
Abstract
Background
Sensitive biomarkers are needed to rapidly identify high-risk infants after hypoxia-ischemia for neuroprotective treatment. Hypotension is a key determinant of hypoxic-ischemic neural injury, and a potent stimulus of humoral pressors including angiotensin-II and arginine vasopressin. We therefore aimed to quantify the relationship between vasopressin and angiotensin-II levels in the latent phase after hypoxia-ischemia induced by umbilical cord occlusion (UCO) with both the severity of preceding hypotension and subsequent neuronal injury.
Methods
Chronically instrumented near-term fetal sheep underwent sham-UCO or UCO for either 15 min or until mean arterial pressure was <8 mmHg. Neuronal injury was assessed after 72 h recovery.
Results
Umbilical cord occlusion was associated with severe hypotension that recovered after UCO; two fetuses developed profound secondary hypotension within 6 h and died. Vasopressin levels but not angiotensin-II were significantly elevated 1–3 h after UCO and were closely associated with the severity of hypotension during UCO and the subsequent severity of neuronal loss in the parasagittal and lateral cortex, caudate nucleus and putamen. The Youden cut-point for vasopressin at 1 h was 180.0 pmol/L, with sensitivity 100% and specificity 92.3% for severe neuronal injury or death.
Conclusion
Vasopressin levels shortly after moderate-severe hypoxia-ischemia may be a useful early biomarker to guide the timely implementation of neuroprotective treatment.
Impact
-
It can be difficuIt to rapidly identify infants who might benefit from therapeutic hypothermia. We investigated whether increases in plasma pressor hormones early after hypoxia-ischemia were biomarkers for neonatal hypoxic-ischemic encephalopathy using near-term fetal sheep.
-
Arginine vasopressin levels were elevated at 1–3 h after hypoxia-ischemia and were predictive of the severity of preceding hypotension and subsequent risk of severe neuronal injury or death after hypoxia-ischemia.
-
Arginine vasopressin may help identify neonates at high risk of hypoxic-ischemic encephalopathy early within the therapeutic window for hypothermia.
Similar content being viewed by others


Introduction
Hypoxia-ischemia (HI) around the time of birth is a major cause of neurodevelopmental disability, occurring in 1–3/1000 live births at term gestation in developed countries.1 Therapeutic hypothermia is now standard care for neonates with moderate-severe HI encephalopathy (HIE).2 Despite hypothermia’s potent neuroprotective effects when initiated within its therapeutic window of opportunity, it can be challenging to identify neonates who may benefit from hypothermia due to a lack of early biomarkers of the risk of HI injury.3
Arterial hypotension during HI is a critical factor triggering brain injury across multiple experimental protocols.4,5 Arginine vasopressin (AVP), renin and angiotensin-II (AII) are key vasoactive humoral factors that support fetal blood pressure during hypoxia and hypotension.6,7,8,9,10,11 AVP is released by the human fetus during labor12 and, in observational studies, levels of AVP and copeptin (which is derived from the same precursor protein and released in equimolar amounts) were associated with acidosis at birth, and with neurodevelopmental outcomes after HIE.13,14 More recent evidence has shown that hypoxia is an additional stimulus of AVP release in the fetus,11 and AVP may have an endogenous neuroprotective role during HI.15 We have recently shown that high AVP levels after HI are correlated with worse electroencephalographic recovery in near-term fetal sheep,16 but the relationship of AVP levels to hypotension and neural injury is unknown.
The purpose of this study was to investigate in a superset of our previously studied cohort whether plasma AVP, renin and AII levels during the first 6 h after HI were correlated with the severity of preceding hypotension during HI induced by reversible umbilical cord occlusion (UCO) and with subsequent neural injury assessed 72 h after UCO in near-term fetal sheep at 0.85 of gestation. At this stage of gestation the fetal sheep is broadly equivalent to the term human baby in terms of neural development.17
Methods
Ethics, subjects and surgical procedures
The subjects of this study represent an overlapping superset of our previous publication that reported changes in heart rate variability, cardiovascular and neurophysiological parameters and the time course of AVP levels.16 All procedures were approved by the Animal Ethics Committee of the University of Auckland following the New Zealand Animal Welfare Act.
