 Timothy M. Smith Jr.
Timothy M. Smith Jr. Anuj Tharakan
Anuj Tharakan Rebecca K. Martin
Rebecca K. Martin- Department of Microbiology and Immunology, School of Medicine, Virginia Commonwealth University, Richmond, VA, United States
Generating inhibitors for A Disintegrin And Metalloproteinase 10 (ADAM10), a zinc-dependent protease, was heavily invested in by the pharmaceutical industry starting over 20 years ago. There has been much enthusiasm in basic research for these inhibitors, with a multitude of studies generating significant data, yet the clinical trials have not replicated the same results. ADAM10 is ubiquitously expressed and cleaves many important substrates such as Notch, PD-L1, EGFR/HER ligands, ICOS-L, TACI, and the “stress related molecules” MIC-A, MIC-B and ULBPs. This review goes through the most recent pre-clinical data with inhibitors as well as clinical data supporting the use of ADAM10 inhibitor use in cancer and autoimmunity. It additionally addresses how ADAM10 inhibitor therapy can be improved and if inhibitor therapy can be paired with other drug treatments to maximize effectiveness in various disease states. Finally, it examines the ADAM10 substrates that are important to each disease state and if any of these substrates or ADAM10 itself is a potential biomarker for disease.
Introduction
A Disintegrin And Metalloproteinase (ADAMs) are type I transmembrane endopeptidases that are a member of the metzincin superfamily which share a zinc-binding consensus motif that is required for catalytic activity. The metzincin superfamily includes ADAMs along with matrix metalloproteases (MMP), and ADAM-thrombospondins (ADAM-TS). ADAM10 is a modular protein comprised of metalloprotease, cysteine-rich, disintegrin, and epidermal growth factor (EGF)-like domains (1, 2). While all functionally active metzincin proteases contain a zinc binding motif, ADAMs are unique in that they also contain a transmembrane domain and are active while membrane bound. ADAM are membrane-anchored and cleave ligands expressed on the surface through a process known as ectodomain shedding. ADAMs also mediate regulated intramembrane proteolysis (RIP) of transmembrane proteins. Following translation of ADAM10 mRNA, the prodomain is cleaved by Furin (3, 4). With the removal of the prodomain, the disintegrin and cysteine rich domains engulf the active metalloproteinase domain. This is thought to be autoinhibitory to add a layer of control to the mature protease (5). Once ADAM10 leaves the endoplasmic reticulum, it can directly associate with all members of a subgroup of tetraspanins, Tspan8, all of which contain eight cysteines in the large extracellular domain (6). These tetraspanins can alter the substrate-specify of ADAM10 through conformational change (6). Synapse-associated protein 97 (SAP97) trafficks ADAM10 to the golgi apparatus (7). ADAM10 either interacts with its substrates at the cell surface or, as with L1, CD44, or CD23, ADAM10 meets these substrates in the endosomal pathway, leading to possible packaging into exosomes (8).
It is now recognized that there are 38 members of the ADAM family that are conserved between invertebrate and vertebrate evolution, with humans harboring 13 proteolytically active ADAMS (9–13). Of all the ADAMs, ADAM17 is the most similar to ADAM10 with regard to structure and function. According to the National Center for Biotechnology Information (NCBI) database, human ADAM10 RNA-seq data from 95 individuals from 27 different tissues showed ubiquitous expression amongst all 27 tissues (14). ADAM10 might be best known historically for its role in Notch signaling, and more recently for cleaving the amyloid precursor protein (APP) associated with the pathophysiology of Alzheimer's disease. It is important to note, however, ADAM10 has over 40 other substrates and counting that are involved in a multitude of biological functions including inflammation, apoptosis, cell adhesion, cell metabolism, cancer proliferation, cancer metastasis, and autoimmunity in addition to other functions [reviewed in (15, 16)]. ADAM10 is crucial for development as mice that are deficient in ADAM10 die around day 9.5 during the early embryonic stages (17). Some of the characterized ligands for ADAM10 in addition to APP and Notch are: E-cadherin, L-selectin, EGF, FASL, CD40L, ligand for inducible T cell costimulatory (ICOS-L), MICA, MICB, and ULBP2 (18–25).
ADAM10, Notch, and the Immune System
ADAM10's ability to regulate Notch signaling has been well-characterized and extensively reviewed (16, 26–29), including a recent review by Lambrecht et al. discussing the role of ADAMs in the immune system (30). Briefly, Notch signaling regulates many different important processes in cellular differentiation, including the development and differentiation of both innate and adaptive immune cells. Generally, the initial cleavage of Notch by furin-like convertase at s1 generates the mature Notch protein as it is transported to the cell membrane (29). Notch engagement with a Notch ligand initiates ADAM10 mediated proteolysis of the extracellular domain of the Notch receptor at the s2 cleavage site. The extracellular domain is released and endocytosed by the adjacent ligand-expressing cells. This cleavage event, produces a substrate that can be cleaved by the γ-secretase complex (S3 cleavage) (31, 32). S3 cleavage releases the Notch intracellular domain (NICD), this translocates to the nucleus where it complexes with transcription factor RBP-Jκ to induce transcription of Notch target genes (31, 33). In addition to ADAM10's importance in Notch signaling, some of ADAM10's most well-characterized substrates are also Notch receptors, such as Delta-like 1–4 and Jagged 1–2 (34).
The loss of ADAM10 in B cell development results in the loss of the marginal zone B cell (MZB) compartment (35). This has been shown to be mediated through Notch2 activation. Overexpression of ADAM10 in hematopoiesis results in the complete loss of the B cell compartment and an overall myeloid expansion that is Notch dependent (32). For B cells, ADAM10 substrates that regulate cell activation and antibody production include but are not limited to, Notch, ICOSL, and CD23 (23, 35, 36).
For T-cells, the strength of Notch signaling has been implicated in both the lineage decision between CD4+ and CD8+ T-cell subsets as well as between αβ and γδ T cell subsets (37–39). In addition, Notch 1 activation through binding of various Notch ligands on antigen presenting cells, such as dendritic cells, skew CD4 cells toward T-helper subsets, induce T-cell proliferation, and control survival of CD4+ memory cells (40, 41). Additional ADAM10 substrates control a wide variety of processes. These include CD44, which alters T cell migration (42, 43), FAS ligand (FASL), which in a soluble form acts as a decoy receptor to reduce activation induced apoptosis (44), and persistent Lymphocyte Activating 3 (LAG3) or T cell immunoglobulin and mucin domain-containing protein 3 (TIM3), which are markers of T cell exhaustion in tumor-infiltrating lymphocytes (45).
ADAM10 is ubiquitously expressed in human tissue with a vast array of substrates. Dysregulation or inhibition of ADAM10 can affect or result in the pathophysiology of a wide range of diseases. This review will focus on characterizing ADAM10 and the potential use of ADAM10 inhibitors in the context of cancer and autoimmunity.
ADAM10 in Cancer
Glioblastoma
Glioblastoma (GBM) is the most aggressive form of glioma resultant from malignant astrocytes in the brain. The link between ADAM10 and GBM disease progression is demonstrated in many studies (46–49). In a study of 50 GBM patients, Kanaya et al. demonstrated that low ADAM10 expression levels in tumor specimens positively correlated with increased survival especially when paired with tumor resection, as opposed to high ADAM10 levels (50). Interestingly, no detectable ADAM10 expression was reported in normal brain tissue (50).
There are several proposed mechanisms of ADAM10's promotion of GBM. Neuroligin-3 (NLGN3) is released from neurons by ADAM10. In multiple recent reports, NLGN3 levels have been linked with high grade GBM (46, 47). In GBM patients NLGN3 levels are high in the deep brain, preparing a pro-GBM tumor microenvironment. This expression in the deep brain is not seen in normal brain tissue (46). NLGN3 acts on GBM, promoting proliferation through the P13K-mTOR pathway, pro-oncogenic gene expression through focal adhesion kinase (FAK), and synapse-related gene expression (46). Reportedly, patient-derived xenografts (PDX) would not grow in NLGN3 knockout mice. Using the ADAM10 inhibitors GI254023X and INCB7839 in mice, the growth of adult and pediatric glioblastoma cell lines, and PDX were inhibited (46, 47). These effects of ADAM10 inhibition were not directly on the tumor, but were shown to be mediated through the blockage of ADAM10 cleavage of NLGN3 from neurons (46, 47).
Another proposed mechanism implicates ADAM10's cleavage of N-cadherin in cell migration and metastasis (49). In vitro experiments where GBM cell lines were treated with an antibody to inhibit ADAM10 (Millipore #AB19026) found decreased tumor growth and migration. This was shown to be driven by cleavage of N-cadherin (49). Musumeci et al. examined 25 grade IV GBM specimens as compared to normal brain tissue controls to identify molecular markers of aggressiveness (48). ADAM10 protein and mRNA were positively correlated with GBM and surface N-cadherin protein was negatively correlated with GBM (48).
Natural killer cells (NKs) have been reported to have an anticancer immune response against GBM and are associated with improved prognosis (51). NK cells recognize GBM by binding ligands for the NKG2D receptor that are expressed in the malignant state. NKG2D binds to MICA, MICB, and ULBP1-6. MICA and ULBP2 are both cleaved by ADAM10 and ADAM17 (52, 53). Loss of NK cell activation of the NKG2D receptor allows GBM escape due to reduced activation of the NK cell cytotoxic effector state (24, 54). Using the ADAM10 specific inhibitor GI254023X, the dual ADAM10/ADAM17 inhibitor GW280264X, or siRNA inhibition of ADAM10 or ADAM17, Wolpert et al. demonstrated increased surface expression of ULBP2 in GBM stem cell lines (55). This subsequently increased the immunogenicity of these GBM stem cell lines (55).
