The Influence of Gas–Wall and Gas–Gas Interactions on the Accommodation Coefficients for Rarefied Gases: A Molecular Dynamics Study


Abstract
:1. Introduction
2. Materials and Methods
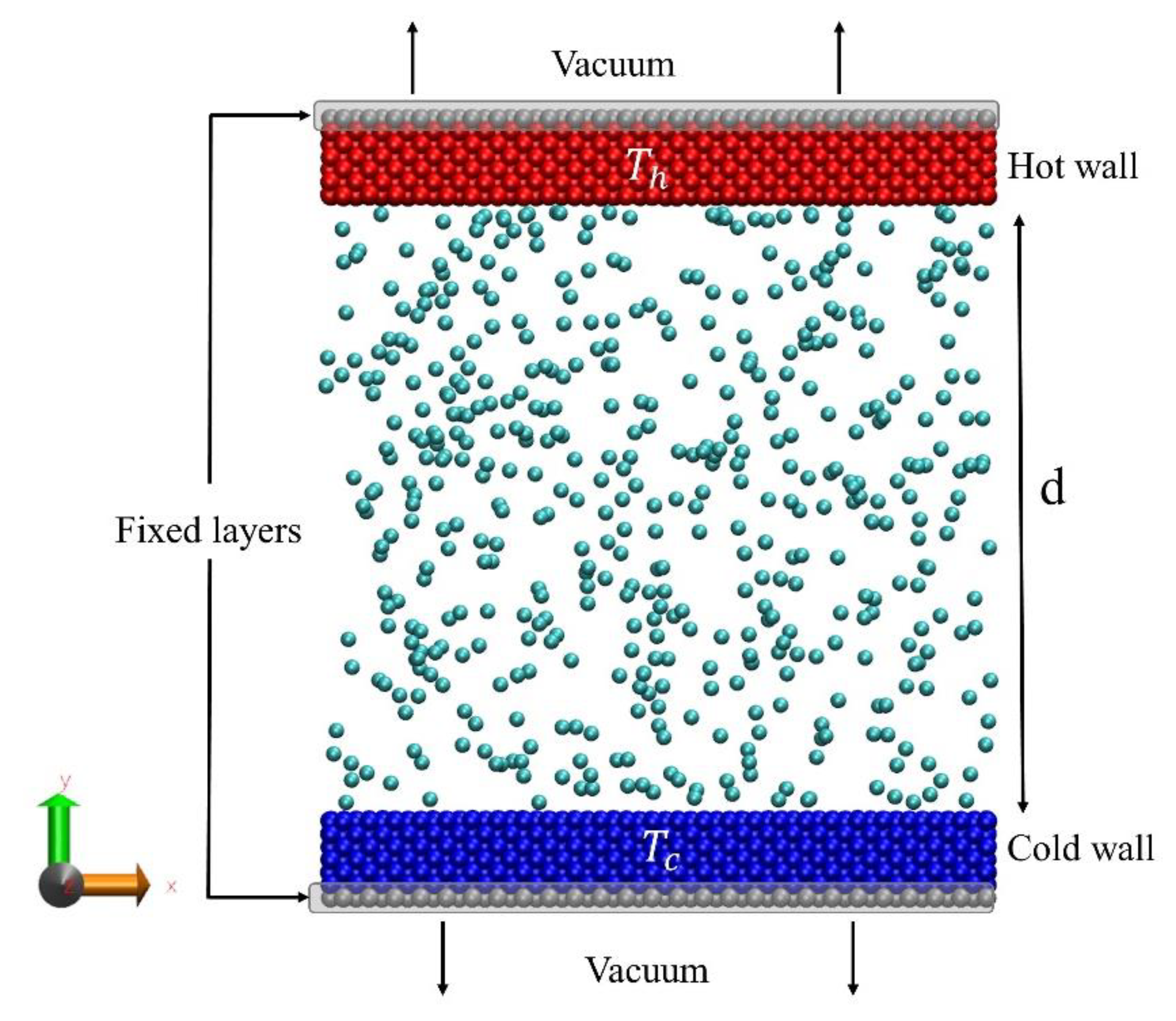
2.1. Molecular Dynamics (MD) Simulations
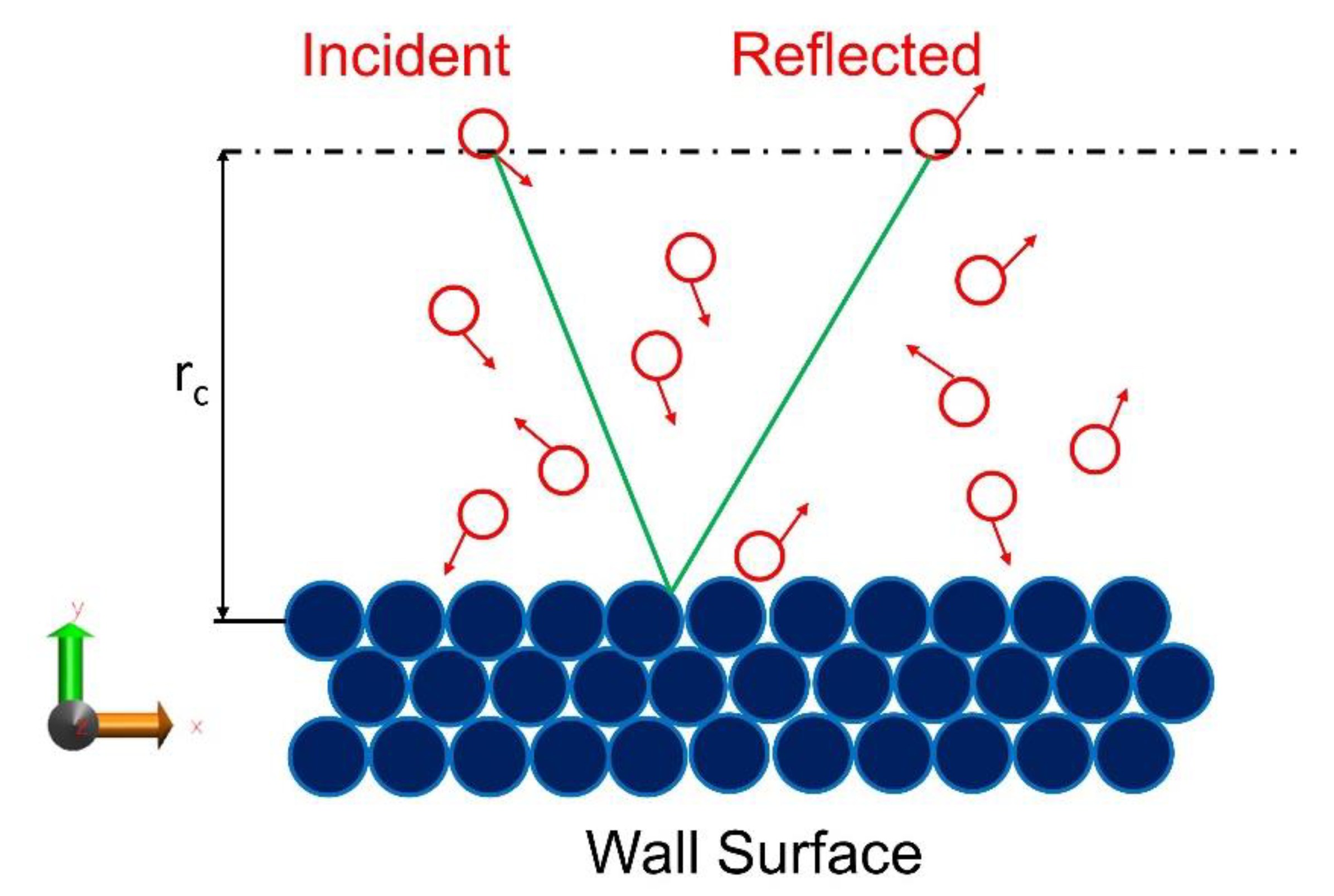
2.2. Computing Accommodation Coefficients
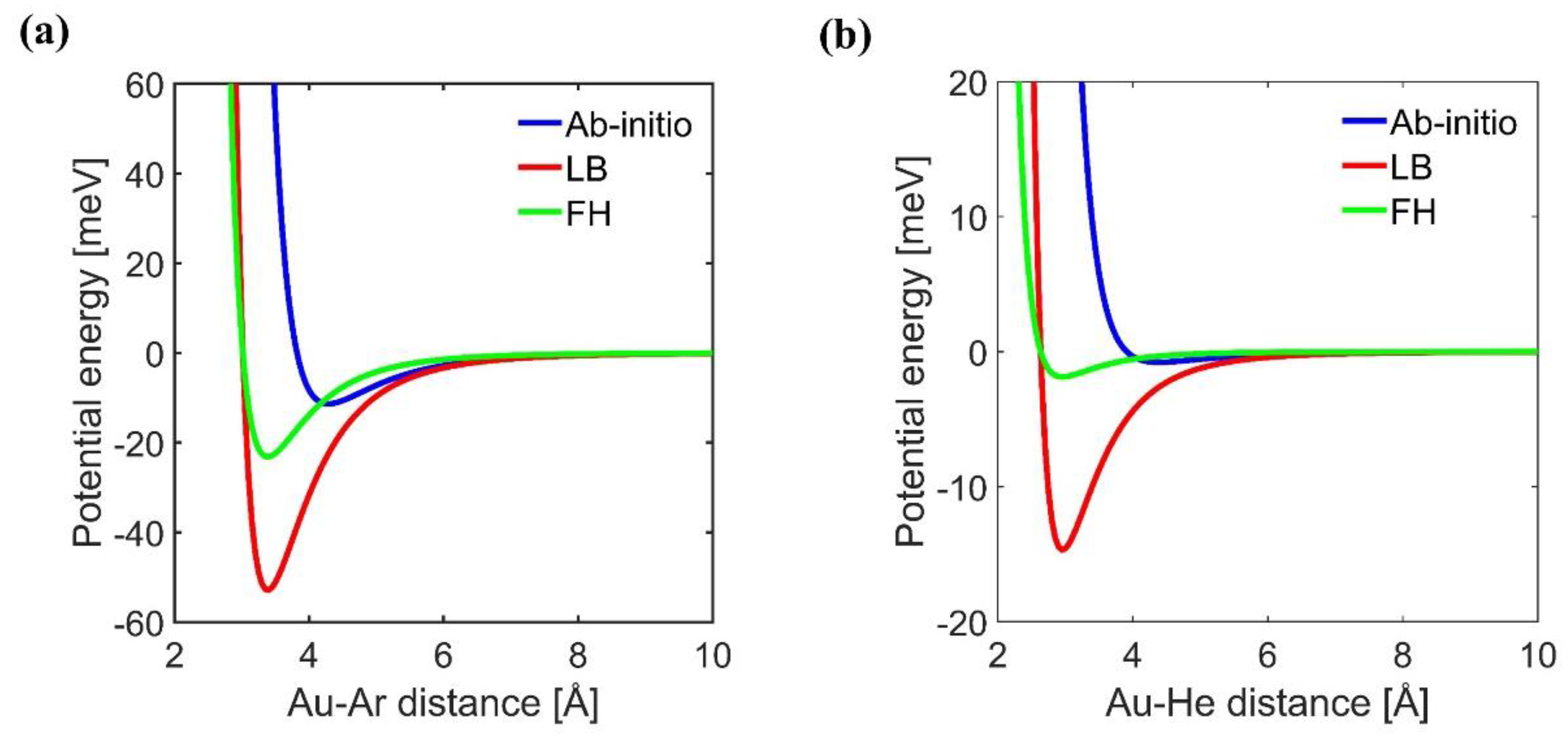
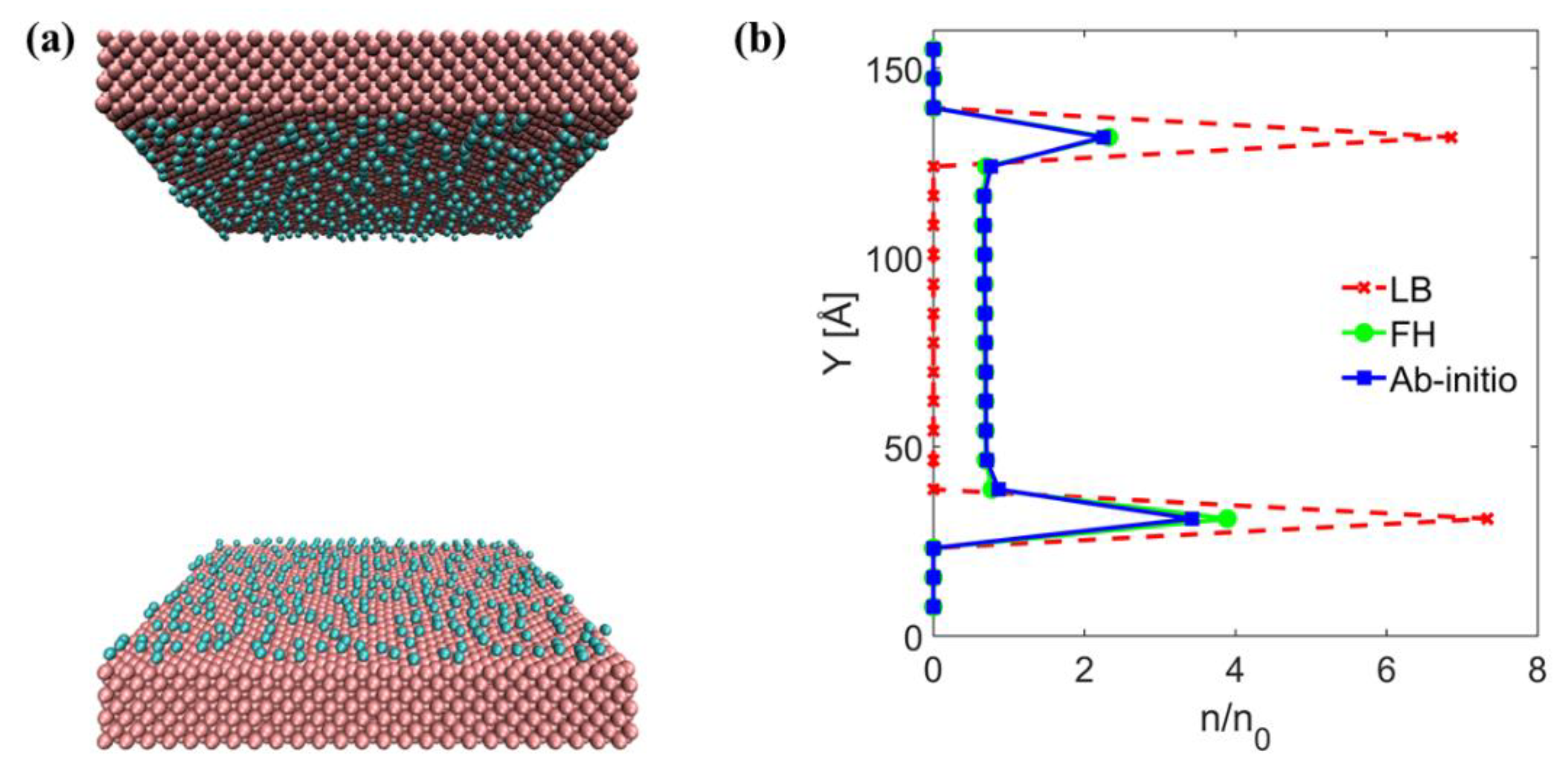
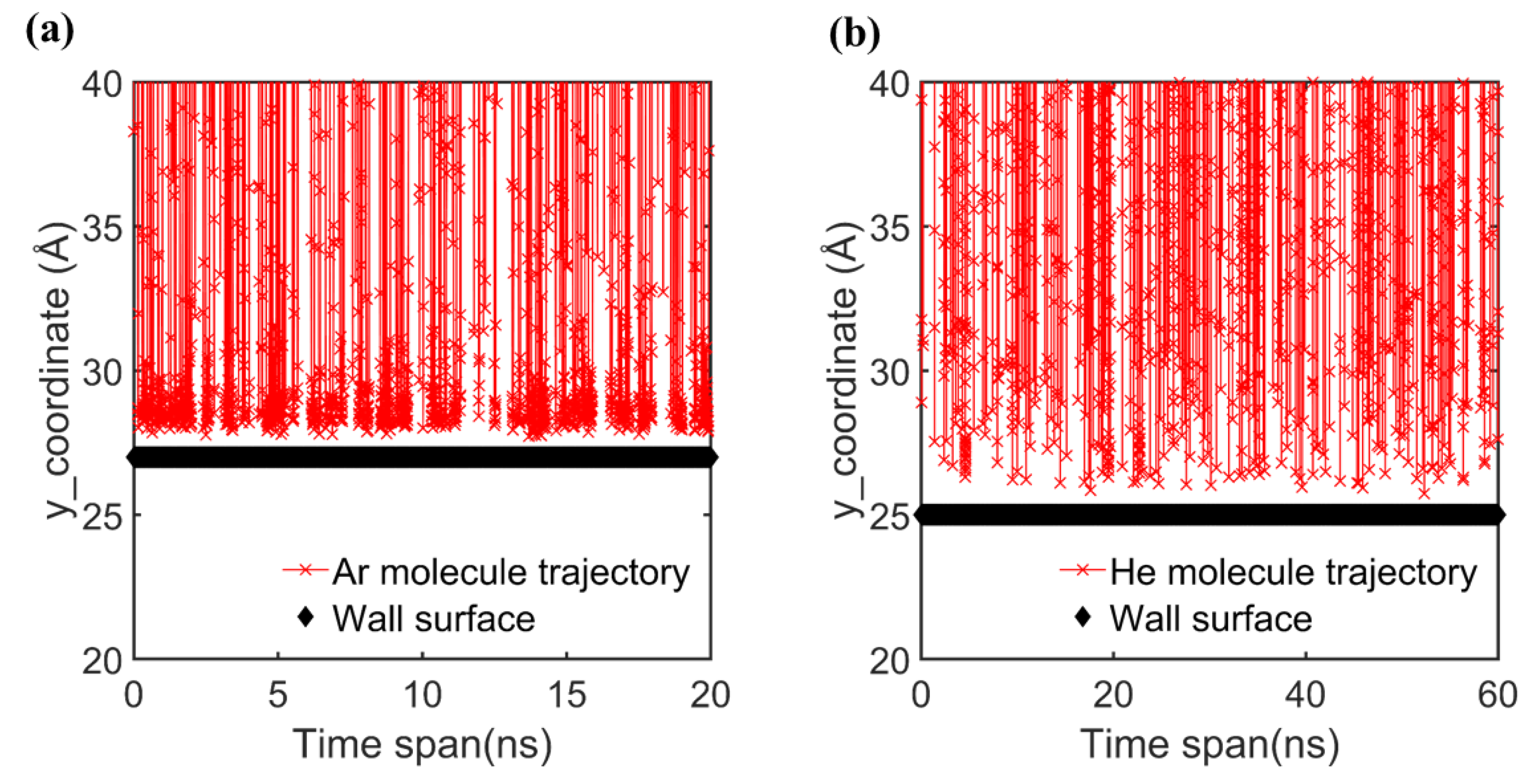
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Karniadakis, G.; Beskok, A.; Aluru, N. Microflows and Nanoflows Fundamentals and Simulation; Springer Science & Business Media: New York, NY, USA, 2006; Volume 29. [Google Scholar]
- Rader, D.J.; Trott, W.M.; Torczynski, J.R.; Gallis, M.A.; Castañeda, J.N.; Grasser, T.W. Microscale Rarefied Gas Dynamics and Surface Interactions for EUVL and MEMS Applications; Department of Energy: Washington, DC, USA, 2004. [Google Scholar]
- Saxena, S.C.; Joshi, R.K. Thermal Accommodation and Adsorption Coefficients of Gases; Hemisphere Publishing: New York, NY, USA, 1989. [Google Scholar]
- Agrawal, A. A comprehensive review on gas flow in microchannels. Int. J. Micro-Nano Scale Transp. 2011, 2, 1–40. [Google Scholar] [CrossRef]