Twenty-one Romney/Suffolk fetal sheep were surgically instrumented at 117–125 d gestational age (term = 147 d), similarly to previous reports.18,19 Ewes were given 5 mL of Streptocin (procaine penicillin (250,000 IU/mL) and dihydrostreptomycin (250 mg/mL, Stockguard Labs, Hamilton, New Zealand) intramuscularly 30 min before the start of surgery. Anesthesia was induced by intravenous injection of propofol (5 mg/kg; AstraZeneca, Auckland, New Zealand), and general anesthesia maintained using 2–3% isoflurane (Medsource, Ashburton, New Zealand) in oxygen. Catheters were placed in the left fetal femoral artery, right brachial artery, and the amniotic sac. Ultrasound flow probes (size 3S, Transonic Systems, Ithaca, NY, USA) were placed around the right femoral artery to measure femoral blood flow (FBF) and left carotid artery to measure carotid artery blood flow (CaBF). CaBF provides an index of cerebral blood flow.20 A pair of electrodes was sewn over the fetal chest to measure the fetal electrocardiogram (ECG). An 18–20-mm diameter inflatable silicone occluder was placed around the umbilical cord (In Vivo Metric, Healdsburg, CA, USA). Eighty mg gentamicin (Rousell, Auckland, New Zealand) was administered into the amniotic sac before closure of the uterus. All fetal leads were exteriorized through the maternal flank and a maternal long saphenous vein was catheterized to provide access for postoperative care.
Postoperative care
Ewes were housed together in separate metabolic cages with ad libitum access to food and water, in temperature-controlled rooms (16 ± 1 °C, humidity 50 ± 10%) with a 12-h light/dark cycle. Daily intravenous antibiotics were administered to the ewe for 4 days (600 mg benzylpenicillin sodium; Novartis, Auckland, New Zealand, and 80 mg gentamicin). Fetal catheters were maintained by continuous infusion of heparinized saline (20 U/mL at 0.2 mL/h).
Experimental recordings
Fetal mean arterial blood pressure (MAP), corrected for maternal movement by subtraction of amniotic fluid pressure (Novatrans II, MX860; Medex, Hilliard, OH, USA), and ECG were recorded continuously from 24 h before until 72 h after UCO. The blood pressure signal was collected at 64 Hz and low-pass filtered at 30 Hz. CaBF and FBF were measured continuously using a two-channel Transonic T-206 Flowmeter (Transonic Systems) and data were low-pass filtered at 10 Hz with a second-order Butterworth filter. The raw ECG signal was analog filtered with a first-order high-pass filter set at 1 Hz and an eight-order low-pass Bessel filter set at 100 Hz, saved at 1024 Hz and then used to derive FHR. Data were collected by computer and stored to disk for offline analysis.
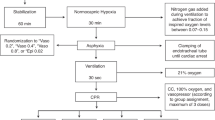
Experimental protocol
Experiments were conducted 4 days after surgery at 121–129 d (0.85) gestation. Fetuses were randomly assigned to HI (n = 16) or sham-HI (n = 5). HI was induced between 0900 and 1000 hours by rapid inflation of the occluder with saline to completely compress the umbilical cord. Successful UCO was confirmed by observation of a rapid onset of bradycardia with a rise in arterial pressure, and by pH and blood gas measurements. UCO was terminated either after 15 min (n = 8) or >15 min at a target MAP of 8 mmHg (n = 8) to represent moderate-severe HI.18 Occlusion was stopped early if MAP fell below 8 mmHg. The occluder was not inflated in shams. Note that in the HI group, one fetus did not have plasma samples, and two fetuses died within 6 h and so did not undergo histological assessment.
Fetal biochemistry
Fetal arterial whole blood and plasma samples were collected before occlusion, at 12 min after the start of occlusion and at 1, 3, 6, 24, 48 and 72 h after occlusion. Whole blood was analyzed for pH, blood gases (Ciba-Corning Diagnostics 845, MA, USA) and glucose and lactate levels (YSI 2300, Yellow Springs, OH, USA). Plasma samples were collected in prechilled vials on ice, centrifuged at 4 °C and stored at −80 °C as rapidly as possible. Samples were stored until completion of the study and underwent batch analysis of AVP, renin and AII levels via radioimmunoassay, as previously described.16,21 The interassay CVs for AVP were: 15.1% at 10.0 pmol/L, 11.9% at 5.1 pmol/L, 23.3% at 1.52 pmol/L.