In GBM, M2 macrophages are correlated with poor prognosis. Gjorgjevski et al. examined tissue from 20 GBM and using qRT-PCR of M1/M2 related genes to various protease genes, including ADAM10 (56). A positive correlation was established between ADAM10 expression and M1-related genes (56). This overall signature then positively correlated with better prognosis. Although this disagrees with the majority of the work done on GBM and ADAM10, the authors attributed the increased survival to the M1-skewed profile (56).
Overall, in GBM, ADAM10 has strong value as a biomarker for prognostic use. A large scale study is warranted to validate ADAM10 as predictive biomarker. ADAM10 appears to be a strong therapeutic candidate to target GBM due to the multiple substrates it cleaves that are implicated in disease progression. Even with very strong pre-clinical evidence, there has yet to be a clinical trial in GBM with ADAM10 inhibitors. This is most likely due to the failures that the ADAM10 inhibitors have been in clinical trials (57). Despite this, the use of ADAM10 inhibitors as a clinical intervention should be carefully evaluated due to ADAM10's role in the cleavage of amyloid plaque precursors (58, 59).
Hodgkin Lymphoma, Non-hodgkin Lymphoma, and Multiple Myeloma
Hodgkin lymphoma (HL) is characterized by a clonal malignant lymphoproliferation in the form of lacunar histiocytes and Reed-Sternberg cells (60). Similar to GBM, ADAM10 promotes an immunosuppressive microenvironment through cleavage of the stress receptors MICB and the ULBP2, resulting in HL that has foregone immune surveillance (61, 62). Zocchi et al. generated two ADAM10 specific inhibitors (LT4 and MN8) (63). They found that treatment with either inhibitor blocked shedding of NKG2D-L in cultured HL samples and HL cell lines developed increased sensitivity to NKG2D-L-mediated killing after inhibitor treatment (63). Multiple studies have described the presence of ADAM10 in extracellular vesicles (EVs) released by the HL cells (64, 65). ADAM10 has additionally been described in EVs released from other tumors, including melanoma, GBM, lung, and colon cancer (66). In both HL studies, CD30 was found to be co-released on these HL EVs. This was proposed to further promotes an immunosuppressive tumor microenvironment (64). Interestingly, following treatment with the ADAM10 inhibitors (LT4 and CAM29), Tosetti et al. report that the inhibitor is additionally secreted in EVs leading to uptake by bystander cells (64). Overall, ADAM10 inhibitor treatment results in the restoration of membrane CD30 levels, which restored sensitivity to anti-CD30 monoclonal therapies used in HL, such as Iratumumab (64).
Non-hodgkin lymphoma (nHL) describes a variety of lymphomas, including Burkitt's lymphoma, diffuse large B cell lymphoma (DLBCL), and marginal zone lymphoma. All of these have in common the lack of Hodgkin cells. The prognosis for nHL can be worse due to the higher frequency of late-stage diagnoses (67). A variant of nHL is DLBCL. Epstein Barr-virus-positive (EBV+) DLBCL, not otherwise specified (NOS) have been shown to have increased expression of the immunosuppressive molecule PD-L1 (68). PD-L1+ DLBCLs can be treated with anti-PD-L1 monoclonal therapy. However, some tumors fail to respond despite being PD-L1+. A correlative study using data from the cancer genome atlas found that DLBCLs with a low PD-L1 protein-to-mRNA ratio while also having higher relative expression levels of ADAM10 or ADAM17 had worse overall survival than high PD-L1 protein-to-mRNA counterparts (68). Two recent studies demonstrated that ADAM10 and ADAM17 can cleave PD-L1 in culture (69). With these data, the regulation of the PDL-1/PD-1 pathway can be better understood. Soluble PD-L1 is not completely understood, thus more work is needed to determine if inhibition of this part of the pathway will be helpful. Then we will know if ADAM inhibition will be helpful or harmful in nHL.
Multiple myeloma (MM) is characterized by a clonal expansion of plasma cells filling the bone marrow volume >30%. These cells often secrete large amounts of monoclonal Ig into the blood. This leads to a loss of structural integrity of the bone resulting in increased risk of bone fracture, as well as kidney damage due to the accumulation of monoclonal Ig deposits (70). In a study utilizing human MM cells, doxorubicin was used to model genotoxic stress. This upregulated ADAM10 and resulted in increased shedding of MICA and MICB. The increased ADAM10 expression in MM was associated with a senescent phenotype (71). This finding suggests that ADAM10 inhibitors may enhance NK cell immunotherapy in multiple myeloma and other cancers.
Breast Cancer
The human epidermal growth factor receptor (HER) family consists of four receptors: EGF receptor (EGFR), HER2, HER3, and HER4. HER2 overexpression occurs in ~10–20% of all invasive ductal mammary carcinomas of no special type (NST) and these are treated with trastuzumab, which is a monoclonal anti-HER2 antibody (72). While patients with HER2+ breast cancers often initially respond well to trastuzumab, it is common for cancer to relapse in a more resistant form (73). ADAM10 is known to cleave HER2. Increased shedding of HER2 into a soluble form, HER2-extra cellular domain (ECD) by ADAM10 has recently been shown to be predictive of reduced progression-free survival (74). In 545 HER2 positive invasive ductal mammary carcinoma (NST) patients, it was found that a high serum HER2-ECD relative to tumor HER2 level was predictive of reduced progression-free survival (74). Feldinger et al. demonstrated that after treatment with trastuzumab, ADAM10 levels increased both in vitro and in vivo using PDX (75). Treatment with the ADAM10 inhibitor INCB8765 increased sensitivity to trastuzumab in vitro, as well as restored responses in trastuzumab-resistant cells (75) (Model Figure 1). A different study confirmed these results in esophageal cancer PDX models and proposed that ADAM10 is conferring this resistance through the cleavage of the HER3 ligand (NRG-1β) (76). When HER2 is lost through trastuzumab treatment, HER3 is upregulated in a compensatory manner (76). The observed increase in ADAM10 then releases the ligand for HER3 and confers trastuzmab resistance. This can be reversed with ADAM10 inhibitor (GI254023X) treatment (76). Further studies are needed to confirm this mechanism in breast cancer as well as to examine the additive effects of ADAM10 inhibitors in combination with monoclonal anti-HER2 therapies, but the cleavage product HER2-ECD shows promise as a prognostic biomarker.
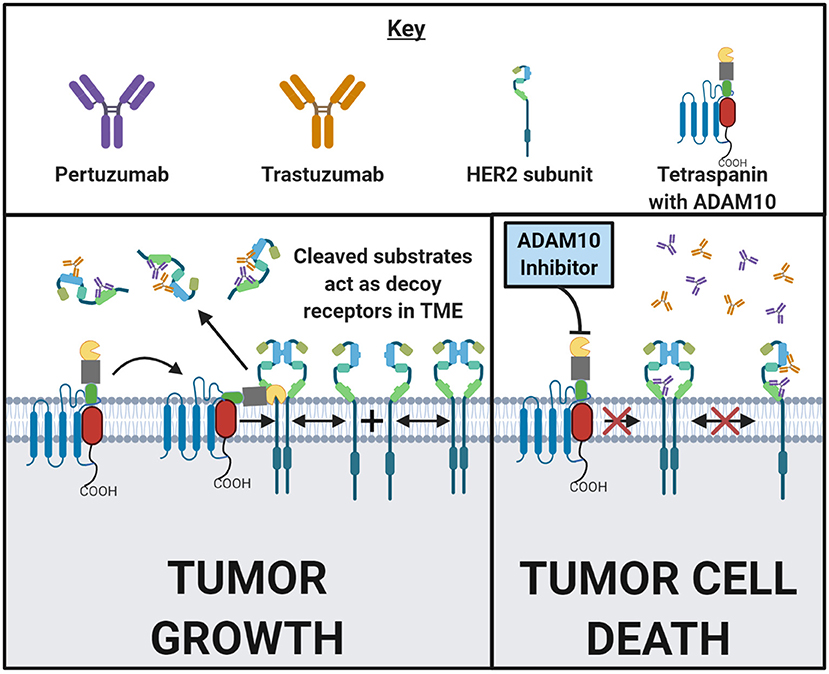
Figure 1. ADAM10 mediates cell surface cleavage of a large repertoire of substrates that can promote a pro-growth environment in malignant tumors. Some of these ADAM10 substrates, such as HER2, are targeted by FDA approved drugs. Following HER2 subunit dimerization with HER- family subunits, and upon ligand-binding or autoactivation, a pro-growth signal cascade is initiated which then drives malignancy. Trastuzumab and pertuzumab are both monoclonal antibodies that bind to two distinct epitopes of HER2 to prevent homodimerizaton and heterodimerization, respectively. In addition to inhibiting dimerization, tumor cells with bound antibody have an increased likelihood of succumbing to antibody dependent cellular cytotoxicity (ADCC) or opsonization. In the left and right panels above, a HER2-positive cancer cell surface in the presence of trastuzumab and pertuzumab is depicted. In the left panel, ADAM10 cleaves the extracellular domain of HER2 from the cell surface into the tumor microenvironment (TME). These cleaved domains then act as decoy receptors, decreasing the amount of trastuzumab or pertuzumab that binds to the tumor cell. The likelihood of ADCC or opsonization is now decreased. In the right panel, treatment with trastuzumab or pertuzumab is combined with the inhibition of ADAM10. Inhibiting ADAM10 results in less HER2 cleavage which reduces the amount of decoy receptors in the TME. Decreasing the decoy receptors in the TME and increasing the amount of HER2 on the tumor cell might enhance trastuzumab's and/or pertuzumab's antitumor effects. This general concept can be applied to other ADAM10 substrates in other disease states. Made in ©BioRender - biorender.com.