- Cao, B.-Y.; Sun, J.; Chen, M.; Guo, Z.-Y. Molecular momentum transport at fluid-solid interfaces in MEMS/NEMS: A review. Int. J. Mol. Sci. 2009, 10, 4638–4706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colin, S. Rarefaction and compressibility effects on steady and transient gas flows in microchannels. Microfluid. Nanofluidics 2005, 1, 268–279. [Google Scholar] [CrossRef]
- Billing, G.D. The dynamics of molecule-surface interaction. Comput. Phys. Rep. 1990, 12, 383–450. [Google Scholar] [CrossRef]
- Barker, J.A.; Auerbach, D.J. Gas-surface interactions and dynamics; Thermal energy atomic and molecular beam studies. Surf. Sci. Rep. 1984, 4, 1–99. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids, 2nd ed.; Oxford University Press: Oxford, UK, 2017; Volume 53. [Google Scholar]
- Maxwell, J.C., III. On stresses in rarefied gases arising from inequalities of temperature. Proc. R. Soc. Lond. 1878, 27, 304–308. [Google Scholar]
- Lord, R.G. Some further extensions of the Cercignani-Lampis gas-surface interaction model. Phys. Fluids 1995, 7, 1159–1161. [Google Scholar] [CrossRef]
- Bird, G.A. Molecular Gas Dynamics and the Direct Simulation of Gas Flows; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Zhang, J. Lattice Boltzmann method for microfluidics: Models and applications. Microfluid. Nanofluidics 2011, 10, 1–28. [Google Scholar] [CrossRef]
- Grad, H. On the kinetic theory of rarefied gases. Commun. Pure Appl. Math. 1949, 2, 331–407. [Google Scholar] [CrossRef]
- Prabha, S.K.; Sathian, S.P. Computational study of thermal dependence of accommodation coefficients in a nano-channel and the prediction of velocity profiles. Comput. Fluids 2012, 68, 47–53. [Google Scholar] [CrossRef]
- Chirita, V.; Pailthorpe, B.A.; Collins, R.E. Non-equilibrium energy and momentum accommodation coefficients of Ar atoms scattered from Ni(001) in the thermal regime: A molecular dynamics study. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 1997, 129, 465–473. [Google Scholar] [CrossRef]
- Sun, J.; Li, Z.-X. Three-dimensional molecular dynamic study on accommodation coefficients in rough nanochannels. Heat Transf. Eng. 2011, 32, 658–666. [Google Scholar] [CrossRef]
- Spijker, P.; Markvoort, A.J.; Nedea, S.V.; Hilbers, P.A.J. Computation of accommodation coefficients and the use of velocity correlation profiles in molecular dynamics simulations. Phys. Rev. E 2010, 81, 