Histology
Seventy-two hours after occlusion, ewes and fetuses were killed by intravenous overdose of sodium pentobarbitone (9 g Pentobarb 300, Provet, Auckland, New Zealand). At post-mortem, fetal brains were perfusion-fixed in situ with 10% phosphate-buffered formalin and immersion fixed for 1 week before paraffin embedding. Coronal forebrain sections (10 µm) were selected from 26 mm anterior to stereotaxic zero to assess the caudate nucleus and putamen, the parasagittal cortex and lateral cortex22 and 17 mm anterior to stereotaxic zero to assess the thalamus, and the cornu ammonis (CA, divided into CA1/2, CA3, CA4 regions) and dentate gyrus (DG) of the dorsal horn of the anterior hippocampus.22 All sections were deparaffinized in xylene, rehydrated and washed in 0.1 mol/L phosphate-buffered saline. Sections were then stained with thionine acetate (Sigma-Aldrich) and acid fuchsin (Sigma-Aldrich) to assess neuronal loss. The sections were then dehydrated and mounted.
Histological analysis
Neuronal loss was scored by light microscopy at ×20 or ×40 magnification on a Nikon 80i microscope with motorized stage and StereoInvestigator (v8, Microbrightfield, Williston, VT, USA) using four fields in the caudate nucleus, three in the putamen, and one field in the hippocampal divisions, lateral cortex, parasagittal cortex, and medial nucleus of the thalamus. The proportion of neurons showing ischemic cell change as shown by nuclear condensation and acid fuchsin staining of the cytoplasm in each region was scored on a six-point scale: 0 = no dead neurons; 5 = 0–9%; 30 = 10–49%; 70 = 50–89%; 95 = 90–99%; and 100 = 100% dead neurons.23 For each animal, average scores for each region were calculated from both hemispheres and from two sections.
Physiological data analysis
Offline analysis of the physiological data was carried out using customized LabVIEW programs (National Instruments). Femoral and carotid vascular resistance (FVR and CaVR) were calculated using the formula mean arterial pressure/carotid blood flow (mmHg/min/mL). Data are presented as 1-h means ± SEM.
Statistical analysis
Data were analyzed using SPSS v25 (IBM, Armonk, NY, USA). Analysis of serial data were performed by two-way ANOVA, with time as a repeated measure and group as the independent factor. The association between the severity of hypotension during HI and neuronal injury was investigated via receiver operating characteristic (ROC) analysis. ROC analysis was also used to investigate the ability of AVP to discriminate between mild-moderate and severe neuronal injury. For these analyses, cases (total n = 8) were fetuses that either developed severe neuronal injury (defined as ≥50% neuronal injury in ≥1 cortical or basal ganglia region, n = 6) or died due to severe hypotension after HI (n = 2). The controls (total n = 13) were the sham group (n = 5) and fetuses that survived with mild-moderate neuronal injury (n = 8).
The severity of hypotension during HI was quantified as the time spent below six different thresholds: 90% of baseline MAP (calculated over 24 h) representing the limit of cerebral autoregulation in near-term fetal sheep,24 20, 15, 12, 10, and 8 mmHg. ROC curves were calculated for each threshold. 15 mmHg demonstrated the highest discriminative ability and was therefore used to investigate the relationship between AVP concentrations, hypotension and neuronal injury. The relationship between AVP concentrations at 1 h after HI and cardiovascular parameters was also investigated. Statistical significance was accepted when p < 0.05.
Results
Fetal characteristics and umbilical cord occlusion
All fetuses were healthy before experiments based on our laboratory standards. The distribution of sex was not significantly different between shams (4 females, 1 male) and HI (11 females, 5 males). UCO was associated with profound acidemia, hypercapnia, hypoxemia, reduced bicarbonate concentrations and increased lactate concentrations (all p < 0.001, Table 1). Profound hypotension was observed by the end of UCO (mean 10.9 ± 0.7 mmHg compared to 43.5 ± 1.4 mmHg in controls, p < 0.001). All fetuses recovered from UCO. Two fetuses (1 female, 1 male) in the HI group subsequently developed severe hypotension and died within 6 h of UCO; they were assigned maximal injury scores and are shown in Figs. 4 and 6 for the purpose of illustration only. The recovery of fetal arterial biochemistry is shown in Table 1.
Cardiovascular physiology after umbilical cord occlusion
Mean arterial blood pressure was increased at 0–2 h (p < 0.05, Fig. 1) and 10–72 h (p < 0.01) after UCO compared to shams. UCO was not associated with any changes to FHR over the 72-h recovery period. CaBF was significantly reduced after UCO from 0 to 72 h (p < 0.05) compared to the sham group, with a significant increase in CaVR from 0 to 72 h (p < 0.05). Continuous FBF and FVR measurements were not available in one sham animal and one UCO animal due to technical problems (final n = 4 shams, n = 13 UCO). FBF was significantly reduced from 0 to 72 h in the HI group compared to the sham group (p < 0.01) in association with a significant increase in FVR from 0 to 72 h in the HI group (p < 0.05).