Triple negative breast cancer (TNBC) is a variant of breast cancer that exhibits little to no expression of the HER2, progesterone receptor (PR), or estrogen receptor (ER) and is often correlated with poor prognosis due to limited treatment options (77). Both ADAM10 and ADAM17 have been found to be expressed in the majority of TNBCs (78). In a study that used RNAi knockdown of ADAM10, or the ADAM10 inhibitor GI254023X in several different TNBC cell lines such as MDA-MB-231 and BT20, it was found that ADAM10 knockdown decreased in vitro cell migration (79). A different study that examined five TNBC cell lines reported finding that microRNA-365, (miR-365) directly interacts with the 3'-UTR of ADAM10 mRNA. Moreover, they reported that re-expression of ADAM10 led to the restoration of the cells ability to proliferate, migrate, and invade which was suppressed when overexpressing miR-365 (80). TNBCs have been reported to have the highest levels of PD-L1 amongst breast cancer types. Much like with DLBCLs, it has been proposed that ADAM10 may play a role in regulating the PD-1/PD-L1 axis (69). Recently, a study done on non-luminal breast cancers (Her2+ and TNBC), published in EBioMedicine, identified soluble APPα that is generated by ADAM10 cleavage of APP, as important in breast cancer tumor migration and proliferation (81). The importance of APP was shown using APP knockdown tumors in vitro and in vivo. This was also shown by knockdown of ADAM10 using RNAi (81). ADAM10 cleavage of APP in Alzheimer's disease has been extensively studied [and is reviewed in (82, 83)]. The expansion of importance in breast cancer emphasizes the examination of ADAM10 cleavage of APP in other cancers.
The xenoestrogens bisphenol-A (BPA) and nonylphenol (NP) are used by the plastics industry in products for human use (84). Urriola-Muñoz et al. demonstrated that both BPA and NP increased ADAM10 and ADAM17 activity, increasing the release of several EGFR ligands (84). Given the variety of important substrates of ADAM10 and ADAM17 in breast cancer as well as other cancers, this study sheds new light on the potential complications associated with these common xenoestrogens. Much more research will need to be done.
Oral Squamous Cell Carcinoma
Oral squamous cell carcinoma (OSCC) is an invasive carcinoma derived from malignantly transformed squamous epithelium lining the oral cavity (85). Based on 80 cases of OSCC samples analyzed, it was determined that OSCC expressed high levels of ADAM10 and that the samples with the highest ADAM10 expression were found in metastatic lesions (86). Tissue inhibitor of metalloproteinase-3 (TIMP-3) has been shown to inhibit OSCC cell growth, angiogenesis, migration, and invasion (87). TIMP-3 is a secreted protein that binds to the ECM and inhibits metalloproteinase activity, with a particular affinity for suppression of ADAM10 activity (87). One study showed that in OSCC, increased hypermethylation of the TIMP-3 promotor led to reduced TIMP-3 mRNA expression. Treatment with a DNA methytransferase inhibitor (DNMTi) or overexpression of TIMP-3 reversed tumor cell migration, proliferation, and reduced the epithelial to mesenchymal transition (87). These effects may be due to loss of ADAM10 activity, but further work would need to be done to show this. Another recent study using HEK293 cells that overexpress TIMP-3 show evidence of reduced ADAM10-specific substrates after secretome analysis by mass spectrometry label free quantification. This study showed that with TIMP-3 overexpression, several ligands for the low-density-lipoprotein receptor-related protein-1 (LRP-1) are upregulated, such as macrophage inhibitor factor (MIF) (88). As TIMP-3 also binds to LRP-1, overexpression of TIMP-3 outcompetes other ligands of LRP-1 and thus the cell compensates by increasing expression (88). The authors of this study warn that the use of TIMP-3 interventions as an ADAM10 regulator could yield these alterations to the secretome and results might be unanticipated.
As in HL and GBM, OSCC are reported to have low expression levels of stress receptor, MICA, which has been shown to be under the regulation of ADAM10. In OSCC, the cleavage of MICA reduces tumor immunogenicity (89). Upon over-expressing MICA in the human squamous cell line SCC-25, increased NK cell killing was observed. This suggests ADAM10 regulation of stress receptors may also be occurring OSCC (89).
ADAM10 and ADAM10 Substrates as Biomarkers in Cancer
A multi-center cross-validation study (1558 enrollment) was conducted to assess Heat shock protein 90α (Hsp90α) as a pan-cancer biomarker (90). A siRNA screen identified ADAM10 as responsible for the release of Hsp90α. This was validated through the use of the ADAM10 inhibitor (GI254023X) on tumor cell lines, where reduced release of Hsp90α was observed (90). The study identified ADAM10 as another potential biomarker in conjunction with Hsp90α, and looked at the pair as an exosomal biomarker (90). Overall, it emphasized the strength of Hsp90α, as a potential pan-cancer biomarker. Another study identified serum ADAM10 levels as a biomarker for disease in colorectal cancer by ELISA. This study also found ADAM10 had a minor, yet significant positive correlation with clinical stage (91). In sacral chordoma, a rare malignant primary bone tumor in the spine, a study spanning seven years positively correlated ADAM10 levels with increased metastasis, disease-free survival, overall survival, and histological type (92). Low ADAM10 expression by histochemical staining in patient tumors equated to longer survival as compared to high ADAM10 expression (92). Many different tumor types have identified ADAMs as potential biomarkers for disease or disease progression, the evidence for ADAM10 as biomarker is building and warrants larger-scale multi-center validation studies in order to be implemented in the clinic.
ADAM10 in Autoimmunity
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a chronic inflammatory disease affecting synovial joints, resulting in synovitis, cartilage destruction, and joint ankylosis. The pathophysiology of RA is largely driven by autoreactive antibodies directed against neoantigens generated by post-translational citrullination and carbamylation of self-peptides (93). These autoantibodies, which can be detected prior to the onset of clinically evident disease, form immune complexes which deposit in synovial joints and induce local inflammatory responses at articular surfaces (93). Given the role of ADAM10 in regulating antibody production and inflammatory responses, it is considered a promising therapeutic target to control RA disease activity and progression.
Cleavage of ADAM10 substrates in synovial tissue is involved in several pro-inflammatory processes. CXCL16, which requires ADAM10 cleavage to exert its biological effects, functions as a chemotactic signal for effector and memory T-helper 1 cells. ADAM10 and CXCL16 are upregulated in synovial joint biopsies from RA patients compared to healthy controls (94). These molecules are co-expressed on the surface of synovial macrophages, where they function to drive the accumulation of effector T-helper 1 cells, thereby promoting local inflammatory processes and exacerbating joint injury (94). In vitro siRNA knockdown of ADAM10 in RA-patient derived synovial fibroblasts also suppressed the release of the proinflammatory cytokines TNF-α, IL-6, and IL-8. These findings indicate that inhibition of ADAM10 may be effective in the treatment of RA by suppressing pro-inflammatory signaling within synovial tissue.
An early histological hallmark of RA pathogenesis is synovial angiogenesis, which permits leukocyte infiltration and progression to synovitis (93). Recent studies demonstrate that ADAM10 is upregulated in endothelial cells and synovial lining fibroblasts in RA tissue biopsies compared to osteoarthritis and healthy patients (95). Additionally, in vitro studies reveal that siRNA knockdown of ADAM10 impairs angiogenesis and suppresses VEGF release in endothelial cell lines (95, 96). Administration of this siRNA in vivo in a murine model of collagen-induced arthritis improved arthritis symptoms and reduced serum levels of the angiogenic cytokine VEGF (97). The involvement of ADAM10 in angiogenic processes in RA progression indicate that early inhibition of ADAM10 may slow or halt disease progression.
An additional, particularly debilitating outcome in RA is bone erosion (98). Erosion of bone in RA is thought to result from a local inflammatory milieu driving exaggerated osteoclast activity and invasion into periosteal regions (93). Endothelial cell ADAM10 has been shown to modulate osteoclast function. Murine models of ADAM10 deletion in endothelial cells led to reduced osteoclast numbers at the chondro-osseous junction and impaired long bone growth, indicating abnormal osteoclast function (99). Thus, inhibition of ADAM10 may restrain osteoclast activity and reduce the incidence of bone erosion in RA.
Though ADAM10 appears to be a promising therapeutic target in the management of RA progression, it may also provide a biomarker for predicting patient responsiveness to biologic therapies. A recent study demonstrated that elevated serum ADAM10 positively predicts treatment responsiveness to tocilizumab, a monoclonal antibody targeting the IL-6 receptor (100). Patients who were responsive to tocilizumab therapy had a roughly 6-fold higher baseline ADAM10 level compared to non-responders. Therefore, routine testing of RA patients for ADAM10 serum levels may offer guidance for the use of targeted therapies for specific patient groups in RA.
To date, the evidence regarding the role of ADAM10 in RA indicates that it would likely be a useful therapeutic target. Many of the existing small molecule inhibitors of ADAM10, such as GI254023X, INCB3619, and INCB7839, have the added benefit of inhibiting the ADAM10 homolog ADAM17. ADAM17 is a sheddase for a variety of pro-inflammatory molecules that are involved in RA pathogenesis (101–103). Thus, these drugs may provide a significant clinical benefit in the management of RA symptoms.
At present, none of the ADAM10 inhibitors have been evaluated for efficacy in clinical trials for RA. Clinical trials of GI254023X were discontinued in phase I/II due to hepatotoxicity following systemic administration (104). Preliminary studies of INCB7839, however, suggest that it is safe and well-tolerated for systemic use indicating that this class of drugs may have promise in the management of RA (105).
Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is a common, multisystem autoimmune disorder that is characterized by aberrant antibody production directed against nuclear antigens such as double-stranded DNA and small nuclear ribonucleoproteins (106). These autoantibodies form immune complexes which deposit within tissues and promote organ dysfunction. Therefore, an effective therapeutic approach to SLE would require suppression of antibody production or the inflammatory reactions to immune complex deposition.
Numerous studies have demonstrated hyperactivity of the B cell activating factor (BAFF)—transmembrane activator and CAML interactor (TACI) system in SLE patients as well as in murine models of SLE (107, 108). Binding of BAFF by TACI on B cells causes excessive proliferation and T-independent activation of low-affinity self-reactive B cells. This activation leads to increased antibody production against harmless self-antigens which promotes disease progression in SLE (109). Recently, TACI was identified as a substrate for cleavage by ADAM10 (110). ADAM10-induced cleavage of TACI induces ectodomain shedding and the generation of soluble TACI (sTACI). This allows sTACI to function as a decoy receptor by binding soluble BAFF and APRIL to prevent T-independent B cell activation (110). These findings indicate that activation of ADAM10 in SLE may suppress aberrant B cell activity and reduce the synthesis of autoreactive antibodies.
ICOSL is another ADAM10 substrate that has been linked to abnormal antibody production (23). B cell ADAM10 is necessary for proper ectodomain shedding of ICOSL. Impaired ICOSL shedding leads to accumulation of B cell surface ICOSL which stimulates internalization of ICOS on T cells. This excessive internalization results in inadequate ICOS signaling, impairing T follicular helper cell maturation, thereby reducing antibody production (23). In a murine model of SLE, ADAM10 deletion in B cells led to suppression of germinal center responses and reduced levels of antibodies directed against ds-DNA (111). This indicates, in contrast to the findings above, that inhibition of ADAM10 may be beneficial in the treatment of SLE.
Activation of macrophages in peripheral tissues is another significant component of SLE pathogenesis (106). Axl is a receptor tyrosine kinase found predominantly on macrophages which, upon binding of its ligand Gas6, inhibits production of pro-inflammatory cytokines (112). ADAM10 promotes shedding of Axl, leading to hyperactivity of tissue macrophages and exacerbation of tissue damage in a murine model of lupus (113). Significantly, the cleaved, soluble form of Axl was found to be increased in serum from patients with active SLE flares compared to SLE patients without active disease and healthy controls (113). This, like the findings of Hoffman et al. indicate that activation of ADAM10 may ameliorate SLE symptoms and slow disease progression.
These studies indicate that ADAM10 may be a relevant therapeutic target in SLE. To date, however, the evidence for modulating ADAM10 in SLE remains unclear. It seems that inhibition of ADAM10 on B cells would reduce TACI cleavage, leading to increased T-independent B cell activation, but may also alter ICOS-ICOSL signaling leading to excessive antibody production. Further study is needed to elucidate the implications of targeting ADAM10 in SLE as a therapeutic strategy. It is likely that the role of ADAM10 in SLE is highly spatio-temporally dependent and the effects of modulating ADAM10 may vary greatly depending upon when intervention occurs in the natural history of the disease.
Psoriasis
Psoriasis is an inflammatory disease that is characterized by hyperproliferative lesions of the skin that are associated with immunological dysregulation and aberrant keratinocyte differentiation (114). Keratinocyte differentiation is critically regulated by Notch signaling which drives sequential maturation from basal stem cells to spinous layer keratinocytes (115, 116). In Notch signaling, Notch ligand binding induces a conformational change in Notch, exposing the negative regulatory region for cleavage by ADAM10. This cleavage event provides a substrate for γ-secretase to perform an additional cleavage to generate the transcriptionally active NICD (17). Epithelial deletion of ADAM10 in adult mice results in hyperproliferation of keratinocytes and dysregulated keratinocyte differentiation due to impaired Notch signaling. Lesions from these mice resemble the architecture of human psoriatic lesions, exemplified by a thickened epidermis and proliferative basal-like cells present in suprabasal epidermal layers (117).
It remains unclear whether psoriatic lesions display derangements in ADAM10 expression or activity. One study demonstrated upregulation of ADAM10 in keratinocytes in psoriatic lesions, with increased levels in deeper layers of the epidermis compared to healthy controls (118). These findings, however, are based on only a single immunohistochemistry study. Thus, further study of the role of ADAM10 in keratinocyte differentiation and propagation of psoriatic inflammation is needed.
Clinically, acitretin is an oral retinoid that is approved for psoriasis treatment (119). Acitretin functions as a retinoic acid receptor agonist that promotes keratinocyte differentiation. Activation of retinoic acid receptors, however, also induces expression of ADAM10 in vitro (120). The upregulation of ADAM10 by acitretin may facilitate Notch signaling in keratinocytes, thereby restoring normal keratinocyte differentiation and epidermal architecture. Tamibarotene, cilostazol, and resveratrol are additional agents that induce the expression of ADAM10 via activation of retinoic acid receptors (121, 122). The effects of these drugs may be comparable to acitretin in the treatment of psoriasis and warrant further study to investigate their efficacy and side effect profiles.
Currently, direct small molecule activators of ADAM10 are not available. The ADAM10 activators that have been described are etazolate, bryostatin, and (–)-epigallocatechin-3-gallate (EGCG) (123). These drugs, however, activate ADAM10 through a mediating receptor. Etazolate activates ADAM10 secondary to activation of the GABAA receptor and therefore displays significant tropism to cells of the central nervous system (124). Bryostatin activates ADAM10 via protein kinase C and can therefore induce ADAM10 activity in a variety of cell types (125, 126). EGCG, a natural occurring compound derived from green tea, has also been described to induce ADAM10 activity (127). At present, the target receptor for EGCG remains unidentified. ADAM10 activation by EGCG, however, is clearly secondary to a tyrosine kinase, as tyrosine kinase inhibitors prevent EGCG induced ADAM10 activation (127). Clinical trials for EGCG are underway and have demonstrated safety and tolerability in studies of prostate cancer and Fragile X syndrome (128, 129). Considering the recent emergence of several biologics for treatment refractory psoriasis, these ADAM10 inducers may provide a valuable and cost-effective first line treatment option for psoriasis.
Bullous Pemphigoid
Bullous pemphigoid (BP) is a severe autoimmune cutaneous blistering condition that is caused by autoantibodies directed against transmembrane collagen XVII (anti-BP180). These antibodies interfere with epidermal basal cell adhesion, leading to separation of the dermis from the epidermis which causes blistering (130). The central role of pathological B cell activation in BP was illustrated in a study by Hall et al. in which patients with recalcitrant disease exhibited a significant reduction in disease activity following B cell depletion by rituximab (131). Analysis of serum and blister fluid revealed elevated levels of semaphorin 4D, which augments production of BP180 antibodies in vitro. Semaphorin 4D release is derived from CD15+ granulocytes and occurred through an ADAM10-dependent mechanism (132). These findings indicate that inhibition of ADAM10 could suppress autoreactive antibody production in BP, thereby reducing disease activity.
Conclusions
The overwhelming data in cancer and autoimmunity, even added within the last 5 years implicates ADAM10 in the progression of disease. The popularity of ADAM inhibition in the 90s is returning, and the larger question is, will it be merely a passing trend? Early, the need for compounds that were specific for ADAM10 was made especially evident following the failure of the first generation metalloproteinase inhibitors, Batimastat, Marimastat, and Neovastat early in clinical trials due to reports of adverse side effects (133, 134) Even with the addition of specific ADAM10 targeting drugs, the other major obstacle was treating ADAM10 at the point of therapeutic intervention. With the ubiquitous nature of ADAM10 throughout human cell types and tissues, this has been extremely difficult. Unless ADAM10 intervention is accompanied by a drug delivery system that allows for a targeted approach, there is almost certainly going to be off-target and on-target effects (through inhibition of Notch cleavage) leading to adverse events.
Even in the absence of a cell- or tissue-specific approach of targeting ADAM10, potential still exists in the right clinical setting. New inhibitor design may also improve off-target and on-target effects, as well as be able to generate a more selective inhibitor between ADAM10 and ADAM17. Recent structural work from Seegar et al. may yield improved inhibitors of ADAM10, as more is now known about the structure and auto-modulation through the disintegrin cysteine-rich domain (5). This, paired with improved knowledge of tetraspanin regulation of ADAM10, may allow generation of better inhibitors that are able to specifically target ADAM10's cleavage of particular substrates (6). Further, protease inhibitors may be surpassed in specificity through the use of monoclonal antibodies that mask the binding pocket for ADAM10, like mAb 8C7 (135). Yet, without the implementation of a personalized medicine approach, especially in cancer, ADAM10 inhibition therapy will most likely continue to fail in clinical trials. Identifying patients who will benefit the most from ADAM10 inhibition therapy should be the priority, as well as applying ADAM10 inhibitors to prevent resistance of the tumor to standard treatments such as anti-CD30 or anti-HER2. In autoimmunity the story remains less clear, yet most autoimmune diseases have relevant targets.
Multiple reviews have been published supporting the potential of ADAM10 protein in platelets and cerebrospinal fluid to serve as a biomarker for Alzheimer's disease diagnoses (82, 136, 137). A study that examined in the urinary vesicles of patients with glomerular kidney diseases found higher levels of ADAM10 (138). ADAM10 is also overexpressed in the synovial tissue of RA patients (95). It has been reported that ADAM10 is correlated with disease activity and regulates monocyte migration and adhesion in RA patient fluids (96, 97). ADAM10 elevation in specific cells or tissues correlates strongly with various disease states. However, ADAM10 is also elevated broadly in many cancers on exosomes. Exosomal ADAM10 was found to be elevated in plasma of cancer patients, and was thought to be an additional pan-cancer marker (90). The use of ADAM10 expression in plasma or ADAM10 substrates as biomarkers in cancer or for disease progression are extremely promising. But, the studies in this review, aside from Hsp90α, need replication in multi-center validation studies prior to use in a clinical setting. The additional complication is the ubiquitous nature of ADAM10 expression and the upregulation in multiple disease states could cause issues with its use as a biomarker in situations with comorbidities.
