome with targeted restriction enzymes. Science. (2008) 321:575–7. doi: 10.1126/science.1160226
90. Kulawiec M, Owens KM, Singh KK. MtDNA G10398A variant in African-American women with breast cancer provides resistance to apoptosis and promotes metastasis in mice. J Hum Genet. (2009) 54:647–54. doi: 10.1038/jhg.2009.89
91. Bellizzi D, Cavalcante P, Taverna D, Rose G, Passarino G, Salvioli S, et al. Gene expression of cytokines and cytokine receptors is modulated by the common variability of the mitochondrial DNA in cybrid cell lines. Genes Cells. (2006) 11:883–91. doi: 10.1111/j.1365-2443.2006.00986.x
92. Wolf DP, Mitalipov N, Mitalipov S. Mitochondrial replacement therapy in reproductive medicine. Trends Mol Med. (2015) 21:68–76. doi: 10.1016/j.molmed.2014.12.001
93. Tachibana M, Sparman M, Sritanaudomchai H, Ma H, Clepper L, Woodward J, et al. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature. (2009) 461:367–72. doi: 10.1038/nature08368
94. Sato A, Kono T, Nakada K, Ishikawa K, Inoue SI, Yonekawa H, et al. Gene therapy for progeny of mito-mice carrying pathogenic mtDNA by nuclear transplantation. Proc Natl Acad Sci USA. (2005) 102:16765–70. doi: 10.1073/pnas.0506197102
95. Wang T, Sha H, Ji D, Zhang HL, Chen D, Cao Y, et al. Polar body genome transfer for preventing the transmission of inherited mitochondrial diseases. Cell. (2014) 157:1591–604. doi: 10.1016/j.cell.2014.04.042
96. Peng T, Sun X, Mumm RH. Optimized breeding strategies for multiple trait integration: I. Minimizing linkage drag in single event introgression. Mol Breed. (2014) 33:89–104. doi: 10.1007/s11032-013-9936-7
97. Kooke R, Wijnker E, Keurentjes JJB. Backcross populations and near isogenic lines. Methods Mol Biol. (2012) 871:3–16. doi: 10.1007/978-1-61779-785-9_1
98. Chen S, Oliveira MT, Sanz A, Kemppainen E, Fukuoh A, Schlicht B, et al. A cytoplasmic suppressor of a nuclear mutation affecting mitochondrial functions in Drosophila. Genetics. (2012) 192:483–93. doi: 10.1534/genetics.112.143719
99. Ballard JWO. Drosophila simulans as a novel model for studying mitochondrial metabolism and aging. Exp Gerontol. (2005) 40:763–73. doi: 10.1016/j.exger.2005.07.014
100. Kurbalija Novičić Z, Immonen E, Jelić M, AnĐelković M, Stamenković-Radak M, Arnqvist G. Within-population genetic effects of mtDNA on metabolic rate in Drosophila subobscura. J Evol Biol. (2015) 28:338–46. doi: 10.1111/jeb.12565
101. Løvlie H, Immonen E, Gustavsson E, Kazancioğlu E, Arnqvist G. The influence of mitonuclear genetic variation on personality in seed beetles. Proc R Soc Lond B Biol Sci. (2014) 281:20141039. doi: 10.1098/rspb.2014.1039
102. Đorđević M, Stojković B, Savković U, Immonen E, Tucić N, Lazarević J, et al. Sex-specific mitonuclear epistasis and the evolution of mitochondrial bioenergetics, ageing, and life history in seed beetles. Evolution. (2017) 71:274–88. doi: 10.1111/evo.13109
103. Riddle NC, Elgin SCR. The Drosophila dot chromosome: where genes flourish amidst repeats. Genetics. (2018) 210:757–72. doi: 10.1534/genetics.118.301146
104. Muller HJ. Genetic variability, twin hybrids and constant hybrids, in a case of balanced lethal factors. Genetics. (1918) 3:422–99.