Data during HI are not shown. *p < 0.05, control vs. HI. Data are 1 h means ± SEM.
Time course of AVP, AII and renin
AVP plasma concentrations were significantly increased during UCO (Fig. 2, p < 0.005) and at 1–6 h after UCO (p < 0.05). There was no effect of UCO on renin concentrations. AII concentrations were significantly increased during UCO (p < 0.01) but resolved by 1 h after UCO. AVP, renin and AII measured during UCO (12 min after the start of UCO) were not associated with the severity of terminal hypotension. AVP levels 1 h after UCO were correlated with higher MAP (p < 0.005, R2 = 0.41, n = 20, Fig. 3), lower FBF p < 0.005, R2 = 0.40, n = 18) and higher FVR (p < 0.001, R2 = 0.63, n = 18) at that time.

The measurements during HI were taken at 12 min after the start of umbilical cord occlusion. *p < 0.05, control vs. HI.

The relationship between plasma arginine vasopressin (AVP) concentrations at 1 h after hypoxia-ischemia (HI) and mean arterial pressure (MAP), femoral blood flow (FBF) and femoral vascular resistance (FVR) at the same time. Open circles (sham controls), closed circles (HI).
Neural injury
No neurons with ischemic cell change were seen in the hippocampus in shams, and <1 cell per field with ischemic cell change were seen in the parasagittal or lateral cortex, caudate nucleus, putamen, and thalamus. HI was associated with mild selective neuronal loss in the parasagittal cortex and lateral cortex (p < 0.05, Figs. 4a and 5a), with moderate to severe neuronal loss in the caudate nucleus (p < 0.05), putamen (p < 0.05), thalamus (p < 0.05) and DG (p < 0.05), and severe neuronal loss in hippocampal CA regions (all p < 0.05, Fig. 5b).

a Histological outcomes after hypoxia-ischemia (HI) quantified as percentage of neurons showing ischemic injury in cortical and subcortical regions. PSC parasagittal cortex, LC lateral cortex, CA cornu ammonis of the hippocampus, DG dentate gyrus. b ROC curve for severe injury or death examining a range of thresholds for hypotension during HI. c Relationship between the severity of hypotension, quantified as the time mean arterial pressure (MAP) was below 15 mmHg, and plasma arginine vasopressin (AVP) concentrations measured at 1 h after HI. Open circles (shams), close circles (HI). d Linear and nonlinear correlations between the severity of hypotension during HI and the severity of neuronal injury in the PSC, LC, caudate nucleus and putamen. Open circles (shams), close circles (HI), crosses (fetuses that died, not included in regression analysis and shown for illustration purposes only).

a Images from the control and hypoxia-ischemia (HI) groups from the parasagittal and lateral cortex and the caudate nucleus and putamen of the striatum. b Images from the CA1/2 region of the hippocampus from the control group, and two examples from the HI group. Note the severe hippocampal injury after both moderate and severe HI. All images were taken at ×40 magnification from sections stained with acid fuchsin and thionine. White arrows indicate examples of healthy neurons. Black arrows indicate examples of dead ischemic neurons with condensed nuclei and acid fuchsin staining of the cytoplasm. Scale bar = 100 µm.
Relationship between hypotension and neural injury
The cumulative duration of MAP below multiple predefined thresholds of hypotension during UCO was highly associated with severe neuronal injury or death. As shown in Fig. 4b, the areas under ROC curves were: 90% of baseline (0.88, 95% CI 0.72–1.00), 20 mmHg (0.91, 95% CI 0.79–1.00), 15 mmHg (1.00, 95% CI 1.00–1.00), 12 mmHg (0.98, 95% CI 0.93–1.00, not shown), 10 mmHg (0.92, 95% CI 0.77–1.00) and 8 mmHg (0.87, 95% CI 0.70–1.00, not shown). 15 mmHg was the optimal threshold for hypotension; the Youden cut-point at 428.5 s showed sensitivity 100% and specificity 100% for developing severe neuronal injury or death.