011203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daun, K.J. Thermal accommodation coefficients between polyatomic gas molecules and soot in laser-induced incandescence experiments. Int. J. Heat Mass Transf. 2009, 52, 5081–5089. [Google Scholar] [CrossRef]
- Reinhold, J.; Veltzke, T.; Wells, B.; Schneider, J.; Meierhofer, F.; Ciacchi, L.C.; Chaffee, A.; Thöming, J. Molecular dynamics simulations on scattering of single Ar, N 2, and CO 2 molecules on realistic surfaces. Comput. Fluids 2014, 97, 31–39. [Google Scholar] [CrossRef]
- Mane, T.; Bhat, P.; Yang, V.; Sundaram, D.S. Energy accommodation under non-equilibrium conditions for aluminum-inert gas systems. Surf. Sci. 2018, 677, 135–148. [Google Scholar] [CrossRef]
- Liao, M.; Grenier, R.; To, Q.-D.; de Lara-Castells, M.P.; Leónard, C. Helium and argon interactions with gold surfaces: Ab initio-assisted determination of the He−Au pairwise potential and its application to accommodation coefficient determination. J. Phys. Chem. C 2018, 122, 14606–14614. [Google Scholar] [CrossRef]
- Yamamoto, K. Slightly rarefied gas flows over a smooth Pt surface. AIP Conf. Proc. 2001, 585, 339–346. [Google Scholar]
- Hyakutake, T.; Yamamoto, K.; Takeuchi, H. Flow of gas mixtures through micro channel. AIP Conf. Proc. 2005, 762, 780–788. [Google Scholar]