Overall, ADAM10 is still extremely targetable. Improvements in drug delivery, reduction in off-target effects, and careful identification of patient populations will be needed to successfully move these drugs into the clinic.
Author Contributions
TS, AT, and RM all participated in the writing, editing, and overall construction of this review.
Funding
This review was supported by the NIH/NIAID R56AI139658.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Marcia L. Moss for her initial involvement in the development of this review article and Timothy F. Martin for his assistance in editing.
References
1. Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. (2005) 6:32–43. doi: 10.1038/nrm1548
2. White JM. ADAMs: modulators of cell-cell and cell-matrix interactions. Curr Opin Cell Biol. (2003) 15:598–606. doi: 10.1016/j.ceb.2003.08.001
3. Wong E, Maretzky T, Peleg Y, Blobel CP, Sagi I. The functional maturation of A Disintegrin and Metalloproteinase (ADAM) 9, 10, and 17 requires processing at a newly identified Proprotein Convertase (PC) cleavage site. J Biol Chem. (2015) 290:12135–46. doi: 10.1074/jbc.M114.624072
4. Moss ML, Bomar M, Liu Q, Sage H, Dempsey P, Lenhart PM, et al. The ADAM10 prodomain is a specific inhibitor of ADAM10 proteolytic activity and inhibits cellular shedding events. J Biol Chem. (2007) 282:35712–21. doi: 10.1074/jbc.M703231200
5. Seegar TCM, Killingsworth LB, Saha N, Meyer PA, Patra D, Zimmerman B, et al. Structural basis for regulated proteolysis by the alpha-secretase ADAM10. Cell. (2017) 171:1638–48.e7. doi: 10.1016/j.cell.2017.11.014
6. Saint-Pol J, Eschenbrenner E, Dornier E, Boucheix C, Charrin S, Rubinstein E. Regulation of the trafficking and the function of the metalloprotease ADAM10 by tetraspanins. Biochem Soc Trans. (2017) 45:937–44. doi: 10.1042/BST20160296
7. Saraceno C, Marcello E, Di Marino D, Borroni B, Claeysen S, Perroy J, et al. SAP97-mediated ADAM10 trafficking from golgi outposts depends on PKC phosphorylation. Cell Death Dis. (2014) 5:e1547. doi: 10.1038/cddis.2014.492
8. Mathews JA, Gibb DR, Chen B-H, Scherle P, Conrad DH. CD23 sheddase a disintegrin and metalloproteinase 10 (ADAM10) is also required for CD23 sorting into B cell-derived exosomes. J Biol Chem. (2010) 285:37531–41. doi: 10.1074/jbc.M110.141556
9. Kuno K, Kanada N, Nakashima E, Fujiki F, Ichimura F, Matsushima K. Molecular cloning of a gene encoding a new type of metalloproteinase-disintegrin family protein with thrombospondin motifs as an inflammation associated gene. J Biol Chem. (1997) 272:556–62. doi: 10.1074/jbc.272.1.556
10. Blobel CP. Metalloprotease-disintegrins: links to cell adhesion and cleavage of TNF alpha and notch. Cell. (1997) 90:589–92. doi: 10.1016/S0092-8674(00)80519-X
11. Fambrough D, Pan D, Rubin GM, Goodman CS. The cell surface metalloprotease/disintegrin kuzbanian is required for axonal extension in Drosophila. Proc Natl Acad Sci USA. (1996) 93:13233–38. doi: 10.1073/pnas.93.23.13233
12. Alfandari D, Wolfsberg TG, White JM, DeSimone DW. ADAM 13: a novel ADAM expressed in somitic mesoderm and neural crest cells during Xenopus laevis development. Dev Biol. (1997) 182:314–30. doi: 10.1006/dbio.1996.8458
13. Weber S, Saftig P. Ectodomain shedding and ADAMs in development. Development. (2012) 139:3693–709. doi: 10.1242/dev.076398
14. Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. (2014) 13:397–406. doi: 10.1074/mcp.M113.035600
15. Crawford HC, Dempsey PJ, Brown G, Adam L, Moss ML. ADAM10 as a therapeutic target for cancer and inflammation. Curr Pharm Des. (2009) 15:2288–99. doi: 10.2174/138161209788682442
16. Gibb DR, Saleem SJ, Chaimowitz NS, Mathews J, Conrad DH. The emergence of ADAM10 as a regulator of lymphocyte development and autoimmunity. Mol Immunol. (2011) 48:1319–27. doi: 10.1016/j.molimm.2010.12.005
17. Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, et al. The disintegrin/metalloprotease ADAM 10 is essential for notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet. (2002) 11:2615–24. doi: 10.1093/hmg/11.21.2615
18. Schirrmeister W, Gnad T, Wex T, Higashiyama S, Wolke C, Naumann M, et al. Ectodomain shedding of E-cadherin and c-Met is induced by Helicobacter pylori infection. Exp Cell Res. (2009) 315:3500–08. doi: 10.1016/j.yexcr.2009.07.029
19. Le Gall SM, Bobé P, Reiss K, Horiuchi K, Niu X-D, Lundell D, et al. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor alpha, L-selectin, and tumor necrosis factor alpha. Mol Biol Cell. (2009) 20:1785–94. doi: 10.1091/mbc.e08-11-1135
20. Sahin U, Weskamp G, Kelly K, Zhou H-M, Higashiyama S, Peschon J, et al. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol. (2004) 164:769–79. doi: 10.1083/jcb.200307137
21. Kirkin V, Cahuzac N, Guardiola-Serrano F, Huault S, Luckerath K, Friedmann E, et al. The Fas ligand intracellular domain is released by ADAM10 and SPPL2a cleavage in T-cells. Cell Death Differ. (2007) 14:1678–87. doi: 10.1038/sj.cdd.4402175
22. Yacoub D, Benslimane N, Al-Zoobi L, Hassan G, Nadiri A, Mourad W. CD154 is released from T-cells by a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) and ADAM17 in a CD40 protein-dependent manner. J Biol Chem. (2013) 288:36083–93. doi: 10.1074/jbc.M113.506220
23. Lownik JC, Luker AJ, Damle SR, Cooley LF, El Sayed R, Hutloff A, et al. ADAM10-mediated ICOS ligand shedding on B Cells is necessary for proper T cell ICOS regulation and T follicular helper responses. J Immunol. (2017) 199:2305–15. doi: 10.4049/jimmunol.1700833
24. Chitadze G, Lettau M, Bhat J, Wesch D, Steinle A, Furst D, et al. Shedding of endogenous MHC class I-related chain molecules A and B from different human tumor entities: heterogeneous involvement of the “a disintegrin and metalloproteases” 10 and 17. Int J cancer. (2013) 133:1557–66. doi: 10.1002/ijc.28174
25. Waldhauer I, Steinle A. Proteolytic release of soluble UL16-binding protein 2 from tumor cells. Cancer Res. (2006) 66:2520–6. doi: 10.1158/0008-5472.CAN-05-2520
26. Saha N, Robev D, Himanen JP, Nikolov DB. ADAM proteases: emerging role and targeting of the non-catalytic domains. Cancer Lett. (2019) 467:50–7. doi: 10.1016/j.canlet.2019.10.003
27. Hsia H-E, Tushaus J, Brummer T, Zheng Y, Scilabra SD, Lichtenthaler SF. Functions of “A disintegrin and metalloproteases (ADAMs)” in the mammalian nervous system. Cell Mol Life Sci. (2019) 76:3055–81. doi: 10.1007/s00018-019-03173-7
28. Kato T, Hagiyama M, Ito A. Renal ADAM10 and 17: their physiological and medical meanings. Front cell Dev Biol. (2018) 6:153. doi: 10.3389/fcell.2018.00153
29. Yuan JS, Kousis PC, Suliman S, Visan I, Guidos CJ. Functions of notch signaling in the immune system: consensus and controversies. Annu Rev Immunol. (2010) 28:343–65. doi: 10.1146/annurev.immunol.021908.132719
30. Lambrecht BN, Vanderkerken M, Hammad H. The emerging role of ADAM metalloproteinases in immunity. Nat Rev Immunol. (2018) 18:745–58. doi: 10.1038/s41577-018-0068-5
31. van Tetering G, van Diest P, Verlaan I, van der Wall E, Kopan R, Vooijs M. Metalloprotease ADAM10 is required for notch1 site 2 cleavage. J Biol Chem. (2009) 284:31018–27. doi: 10.1074/jbc.M109.006775
32. Gibb DR, Saleem SJ, Kang D-J, Subler MA, Conrad DH. ADAM10 overexpression shifts lympho- and myelopoiesis by dysregulating site 2/site 3 cleavage products of notch. J Immunol. (2011) 186:4244–52. doi: 10.4049/jimmunol.1003318
33. Chastagner P, Rubinstein E, Brou C. Ligand-activated notch undergoes DTX4-mediated ubiquitylation and bilateral endocytosis before ADAM10 processing. Sci Signal. (2017) 10:483 doi: 10.1126/scisignal.aag2989
34. Tousseyn T, Thathiah A, Jorissen E, Raemaekers T, Konietzko U, Reiss K, et al. ADAM10, the rate-limiting protease of regulated intramembrane proteolysis of notch and other proteins, is processed by ADAMS-9, ADAMS-15, and the gamma-secretase. J Biol Chem. (2009) 284:11738–747. doi: 10.1074/jbc.M805894200
35. Gibb DR, El Shikh M, Kang D-J, Rowe WJ, El Sayed R, Cichy J, et al. ADAM10 is essential for notch2-dependent marginal zone B cell development and CD23 cleavage in vivo. J Exp Med. (2010) 207:623–35. doi: 10.1084/jem.20091990
36. Weskamp G, Ford JW, Sturgill J, Martin S, Docherty AJP, Swendeman S, et al. ADAM10 is a principal “sheddase” of the low-affinity immunoglobulin E receptor CD23. Nat Immunol. (2006) 7:1293–8. doi: 10.1038/ni1399
37. Yasutomo K, Doyle C, Miele L, Fuchs C, Germain RN. The duration of antigen receptor signalling determines CD4+ versus CD8+ T-cell lineage fate. Nature. (2000) 404:506–10. doi: 10.1038/35006664
38. Washburn T, Schweighoffer E, Gridley T, Chang D, Fowlkes BJ, Cado D, et al. Notch activity influences the alphabeta versus gammadelta T cell lineage decision. Cell. (1997) 88:833–43. doi: 10.1016/S0092-8674(00)81929-7
39. Robey E, Chang D, Itano A, Cado D, Alexander H, Lans D, et al. An activated form of notch influences the choice between CD4 and CD8 T cell lineages. Cell. (1996) 87:483–92. doi: 10.1016/S0092-8674(00)81368-9