105. Clancy DJ. Variation in mitochondrial genotype has substantial lifespan effects which may be modulated by nuclear background. Aging Cell. (2008) 7:795–804. doi: 10.1111/j.1474-9726.2008.00428.x
106. Aw WC, Correa CC, Clancy DJ, Ballard JWO. Mitochondrial DNA variants in Drosophila melanogaster are expressed at the level of the organismal phenotype. Mitochondrion. (2011) 11:756–63. doi: 10.1016/j.mito.2011.06.012
107. Camus MF, Wolf JBW, Morrow EH, Dowling DK. Single nucleotides in the mtDNA sequence modify mitochondrial molecular function and are associated with sex-specific effects on fertility and aging. Curr Biol. (2015) 25:2717–22. doi: 10.1016/j.cub.2015.09.012
108. Wolff JN, Pichaud N, Camus MF, Côté G, Blier PU, Dowling DK. Evolutionary implications of mitochondrial genetic variation: mitochondrial genetic effects on OXPHOS respiration and mitochondrial quantity change with age and sex in fruit flies. J Evol Biol. (2016) 29:736–47. doi: 10.1111/jeb.12822
109. Correa CC, Aw WC, Melvin RG, Pichaud N, Ballard JWO. Mitochondrial DNA variants influence mitochondrial bioenergetics in Drosophila melanogaster. Mitochondrion. (2012) 12:459–64. doi: 10.1016/j.mito.2012.06.005
110. Angajala A, Lim S, Phillips JB, Kim J-H, Yates C, You Z, et al. Diverse roles of mitochondria in immune responses: novel insights into immuno-metabolism. Front Immunol. (2018) 9:1605. doi: 10.3389/fimmu.2018.01605
111. Kapnick SM, Pacheco SE, McGuire PJ. The emerging role of immune dysfunction in mitochondrial diseases as a paradigm for understanding immunometabolism. Metabolism. (2018) 81:97–112. doi: 10.1016/j.metabol.2017.11.010
112. Bevers RPJ, Litovchenko M, Kapopoulou A, Braman VS, Robinson MR, Auwerx J, et al. Mitochondrial haplotypes affect metabolic phenotypes in the Drosophila genetic reference panel. Nat Metab. (2019) 1:1226–42. doi: 10.1038/s42255-019-0147-3
113. Eyre-Walker A. Mitochondrial replacement therapy: are mito-nuclear interactions likely to be a problem? Genetics. (2017) 205:1365–72. doi: 10.1534/genetics.116.196436
114. Buchon N, Silverman N, Cherry S. Immunity in Drosophila melanogaster–from microbial recognition to whole-organism physiology. Nat Rev Immunol. (2014) 14:796–810. doi: 10.1038/nri3763
115. Rosetto M, Engström Y, Baldari CT, Telford JL, Hultmark D. Signals from the IL-1 receptor homolog, Toll, can activate an immune response in a Drosophila hemocyte cell line. Biochem Biophys Res Commun. (1995) 209:111–6. doi: 10.1006/bbrc.1995.1477
116. Tanji T, Hu X, Weber ANR, Ip YT. Toll and IMD pathways synergistically activate an innate immune response in Drosophila melanogaster. Mol Cell Biol. (2007) 27:4578–88. doi: 10.1128/MCB.01814-06
117. Valanne S, Wang J-H, Ramet M. The Drosophila Toll signaling pathway. J Immunol. (2011) 186:649–56. doi: 10.4049/jimmunol.1002302
118. Myllymaki H, Valanne S, Ramet M. The Drosophila Imd signaling pathway. J Immunol. (2014) 192:3455–62. doi: 10.4049/jimmunol.1303309
119. Hanson MA, Dostálová A, Ceroni C, Poidevin M, Kondo S, Lemaitre B. Synergy and remarkable specificity of antimicrobial peptides in vivo using a systematic knockout approach. eLife. (2019) 8:e44341. doi: 10.7554/eLife.44341
120. Dostert C, Jouanguy E, Irving P, Troxler L, Galiana-Arnoux D, Hetru C, et al. The Jak-STAT signaling pathway is required but not sufficient for the antiviral response of Drosophila. Nat Immunol. (2005) 6:946–53. doi: 10.1038/ni1237
121. Zambon RA, Vakharia VN, Wu LP. RNAi is an antiviral immune response against a dsRNA virus in Drosophila melanogaster. Cell Microbiol. (2006) 8:880–9. doi: 10.1111/j.1462-5822.2006.00688.x
122. Obbard DJ, Gordon KHJ, Buck AH, Jiggins FM. The evolution of RNAi as a defence against viruses and transposable elements. Philos Trans R Soc Lond B Biol Sci. (2009) 364:99–115. doi: 10.1098/rstb.2008.0168
123. Parsons B, Foley E. Cellular immune defenses of Drosophila melanogaster. Dev Comp Immunol. (2016) 58:95–101. doi: 10.1016/j.dci.2015.12.019
124. Anderl I, Vesala L, Ihalainen TO, Vanha-aho L-M, Andó I, Rämet M, et al. Transdifferentiation and proliferation in two distinct hemocyte lineages in Drosophila melanogaster larvae after wasp infection. PLoS Pathog. (2016) 12:e1005746. doi: 10.1371/journal.ppat.1005746
125. Nappi AJ, Vass E. Cytotoxic reactions associated with insect immunity. Adv Exp Med Biol. (2001) 484:329–48.
Keywords: Drosophila melanogaster, cybrid, infection, innate immunity, mitochondria, mtDNA, oxidative phosphorylation, reactive oxygen species
Citation: Salminen TS and Vale PF (2020) Drosophila as a Model System to Investigate the Effects of Mitochondrial Variation on Innate Immunity. Front. Immunol. 11:521. doi: 10.3389/fimmu.2020.00521
Received: 23 October 2019; Accepted: 06 March 2020;
Published: 25 March 2020.
Edited by:
Nicolas Bertho, INRA Biologie, Épidémiologie et Analyse de Risque en Santé Animale (BIOEPAR), FranceReviewed by:
Kristi L. Montooth, University of Nebraska-Lincoln, United StatesJavier Santander, Memorial University of Newfoundland, Canada
Copyright © 2020 Salminen and Vale. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tiina S. Salminen, tiina.susanna.salminen@gmail.com; tiina.s.salminen@tuni.fi