- Daun, K.J.; Sipkens, T.A.; Titantah, J.T.; Karttunen, M. Thermal accommodation coefficients for laser-induced incandescence sizing of metal nanoparticles in monatomic gases. Appl. Phys. B 2013, 112, 409–420. [Google Scholar] [CrossRef]
- Cengel, Y.A.; Boles, M.A. Thermodynamics: An Engineering Approach, 8th ed.; McGraw-Hill Education: New York, NY, USA, 2015. [Google Scholar]
- Sheng, H.W.; Kramer, M.J.; Cadien, A.; Fujita, T.; Chen, M.W. Highly optimized embedded-atom-method potentials for fourteen fcc metals. Phys. Rev. B 2011, 83, 134118. [Google Scholar] [CrossRef] [Green Version]
- Grenier, R.; To, Q.-D.; de Lara-Castells, M.P.; Leónard, C. Argon interaction with gold surfaces: Ab initio-assisted determination of pair Ar−Au potentials for molecular dynamics simulations. J. Phys. Chem. A 2015, 119, 6897–6908. [Google Scholar] [CrossRef] [PubMed]
- Heinz, H.; Vaia, R.A.; Farmer, B.L.; Naik, R.R. Accurate simulation of surfaces and interfaces of face-centered cubic metals using 12-6 and 9-6 lennard-jones potentials. J. Phys. Chem. C 2008, 112, 17281–17290. [Google Scholar] [CrossRef]