40. Guy CS, Vignali KM, Temirov J, Bettini ML, Overacre AE, Smeltzer M, et al. Distinct TCR signaling pathways drive proliferation and cytokine production in T cells. Nat Immunol. (2013) 14:262–70. doi: 10.1038/ni.2538
41. Maekawa Y, Ishifune C, Tsukumo S, Hozumi K, Yagita H, Yasutomo K. Notch controls the survival of memory CD4+ T cells by regulating glucose uptake. Nat Med. (2015) 21:55–61. doi: 10.1038/nm.3758
42. Baaten BJG, Tinoco R, Chen AT, Bradley LM. Regulation of antigen-experienced T cells: lessons from the quintessential memory marker CD44. Front Immunol. (2012) 3:23. doi: 10.3389/fimmu.2012.00023
43. Nagano O, Saya H. Mechanism and biological significance of CD44 cleavage. Cancer Sci. (2004) 95:930–5. doi: 10.1111/j.1349-7006.2004.tb03179.x
44. Ebsen H, Lettau M, Kabelitz D, Janssen O. Subcellular localization and activation of ADAM proteases in the context of FasL shedding in T lymphocytes. Mol Immunol. (2015) 65:416–28. doi: 10.1016/j.molimm.2015.02.008
45. Woo S-R, Turnis ME, Goldberg M V, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. (2012) 72:917–27. doi: 10.1158/0008-5472.CAN-11-1620
46. Venkatesh HS, Tam LT, Woo PJ, Lennon J, Nagaraja S, Gillespie SM, et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature. (2017) 549:533–7. doi: 10.1038/nature24014
47. Liu R, Qin X-P, Zhuang Y, Zhang Y, Liao H-B, Tang J-C, et al. Glioblastoma recurrence correlates with NLGN3 levels. Cancer Med. (2018) 7:2848–59. doi: 10.1002/cam4.1538
48. Musumeci G, Magro G, Cardile V, Coco M, Marzagalli R, Castrogiovanni P, et al. Characterization of matrix metalloproteinase-2 and−9, ADAM-10 and N-cadherin expression in human glioblastoma multiforme. Cell Tissue Res. (2015) 362:45–60. doi: 10.1007/s00441-015-2197-5
49. Kohutek ZA, Charles G, Redpath GT, Hussaini IM. ADAM-10-mediated N-Cadherin cleavage is protein kinase C-alpha dependent and promotes glioblastoma cell migration. (2009) 29:4605–15. doi: 10.1523/JNEUROSCI.5126-08.2009
50. Kanaya K, Sakai K, Hongo K, Fukushima M, Kawakubo M. High expression of ADAM10 predicts a poor prognosis for patients with glioblastoma. (2017) 10:618–24.
51. Wang J, Matosevic S. NT5E/CD73 as correlative factor of patient survival and natural killer cell infiltration in glioblastoma. J Clin Med. (2019) 8:1526. doi: 10.3390/jcm8101526
52. Thompson TW, Kim AB, Li PJ, Wang J, Jackson BT, Huang KTH, et al. Endothelial cells express NKG2D ligands and desensitize antitumor NK responses. Elife. (2017) 6:e30881. doi: 10.7554/eLife.30881
53. Lee Y-S, Heo W, Son C-H, Kang C-D, Park Y-S, Bae J. Upregulation of Myc promotes the evasion of NK cellmediated immunity through suppression of NKG2D ligands in K562 cells. Mol Med Rep. (2019) 20:3301–7. doi: 10.3892/mmr.2019.10583
54. Kohga K, Takehara T, Tatsumi T, Miyagi T, Ishida H, Ohkawa K, et al. Anticancer chemotherapy inhibits MHC class I-related chain a ectodomain shedding by downregulating ADAM10 expression in hepatocellular carcinoma. Cancer Res. (2009) 69:8050–7. doi: 10.1158/0008-5472.CAN-09-0789
55. Wolpert F, Tritschler I, Steinle A, Weller Michael, Eisele G. A disintegrin and metalloproteinases 10 and 17 modulate the immunogenicity of glioblastoma-initiating cells. Neuro Oncol. (2013) 16:382–91. doi: 10.1093/neuonc/not232
56. Gjorgjevski M, Hannen R, Carl B, Li Y, Landmann E, Buchholz M, et al. Molecular profiling of the tumor microenvironment in glioblastoma patients: correlation of microglia/macrophage polarization state with metalloprotease expression profiles and survival. Biosci Rep. (2019) 39:BSR20182361. doi: 10.1042/BSR20182361
57. Pavlaki M, Zucker S. Matrix metalloproteinase inhibitors (MMPIs): the beginning of phase I or the termination of phase III clinical trials. Cancer Metastasis Rev. (2003) 22:177–203. doi: 10.1023/A:1023047431869
58. Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, et al. Constitutive and regulated α-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA. (1999) 96:3922–7. doi: 10.1073/pnas.96.7.3922
59. Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, et al. ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. (2010) 29:3020–32. doi: 10.1038/emboj.2010.167
60. Moy RH, Younes A. Immune checkpoint inhibition in hodgkin lymphoma. HemaSphere. (2018) 2:e20. doi: 10.1097/HS9.0000000000000020
61. Ghadially H, Brown L, Lloyd C, Lewis L, Lewis A, Dillon J, et al. MHC class I chain-related protein A and B (MICA and MICB) are predominantly expressed intracellularly in tumour and normal tissue. Br J Cancer. (2017) 116:1208–17. doi: 10.1038/bjc.2017.79
62. Mullooly M, McGowan PM, Crown J, Duffy MJ. The ADAMs family of proteases as targets for the treatment of cancer. Cancer Biol Ther. (2016) 17:870–80. doi: 10.1080/15384047.2016.1177684
63. Zocchi MR, Camodeca C, Nuti E, Rossello A, Venè R, Tosetti F, et al. ADAM10 new selective inhibitors reduce NKG2D ligand release sensitizing hodgkin lymphoma cells to NKG2D-mediated killing. Oncoimmunology. (2015) 5:e1123367. doi: 10.1080/2162402X.2015.1123367
64. Tosetti F, Venè R, Camodeca C, Nuti E, Rossello A, D'Arrigo C, et al. Specific ADAM10 inhibitors localize in exosome-like vesicles released by hodgkin lymphoma and stromal cells and prevent sheddase activity carried to bystander cells. Oncoimmunology. (2018) 7:e1421889. doi: 10.1080/2162402X.2017.1421889
65. Hansen HP, Trad A, Dams M, Zigrino P, Moss M, Tator M, et al. CD30 on extracellular vesicles from malignant hodgkin cells supports damaging of CD30 ligand-expressing bystander cells with brentuximab-Vedotin, in vitro. Oncotarget. (2016) 7:30523–35. doi: 10.18632/oncotarget.8864
66. Yamamoto M, Harada Y, Suzuki T, Fukushige T, Yamakuchi M, Kanekura T, et al. Application of high-mannose-type glycan-specific lectin from oscillatoria agardhii for affinity isolation of tumor-derived extracellular vesicles. Anal Biochem. (2019) 580:21–9. doi: 10.1016/j.ab.2019.06.001
67. Chihara D, Nastoupil LJ, Williams JN, Lee P, Koff JL, Flowers CR. New insights into the epidemiology of non-Hodgkin lymphoma and implications for therapy. Expert Rev Anticancer Ther. (2015) 15:531–44. doi: 10.1586/14737140.2015.1023712
68. Orme J, Villasboas J, Dong H. Tumor ADAM10/ADAM17-Mediated PD-L1 loss may predict poor outcomes in diffuse large B cell lymphoma. Blood. (2019) 134:4120. doi: 10.1182/blood-2019-124189
69. Romero Y, Wise R, Zolkiewska A. Proteolytic processing of PD-L1 by ADAM proteases in breast cancer cells. Cancer Immunol Immunother. (2019) 69 :43–55 doi: 10.1007/s00262-019-02437-2
70. Landgren O. Shall we treat smoldering multiple myeloma in the near future? Hematol Am Soc Hematol Educ Progr. (2017) 2017:194–204. doi: 10.1182/asheducation-2017.1.194
71. Zingoni A, Cecere F, Vulpis E, Fionda C, Molfetta R, Soriani A, et al. Genotoxic stress induces senescence-associated ADAM10-dependent release of NKG2D MIC ligands in multiple myeloma cells. J Immunol. (2015) 195:736–48. doi: 10.4049/jimmunol.1402643
72. Baker JHE, Kyle AH, Reinsberg SA, Moosvi F, Patrick HM, Cran J, et al. Heterogeneous distribution of trastuzumab in HER2-positive xenografts and metastases: role of the tumor microenvironment. Clin Exp Metastasis. (2018) 35:691–705. doi: 10.1007/s10585-018-9929-3
73. Liu J, Pan C, Guo L, Wu M, Guo J, Peng S, et al. A new mechanism of trastuzumab resistance in gastric cancer: MACC1 promotes the warburg effect via activation of the PI3K/AKT signaling pathway. J Hematol Oncol. (2016) 9:76. doi: 10.1186/s13045-016-0302-1
74. Zheng H, Zhong A, Xie S, Wang Y, Sun J, Zhang J, et al. Elevated serum HER-2 predicts poor prognosis in breast cancer and is correlated to ADAM10 expression. Cancer Med. (2019) 8:679–85. doi: 10.1002/cam4.1859
75. Feldinger K, Generali D, Kramer-Marek G, Gijsen M, Ng TB, Wong JH, et al. ADAM10 mediates trastuzumab resistance and is correlated with survival in HER2 positive breast cancer. Oncotarget. (2014) 5:6633–46. doi: 10.18632/oncotarget.1955
76. Ebbing EA, Medema JP, Damhofer H, Meijer SL, Krishnadath KK, van Berge Henegouwen MI, et al. ADAM10-mediated release of heregulin confers resistance to trastuzumab by activating HER3. Oncotarget. (2016) 7:10243–54. doi: 10.18632/oncotarget.7200
77. Sharma P. Biology and management of patients with triple-negative breast cancer. Oncologist. (2016) 21:1050–62. doi: 10.1634/theoncologist.2016-0067
78. Mullooly M, McGowan PM, Sukor SU, Madden SF, McDermott E, Crown J, et al. ADAMs as new therapeutic targets for triple-negative breast cancer. J Clin Oncol. (2011) 29:1062. doi: 10.1200/jco.2011.29.15_suppl.1062
79. Mullooly M, McGowan PM, Kennedy SA, Madden SF, Crown J, O' Donovan N, et al. ADAM10: a new player in breast cancer progression? Br J Cancer. (2015) 113:945–51. doi: 10.1038/bjc.2015.288
80. Liu F, Zhuang L, Wu R, Li D. miR-365 inhibits cell invasion and migration of triple negative breast cancer through ADAM10. J BUON. (2019) 24:1905–12.