The duration of MAP < 15 mmHg showed a significant linear relationship with the severity of neuronal injury in the parasagittal cortex (p < 0.005, R2 = 0.48, n = 19, Fig. 4d) and a sigmoidal relationship with the severity of neuronal injury in the lateral cortex (R2 = 0.82, n = 19), caudate nucleus (R2 = 0.77, n = 19) and putamen (R2 = 0.78, n = 19). By contrast, the duration of MAP < 15 mmHg was not associated with neuronal injury in the thalamus or hippocampal regions.
Relationship between AVP and neural injury
AVP levels at 1 h were linearly associated with the duration of MAP < 15 mmHg during UCO (p < 0.001, R2 = 0.67, n = 20, Fig. 4c). Similarly, AVP levels at 3 h were also associated with the duration of MAP < 15 mmHg (p < 0.05, R2 = 0.24, n = 20, data not shown). By contrast, renin and AII concentrations after HI were not associated with the severity of hypotension or simultaneous AVP concentrations. AVP levels at 1 h showed a close relationship with risk of severe neuronal injury or death, with an area under the ROC curve of 0.98 (95% CI 0.95–1.00, Fig. 6a). The Youden cut-point at 180.0 pmol/L showed sensitivity 100% and specificity 92.3%. Plasma AVP concentrations at 1 h showed a significant linear relationship with the severity of neuronal injury in the parasagittal cortex (p < 0.001, R2 = 0.58, n = 18, Fig. 6b), lateral cortex (p < 0.001, R2 = 0.68, n = 18), caudate nucleus (p < 0.001, R2 = 0.58, n = 18) and putamen (p < 0.001, R2 = 0.62, n = 18), but were not associated with neuronal injury in hippocampal regions or the thalamus (data not shown).

ROC curves of plasma arginine vasopressin (AVP) at 1 h (a) and 3 h (c) after hypoxia-ischemia (HI) for severe neuronal injury or death. Additionally, the relationships between AVP concentrations at 1 h (b) and 3 h (d) after HI and the severity of neuronal injury in the parasagittal cortex, lateral cortex, caudate nucleus and putamen are shown. The dashed line indicates the Youden cut-points at 180.0 and 39.9 pmol/L at 1 and 3 h after HI, respectively. Open circles (shams), close circles (HI), crosses (fetuses that died, not included in regression analysis, shown for illustration purposes only).
Although AVP levels at 3 h were substantially lower than at 1 h, they were still predictive of severe neuronal injury or death with an area under the ROC curve of 0.92 (95% CI 0.80–1.00, Fig. 6c). The Youden cut-point at 39.9 pmol/L showed sensitivity 85.7% and specificity 84.6%. AVP levels at 3 h were linearly associated with neuronal injury in the parasagittal cortex (p < 0.001, R2 = 0.60, n = 18, Fig. 6d), lateral cortex (p < 0.005, R2 = 0.47, n = 18), caudate nucleus (p < 0.01, R2 = 0.38, n = 18) and putamen (p < 0.01, R2 = 0.38, n = 18) but were not associated with neuronal injury in hippocampal regions or the thalamus. Renin and AII levels were not associated with the severity of neuronal injury (data not shown).
Discussion
This study demonstrates that plasma AVP but not renin or AII remain elevated for at least 3 h after moderate-severe HI induced by reversible UCO. AVP levels at 1 and 3 h after HI were associated with both the severity of hypotension during UCO and subsequent neuronal injury after 3 days in the parasagittal and lateral cortex, the caudate nucleus and putamen but not the hippocampus. The Youden cut-point at 180.0 pmol/L for AVP levels at 1 h identified cases that developed severe neuronal injury or died with 100% sensitivity and 92.3% specificity. Similarly, although AVP levels fell steadily after UCO, at 3 h the Youden cut-point of 39.9 pmol/L offered 85.7% sensitivity and 84.6% specificity. Thus, AVP levels seem to be a sensitive blood biomarker of the severity of HI injury within the critical early latent phase.
During UCO, AVP and AII increased markedly, consistent with previous evidence that these vasoactive factors help to attenuate the fall in arterial pressure during HI.7,8,25 The primary stimulus for AVP release is most likely the profound hypotension during UCO.26 Consistent with this, the duration that MAP was <15 mmHg during UCO was closely associated with AVP concentrations at 1 h. Other factors that may have further stimulated AVP release include profound fetal hypoxia and hypercapnia during UCO.11 AVP levels increased further by 1 h compared to levels during UCO. Unfortunately, for technical reasons, we did not have samples from immediately after UCO. Considering that AVP is potently released in response to arterial hypotension,26 it is likely that additional release of AVP both in the final minutes of UCO (i.e. after the sample at 12 min) when hypotension was worsening, and during the immediate reperfusion period contributed to the higher concentrations observed 1 h after HI.