- Schroeder, D.V. Interactive molecular dynamics. Am. J. Phys. 2015, 83, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Thomas, L.B.; Brown, R.E. The accommodation coefficients of gases on platinum as a function of pressure. J. Chem. Phys. 1950, 18, 1367–1372. [Google Scholar] [CrossRef]
- Markvoort, A.J.; Hilbers, P.A.J.; Nedea, S.V. Molecular dynamics study of the influence of wall-gas interactions on heat flow in nanochannels. Phys. Rev. E 2005, 71, 066702. [Google Scholar] [CrossRef] [Green Version]
- Trott, W.M.; Castaeda, J.N.; Torczynski, J.R.; Gallis, M.A.; Rader, D.J. An experimental assembly for precise measurement of thermal accommodation coefficients. Rev. Sci. Instrum. 2011, 82, 621. [Google Scholar] [CrossRef]
- Agrawal, A.; Prabhu, S.V. Survey on measurement of tangential momentum accommodation coefficient. J. Vac. Sci. 2008, 26, 634–645. [Google Scholar] [CrossRef]
- Mann, W. The exchange of energy between a platinum surface and gas molecules. Proc. R. Soc. Lond. Ser. A Contain. Pap. A Math. Phys. Character 1934, 146, 776–791. [Google Scholar]
- Thomas, L.B.; Olmer, F. The accommodation coefficients of He, Ne, A, H2, D2, 02, C02, and Hg on platinum as a function of temperature. J. Am. Chem. Soc. 1943, 65, 1036–1043. [Google Scholar] [CrossRef]