81. Tsang JYS, Lee MA, Chan TH, Li J, Ni YB, Shao Y, et al. Proteolytic cleavage of amyloid precursor protein by ADAM10 mediates proliferation and migration in breast cancer. EBioMed. (2018) 38:89–99. doi: 10.1016/j.ebiom.2018.11.012
82. Yuan X-Z, Sun S, Tan C-C, Yu J-T, Tan L. The role of ADAM10 in Alzheimer's disease. J Alzheimers Dis. (2017) 58:303–22. doi: 10.3233/JAD-170061
83. Manzine PR, Ettcheto M, Cano A, Busquets O, Marcello E, Pelucchi S, et al. ADAM10 in Alzheimer's disease: pharmacological modulation by natural compounds and its role as a peripheral marker. Biomed Pharmacother. (2019) 113:108661. doi: 10.1016/j.biopha.2019.108661
84. Urriola-Muñoz P, Li X, Maretzky T, McIlwain DR, Mak TW, Reyes JG, et al. The xenoestrogens biphenol-A and nonylphenol differentially regulate metalloprotease-mediated shedding of EGFR ligands. J Cell Physiol. (2018) 233:2247–56. doi: 10.1002/jcp.26097
85. Peltanova B, Raudenska M, Masarik M. Effect of tumor microenvironment on pathogenesis of the head and neck squamous cell carcinoma: a systematic review. Mol Cancer. (2019) 18:63. doi: 10.1186/s12943-019-0983-5
86. Stasikowska-Kanicka O, Wagrowska-Danilewicz M, Kulicka P, Danilewicz M. Overexpression of ADAM10 in oral squamous cell carcinoma with metastases. Pol J Pathol. (2018) 69:67–72. doi: 10.5114/pjp.2018.75339
87. Su CW, Chang YC, Chien MH, Hsieh YH, Chen MK, Lin CW, et al. Loss of TIMP3 by promoter methylation of Sp1 binding site promotes oral cancer metastasis. Cell Death Dis. (2019) 10:793. doi: 10.1038/s41419-019-2016-0
88. Scilabra SD, Pigoni M, Pravatá V, Schätzl T, Müller SA, Troeberg L, et al. Increased TIMP-3 expression alters the cellular secretome through dual inhibition of the metalloprotease ADAM10 and ligand-binding of the LRP-1 receptor. Sci Rep. (2018) 8:14697. doi: 10.1038/s41598-018-32910-4
89. Chen S, Ying M, Lin X, Zheng X, Liu C, Liu H. Expression of MICA in oral squamous carcinoma cells and its effect on NK cells. Int J Clin Exp Med. (2015) 8:18208–12.
90. Liu W, Li J, Zhang P, Hou Q, Feng S, Liu L, et al. A novel pan-cancer biomarker plasma heat shock protein 90alpha and its diagnosis determinants in clinic. Cancer Sci. (2019) 110:2941–59. doi: 10.1111/cas.14143
91. Walkiewicz K, Strzelczyk J, Waniczek D, Biernacki K, Muc-Wierzgon M, Copija A, et al. Adamalysines as biomarkers and a potential target of therapy in colorectal cancer patients: preliminary results. Dis Markers. (2019) 2019:5035234. doi: 10.1155/2019/5035234
92. Ding Y, Bui MM, Wang Q, Sun X, Zhang M, Niu X, et al. ADAM10 is a potential novel prognostic biomarker for sacral chordoma. Ann Clin Lab Sci. (2019) 49:309–16.
93. Mcinnes IB, Schett G. The pathogenesis of rheumatoid arthritis. (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
94. Voort R Van Der, Lieshout AWT Van, Toonen LWJ, Slo AW, Berg WB Van Den, Figdor CG, et al. Elevated CXCL16 expression by synovial macrophages recruits memory T cells into rheumatoid joints. (2005) 52:1381–91. doi: 10.1002/art.21004
95. Isozaki T, Rabquer BJ, Ruth JH Iii, GKH, Koch AE. ADAM-10 is overexpressed in rheumatoid arthritis synovial tissue and mediates angiogenesis. (2013) 65:98–108. doi: 10.1002/art.37755
96. Isozaki T, Ishii S, Nishimi S, Nishimi A, Oguro N, Seki S, et al. A disintegrin and metalloprotease-10 is correlated with disease activity and mediates monocyte migration and adhesion in rheumatoid arthritis. Transl Res. (2015) 166:244–53. doi: 10.1016/j.trsl.2015.02.005
97. Li DAN, Xiao Z, Wang G, Song X. Knockdown of ADAM10 inhibits migration and invasion of fibroblast-like synoviocytes in rheumatoid arthritis. (2015) 5517–23. doi: 10.3892/mmr.2015.4011
98. Mcinnes IB, Schett G. Targeted treatments for rheumatoid arthritis 1 pathogenetic insights from the treatment of rheumatoid arthritis. Lancet. (2017) 389:2328–37. doi: 10.1016/S0140-6736(17)31472-1
99. Zhao R, Wang A, Hall KC, Otero M, Weskamp G, Zhao B, et al. Lack of ADAM10 in endothelial cells affects osteoclasts at the chondro-osseus junction. (2014) 224–230. doi: 10.1002/jor.22492
100. Isozaki T, Nishimi S, Nishimi A, Saito M, Miwa Y, Toyoshima Y, et al. A disintegrin and metalloproteinase (ADAM)-10 as a predictive factor for tocilizumab effectiveness in rheumatoid arthritis. Mod Rheumatol. (2017) 27:782–6. doi: 10.1080/14397595.2016.1256025
101. Ishii S, Isozaki T, Furuya H, Takeuchi H, Tsubokura Y, Inagaki K, et al. ADAM-17 is expressed on rheumatoid arthritis fibroblast-like synoviocytes and regulates proinflammatory mediator expression and monocyte adhesion. Arthritis Res Ther. (2018) 20:159. doi: 10.1186/s13075-018-1657-1
102. Pu Y, Cao D, Xie C, Pei H, Li D, Tang M, et al. Anti-arthritis effect of a novel quinazoline derivative through inhibiting production of TNF-α mediated by TNF-α converting enzyme in murine collagen-induced arthritis model. Biochem Biophys Res Commun. (2015) 462:288–93. doi: 10.1016/j.bbrc.2015.04.111
103. Nielsen MA, Andersen T, Etzerodt A, Kragstrup TW, Rasmussen TK, Stengaard-Pedersen K, et al. A disintegrin and metalloprotease-17 and galectin-9 are important regulators of local 4-1BB activity and disease outcome in rheumatoid arthritis. Rheumatol (oxford). (2016) 55:1871–9. doi: 10.1093/rheumatology/kew237
104. Dreymueller D, Uhlig S, Ludwig A. Adam-family metalloproteinases in lung inflammation: potential therapeutic targets. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L325–43. doi: 10.1152/ajplung.00294.2014
105. Fridman JS, Caulder E, Hansbury M, Liu X, Yang G, Wang Q, et al. Selective inhibition of ADAM metalloproteases as a novel approach for modulating ErbB pathways in cancer. Clin Cancer Res. (2007) 13:1892–902. doi: 10.1158/1078-0432.CCR-06-2116
106. Pisetsky DS. Evolving story of autoantibodies in systemic lupus erythematosus. J Autoimmun. (2019) 102356. doi: 10.1016/j.jaut.2019.102356
107. Petri M, Stohl W, Chatham W, McCune WJ, Chevrier M, Ryel J, et al. Association of plasma B lymphocyte stimulator levels and disease activity in systemic lupus erythematosus. Arthritis Rheum. (2008) 58:2453–9. doi: 10.1002/art.23678
108. Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, et al. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. (1999) 190:1697–710. doi: 10.1084/jem.190.11.1697
109. MacKay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol. (2009) 9:491–502. doi: 10.1038/nri2572
110. Hoffmann FS, Kuhn P-H, Laurent SA, Hauck SM, Berer K, Wendlinger SA, et al. The immunoregulator soluble TACI is released by ADAM10 and reflects B-cell activation in autoimmunity. J Immunol. (2015) 194:542–52. doi: 10.4049/jimmunol.1402070
111. Lownik JC, Wimberly JL, Conrad DH, Martin RK. B cell ADAM10 controls murine lupus progression through regulation of the ICOS:ICOS ligand axis. J Immunol. (2019) 308:L325–43. doi: 10.4049/jimmunol.1801207