Moreover, the very prolonged elevation of AVP in the present study supports the hypothesis that AVP continued to be released in the early hours after HI. The half-life of AVP in adult humans is just 24 min.27 Although it is possible that impaired hepatic and renal clearance of AVP after HI could have partly contributed to the prolonged elevation in AVP,28,29 AVP clearance is considerably faster in the near-term fetal sheep than in adult sheep or humans, with a half-life of 2.8 min.30 Even more rapid clearance has been observed in newborn sheep.31 In the present study, AVP concentrations on average fell from a peak of 248.2 ± 59.3 pmol/L at 1 h after HI to 80.3 ± 21.3 pmol/L at 3 h after HI, considerably slower than would be expected from previous findings.30 This suggests that AVP continued to be released during the early hours after HI, likely to provide arterial pressure support at a time of impaired cardiac function. Supporting this postulate, AVP concentrations at 1 h after HI were correlated with the simultaneous hypertension, reduction in FBF and increase in FVR (Fig. 3). These data suggest that AVP is at least one of the humoral factors that contribute to blood pressure support after severe HI. Naturally, additional vasoactive factors are likely important, such as catecholamines.32 By contrast, AII levels fell to sham control values by 1 h after UCO, suggesting that AII was not critical to sustain blood pressure after HI.
After the first 6 h of recovery, the HI group showed a continuing increase in vascular tone and reduced blood flow in both the carotid and femoral arteries. This was an important contributor to the secondary increase in arterial pressure between 10 and 72 h. AVP (and AII) had returned to control levels by this time, suggesting that additional vasoactive factors other than AVP and AII are critical mediators of late, sustained vasoconstriction.
There is considerable preclinical evidence that the severity and duration of hypotension during HI are key determining factors for the subsequent severity of brain injury.5 In the present study we defined hypotension using formal ROC analysis (Fig. 2b). The duration that MAP was <15 mmHg during HI showed the strongest association with severe injury or death, and showed a sigmoidal relationship with the severity of neuronal loss in the lateral cortex, caudate nucleus and putamen and a linear relationship with neuronal loss in the parasagittal cortex. This relationship confirms that there is a strikingly narrow threshold during HI for severe neuronal injury to develop in these regions. This indicates that it was the short, terminal period of severe hypotension that was most associated with adverse outcome. In turn this supports the hypothesis that it is not simply the presence of hypotension that is important, but rather that even short periods of severe hypotension during profound hypoxia critically extend and exacerbate brain injury.
The duration that MAP was <15 mmHg was associated with the concentration of AVP at 1 h (Fig. 2c) and in turn AVP levels at 1 h predicted severe neuronal injury or death based on ROC analysis. Regional analysis showed AVP levels were associated with the severity of neuronal injury in the parasagittal and lateral cortex, as well as the caudate nucleus and putamen. Similar but weaker associations were found between AVP levels and neuronal injury at 3 h. Considering that the severity of hypotension was associated with both AVP concentrations at 1 h and the severity of neuronal injury at 72 h, the association between AVP and neuronal injury is therefore most likely indirect, via its association with hypotension.
By contrast, AVP levels were not correlated with hippocampal injury, reflecting the relatively severe hippocampal damage even in fetuses with little basal ganglia injury (Fig. 5b). Previous studies have shown that the hippocampus is exceptionally vulnerable to prolonged periods of HI at this gestation, such that extensive cell loss is seen after just 10 min of HI, a much shorter period than in the present study.33,34 Considering this, the results of the present study suggest that AVP levels 1 h after HI are unlikely to be a sensitive marker of mild HI injury resulting from more mild hypotension. Alternatively, copeptin is released in equimolar amounts to AVP but has a longer half-life allowing it to be measured as a surrogate for AVP.11,14 It is therefore possible that copeptin levels after HI may provide better stratification of the severity of HI, including milder injury, than observed with AVP.11
A key strength of the present study is the clinically relevant paradigm of systemic HI, consistent with a severe sentinel event. Such events account for approximately 25% of cases of HIE.35,36,37 Further studies will help confirm whether AVP levels show similar changes during episodes of intermittent labor-like hypoxia and to assess the predictive value of AVP or copeptin levels immediately after HI. In the present study, plasma samples were stored at −80 °C until they were batch analyzed at the completion of the study. AVP is stable at −80 °C;38 however, it is possible that due to sample handling after collection our present findings may represent an underestimate of absolute AVP levels after HI.