- Thomas, L.B.; Schofield, E.B. Thermal accommodation coefficient of helium on a bare tungsten surface. J. Chem. Phys. 1955, 23, 861–866. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom Type | εii (meV) | σii (Å) | MW (a.m.u) |
|---|---|---|---|
| Au [29] | 229.4 | 2.63 | 196.96 |
| Ar [30] | 12.2 | 3.35 | 39.94 |
| He [30] | 0.94 | 2.64 | 4.00 |
| Parameter | Value |
|---|---|
| 11.36 (meV) | |
| 3.819 (Å) | |
| 0.787 (meV) | |
| 4.342 (Å) |
| System | Pressure (MPa) | Number Density (1/nm3) | MFP (nm) | EAC | MAC | MD Simulations time (ns) * |
|---|---|---|---|---|---|---|
| Au–Ar | 2.75 | 0.59 | 2.63 | 0.874 | 0.883 | 20 |
| 1.27 | 0.27 | 5.71 | 0.832 | 0.846 | 50 | |
| 0.84 | 0.18 | 8.57 | 0.816 | 0.822 | 70 | |
| 0.42 | 0.09 | 17.14 | 0.783 | 0.791 | 100 | |
| Au–He | 0.21 | 0.048 | 58.73 | 0.048 | 0.059 | 60 |
| 0.13 | 0.029 | 97.89 | 0.046 | 0.057 | 90 | |
| 0.08 | 0.019 | 146.84 | 0.043 | 0.052 | 150 | |
| 0.04 | 0.009 | 293.70 | 0.042 | 0.049 | 250 |
| System | Pair potential | αx | αy | αz | αE |
|---|---|---|---|---|---|
| Au–Ar | Ab-initio (Parallel walls) | 0.824 | 0.913 | 0.832 | 0.874 |
| Ab-initio (Molecular beam) [22] | 0.40 | 0.77 | 0.40 | 0.56 | |
| Fender Halsey | 0.915 | 0.934 | 0.913 | 0.913 | |