112. Sharif MN, Šošić D, Rothlin C V, Kelly E, Lemke G, Olson E, et al. Twist mediates suppression of inflammation by type I IFNs and Axl. J Exp Med. (2006) 203:1891–901. doi: 10.1084/jem.20051725
113. Orme JJ, Du Y, Vanarsa K, Mayeux J, Li L, Mutwally A, et al. Heightened cleavage of Axl receptor tyrosine kinase by ADAM metalloproteases may contribute to disease pathogenesis in SLE. Clin Immunol. (2016) 169:58–68. doi: 10.1016/j.clim.2016.05.011
114. Parisi R, Symmons DPM, Griffiths CEM, Ashcroft DM. Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Invest Dermatol. (2013) 133:377–85. doi: 10.1038/jid.2012.339
115. Whitfield JF. Taming psoriatic keratinocytes - PTHs' uses go up another notch. J Cell Biochem. (2004) 93:251–6. doi: 10.1002/jcb.20216
116. Nickoloff BJ, Qin JZ, Chaturvedi V, Denning MF, Bonish B, Miele L. Jagged-1 mediated activation of notch signaling induces complete maturation of human keratinocytes through NF-κB and PPARγ. Cell Death Differ. (2002) 9:842–55. doi: 10.1038/sj.cdd.4401036
117. Weber S, Niessen MT, Prox J, Lüllmann-Rauch R, Schmitz A, Schwanbeck R, et al. The disintegrin/metalloproteinase Adam10 is essential for epidermal integrity and notch-mediated signaling. Development. (2011) 138:495–505. doi: 10.1242/dev.055210
118. Oh ST, Schramme A, Stark A, Tilgen W, Gutwein P. Reichrath J. Overexpression of ADAM 10 and ADAM 12 in lesional psoriatic skin. (2008) 158:1371–3. doi: 10.1111/j.1365-2133.2008.08513.x
119. Pang M, Murase JE, Koo J. An updated review of acitretin–a systemic retinoid for the treatment of psoriasis. Expert Opin Drug Metab Toxicol. (2008) 4:953–64. doi: 10.1517/17425255.4.7.953
120. Tippmann F, Hundt J, Schneider A, Endres K, Fahrenholz F. Up-regulation of the alpha-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J. (2019) 23:1643–54. doi: 10.1096/fj.08-121392
121. Lee HR, Shin HK, Park SY, Kim HY, Lee WS, Rhim BY, et al. Cilostazol suppresses β-amyloid production by activating a disintegrin and metalloproteinase 10 via the upregulation of SIRT1-coupled retinoic acid receptor-β. J Neurosci Res. (2014) 92:1581–90. doi: 10.1002/jnr.23421
122. Fukasawa H, Nakagomi M, Yamagata N, Katsuki H, Kawahara K, Kitaoka K, et al. Tamibarotene: a candidate retinoid drug for Alzheimer's disease. Biol Pharm Bull. (2012) 35:1206–12. doi: 10.1248/bpb.b12-00314
123. Wetzel S, Seipold L, Saftig P. The metalloproteinase ADAM10: a useful therapeutic target? Biochim Biophys Acta Mol Cell Res. (2017) 1864:2071–81. doi: 10.1016/j.bbamcr.2017.06.005
124. Marcade M, Bourdin J, Loiseau N, Peillon H, Rayer A, Drouin D, et al. Etazolate, a neuroprotective drug linking GABAA receptor pharmacology to amyloid precursor protein processing. J Neurochem. (2008) 106:392–404. doi: 10.1111/j.1471-4159.2008.05396.x
125. Zuo L, Li J, Ge S, Ge Y, Shen M, Wang Y, et al. Bryostatin-1 ameliorated experimental colitis in Il-10−/− mice by protecting the intestinal barrier and limiting immune dysfunction. J Cell Mol Med. (2019) 23:5588–99. doi: 10.1111/jcmm.14457
126. Zeng N, Xu Y, Wu Y, Hongbo T, Wu M. Bryostatin 1 causes attenuation of TPA-mediated tumor promotion in mouse skin. Mol Med Rep. (2018) 17:1077–82. doi: 10.3892/mmr.2017.7993
127. Obregon DF, Rezai-Zadeh K, Bai Y, Sun N, Hou H, Ehrhart J, et al. ADAM10 activation is required for green tea (-)-epigallocatechin-3-gallate- induced alpha-secretase cleavage of amyloid precursor protein. J Biol Chem. (2006) 281:16419–27. doi: 10.1074/jbc.M600617200
128. de la Torre R, de Sola S, Farré M, Xicota L, Cuenca-Royo A, Rodriguez J, et al. A phase 1, randomized double-blind, placebo controlled trial to evaluate safety and efficacy of epigallocatechin-3-gallate and cognitive training in adults with fragile X syndrome. Clin Nutr. (2019) 39:378–87. doi: 10.1016/j.clnu.2019.02.028
129. Athene Lane J, Er V, Avery KNL, Horwood J, Cantwell M, Caro GP, et al. Prodiet: a phase II randomized placebo-controlled trial of green tea catechins and lycopene in men at increased risk of prostate cancer. Cancer Prev Res. (2018) 11:687–96. doi: 10.1158/1940-6207.CAPR-18-0147
130. Nishie W. Update on the pathogenesis of bullous pemphigoid: an autoantibody-mediated blistering disease targeting collagen XVII. J Dermatol Sci. (2014) 73:179–86. doi: 10.1016/j.jdermsci.2013.12.001
131. Hall RP, Streilein RD, Hannah DL, McNair PD, Fairley JA, Ronaghy A, et al. Association of serum b-cell activating factor level and proportion of memory and transitional b cells with clinical response after rituximab treatment of bullous pemphigoid patients. J Invest Dermatol. (2013) 133:2786–8. doi: 10.1038/jid.2013.236
132. Shen S, Ke Y, Dang E, Fang H, Chang Y, Zhang J, et al. Semaphorin 4D from CD15 + granulocytes via ADAM10-induced cleavage contributes to antibody production in bullous pemphigoid. J Invest Dermatol. (2018) 138:588–97. doi: 10.1016/j.jid.2017.09.037
133. Drummond AH, Beckett P, Brown PD, Bone EA, Davidson AH, Galloway WA, et al. Preclinical and clinical studies of MMP inhibitors in cancer. Ann N Y Acad Sci. (1999) 878:228–35. doi: 10.1111/j.1749-6632.1999.tb07688.x
134. Macaulay VM, O'Byrne KJ, Saunders MP, Braybrooke JP, Long L, Gleeson F, et al. Phase I study of intrapleural batimastat (BB-94), a matrix metalloproteinase inhibitor, in the treatment of malignant pleural effusions. Clin cancer Res. (1999) 5:513–20.
135. Atapattu L, Saha N, Chheang C, Eissman MF, Xu K, Vail ME, et al. An activated form of ADAM10 is tumor selective and regulates cancer stem-like cells and tumor growth. J Exp Med. (2016) 213:1741–57. doi: 10.1084/jem.20151095
136. Manzine PR, de Franca Bram JM, Barham EJ, do Vale F de AC, Selistre-de-Araujo HS, Cominetti MR, et al. ADAM10 as a biomarker for Alzheimer's disease: a study with Brazilian elderly. Dement Geriatr Cogn Disord. (2013) 35:58–66. doi: 10.1159/000345983
137. Sogorb-Esteve A, García-Ayllón M-S, Gobom J, Alom J, Zetterberg H, Blennow K, et al. Levels of ADAM10 are reduced in Alzheimer's disease CSF. J Neuroinflammation. (2018) 15:213. doi: 10.1186/s12974-018-1255-9
Keywords: metalloproteases, ADAM10 inhibitors, NKG2D, hodgkin lymphoma, glioblastoma, breast cancer, systemic lupus erythematosus, rheumatoid arthritis
Citation: Smith TM Jr, Tharakan A and Martin RK (2020) Targeting ADAM10 in Cancer and Autoimmunity. Front. Immunol. 11:499. doi: 10.3389/fimmu.2020.00499
Received: 16 December 2019; Accepted: 04 March 2020;
Published: 24 March 2020.
Edited by:
Maria Raffaella Zocchi, San Raffaele Scientific Institute (IRCCS), ItalyReviewed by:
Alessandra Zingoni, Sapienza University of Rome, ItalyPeer-Hendrik Kuhn, Technical University of Munich, Germany
Copyright © 2020 Smith, Tharakan and Martin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rebecca K. Martin, rebecca.martin@vcuhealth.org