Overall, these findings compliment previous clinical studies. Both AVP and copeptin have been shown to be associated with the severity of acidemia at birth.13,39,40 More importantly, copeptin levels at 6 and 12 h after birth in infants with HIE in the Total Body Hypothermia trial were correlated with the severity of neurodevelopmental impairment at 2 years.41 These time points are too late for timely initiation of therapeutic hypothermia, but given the long half-life of copeptin, this finding strongly supports further studies to evaluate copeptin levels at or shortly after birth to rapidly identify moderate to severe HIE. Moreover, it is reasonable to note that sentinel events represent only approximately 25% of cases of HIE,35,36,37 and so the association between copeptin levels and neurodevelopmental outcome41 suggests that copeptin must have been elevated after a range of HI insults. Regardless, it will be important for future studies to investigate whether the pattern of HI influences the utility of AVP and copeptin as biomarkers. Moreover, it will be important to investigate how factors such as the mode of delivery, maternal analgesia and hypothermic treatment affect AVP and copeptin levels, before clinical translation of these findings can be considered. Notwithstanding these important limitations, the present study shows that AVP levels in the critical early latent phase of injury after HI are highly associated with the severity of subsequent cortical and subcortical neuronal loss, supporting utility to identify high-risk infants who would benefit from neuroprotective therapy.
References
Lee, A. C. et al. Intrapartum-related neonatal encephalopathy incidence and impairment at regional and global levels for 2010 with trends from 1990. Pediatr. Res. 74, 50–72 (2013).
Jacobs, S. E. et al. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst. Rev. 1, CD003311 (2013).
Graham, E. M., Everett, A. D., Delpech, J. C. & Northington, F. J. Blood biomarkers for evaluation of perinatal encephalopathy: state of the art. Curr. Opin. Pediatr. 30, 199–203 (2018).
Wassink, G. et al. A working model for hypothermic neuroprotection. J. Physiol. 596, 5641–5654 (2018).
Gunn, A. J., Parer, J. T., Mallard, E. C., Williams, C. E. & Gluckman, P. D. Cerebral histologic and electrocorticographic changes after asphyxia in fetal sheep. Pediatr. Res. 31, 486–491 (1992).
Zubrow, A. B., Daniel, S. S., Stark, R. I., Husain, M. K. & James, L. S. Plasma vasopressin, renin, and catecholamines during nitroprusside-induced maternal and fetal hypotension in sheep. Pediatr. Res. 24, 73–78 (1988).
Giussani, D. A. et al. Effect of carotid denervation on plasma vasopressin levels during acute hypoxia in the late-gestation sheep fetus. J. Physiol. 477, 81–87 (1994).
Giussani, D. A. et al. Adrenergic and vasopressinergic contributions to the cardiovascular response to acute hypoxaemia in the llama fetus. J. Physiol. 515, 233–241 (1999).
Perez, R., Espinoza, M., Riquelme, R., Parer, J. T. & Llanos, A. J. Arginine vasopressin mediates cardiovascular responses to hypoxemia in fetal sheep. Am. J. Physiol. 256, R1011–R1018 (1989).
Lumbers, E. R. et al. Nonimmune hydrops fetalis and activation of the renin-angiotensin system after asphyxia in preterm fetal sheep. Am. J. Physiol. Regul. Integr. Comp. Physiol. 280, R1045–R1051 (2001).
Summanen, M., Back, S., Voipio, J. & Kaila, K. Surge of peripheral Arginine Vasopressin in a rat model of birth asphyxia. Front. Cell. Neurosci. 12, 2 (2018).
Chard, T., Hudson, C. N., Edwards, C. R. & Boyd, N. R. Release of oxytocin and vasopressin by the human foetus during labour. Nature 234, 352–354 (1971).
Summanen, M. et al. Comparison of umbilical serum copeptin relative to erythropoietin and S100B as asphyxia biomarkers at birth. Neonatology 112, 60–66 (2017).
Evers, K. S. & Wellmann, S. Arginine Vasopressin and Copeptin in perinatology. Front. Pediatr. 4, 75 (2016).
Spoljaric, A. et al. Vasopressin excites interneurons to suppress hippocampal network activity across a broad span of brain maturity at birth. Proc. Natl. Acad. Sci. USA 114, E10819–E10828 (2017).