| Experimental results: αE = 0.85 [34] ; TMAC(αx, αz) = 0.893 [35] * | |||||
| Au–He | Ab-initio (Parallel walls) | 0.036 | 0.113 | 0.038 | 0.048 |
| Ab-initio (Molecular beam) [22] | 0.013 | 0.046 | 0.014 | 0.017 | |
| Fender Halsey | 0.245 | 0.347 | 0.221 | 0.069 | |
| Lorentz-Berthelot | 0.642 | 0.748 | 0.653 | 0.187 | |
| Experimental result: αE = 0.31 [34] | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammad Nejad, S.; Nedea, S.; Frijns, A.; Smeulders, D. The Influence of Gas–Wall and Gas–Gas Interactions on the Accommodation Coefficients for Rarefied Gases: A Molecular Dynamics Study. Micromachines 2020, 11, 319. https://doi.org/10.3390/mi11030319
Mohammad Nejad S, Nedea S, Frijns A, Smeulders D. The Influence of Gas–Wall and Gas–Gas Interactions on the Accommodation Coefficients for Rarefied Gases: A Molecular Dynamics Study. Micromachines. 2020; 11(3):319. https://doi.org/10.3390/mi11030319
Chicago/Turabian StyleMohammad Nejad, Shahin, Silvia Nedea, Arjan Frijns, and David Smeulders. 2020. "The Influence of Gas–Wall and Gas–Gas Interactions on the Accommodation Coefficients for Rarefied Gases: A Molecular Dynamics Study" Micromachines 11, no. 3: 319. https://doi.org/10.3390/mi11030319