Kasai, M. et al. Early sinusoidal heart rate patterns and heart rate variability to assess hypoxia-ischaemia in near-term fetal sheep. J. Physiol. 597, 5535–5548 (2019).
Barlow, R. M. The foetal sheep: morphogenesis of the nervous system and histochemical aspects of myelination. J. Comp. Neurol. 135, 249–262 (1969).
Drury, P. P. et al. Status epilepticus after prolonged umbilical cord occlusion is associated with greater neural injury in fetal sheep at term-equivalent. PLoS ONE 9, e96530 (2014).
Lear, C. A. et al. Sympathetic neural activation does not mediate heart rate variability during repeated brief umbilical cord occlusions in near-term fetal sheep. J. Physiol. 594, 1265–1277 (2016).
van Bel, F., Roman, C., Klautz, R. J., Teitel, D. F. & Rudolph, A. M. Relationship between brain blood flow and carotid arterial flow in the sheep fetus. Pediatr. Res. 35, 329–333 (1994).
Rademaker, M. T., Charles, C. J., Nicholls, M. G. & Richards, A. M. Interactions of enhanced urocortin 2 and mineralocorticoid receptor antagonism in experimental heart failure. Circ. Heart Fail. 6, 825–832 (2013).
Gluckman, P. D. & Parsons, Y. Stereotaxic method and atlas for the ovine fetal forebrain. J. Dev. Physiol. 5, 101–128 (1983).
Gunn, A. J., Gunn, T. R., de Haan, H. H., Williams, C. E. & Gluckman, P. D. Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J. Clin. Invest. 99, 248–256 (1997).
Papile, L. A., Rudolph, A. M. & Heymann, M. A. Autoregulation of cerebral blood flow in the preterm fetal lamb. Pediatr. Res. 19, 159–161 (1985).
Green, L. R., McGarrigle, H. H., Bennet, L. & Hanson, M. A. Angiotensin II and cardiovascular chemoreflex responses to acute hypoxia in late gestation fetal sheep. J. Physiol. 507, 857–867 (1998).
Holmes, C. L., Patel, B. M., Russell, J. A. & Walley, K. R. Physiology of vasopressin relevant to management of septic shock. Chest 120, 989–1002 (2001).
Baumann, G. & Dingman, J. F. Distribution, blood transport, and degradation of antidiuretic hormone in man. J. Clin. Invest. 57, 1109–1116 (1976).
Sarkar, S. et al. Relationship between acute kidney injury and brain MRI findings in asphyxiated newborns after therapeutic hypothermia. Pediatr. Res. 75, 431–435 (2014).
Lee, J. K. et al. Relationships between cerebral autoregulation and markers of kidney and liver injury in neonatal encephalopathy and therapeutic hypothermia. J. Perinatol. 37, 938–942 (2017).
Wiriyathian, S., Porter, J. C., Naden, R. P. & Rosenfeld, C. R. Cardiovascular effects and clearance of arginine vasopressin in the fetal lamb. Am. J. Physiol. 245, E24–E31 (1983).
Miao, D. C., Velaphi, S. C., Roy, T., Despain, K. & Rosenfeld, C. R. Metabolism and synthesis of arginine vasopressin in conscious newborn sheep. Am. J. Physiol. Endocrinol. Metab. 295, E672–E677 (2008).
Quaedackers, J. S., Roelfsema, V., Heineman, E., Gunn, A. J. & Bennet, L. The role of the sympathetic nervous system in post-asphyxial intestinal hypoperfusion in the preterm sheep fetus. J. Physiol. 557, 1033–1044 (2004).
Williams, C. E., Gunn, A. J., Mallard, C. & Gluckman, P. D. Outcome after ischemia in the developing sheep brain: an electroencephalographic and histological study. Ann. Neurol. 31, 14–21 (1992).
Hunter, C. J. et al. Key neuroprotective role for endogenous adenosine A1 receptor activation during asphyxia in the fetal sheep. Stroke 34, 2240–2245 (2003).
Jonsson, M., Agren, J., Norden-Lindeberg, S., Ohlin, A. & Hanson, U. Neonatal encephalopathy and the association to asphyxia in labor. Am. J. Obstet. Gynecol. 211, 667 e661–667.e668 (2014).
West, C. R. et al. Antenatal antecedents of moderate or severe neonatal encephalopathy in term infants - a regional review. Aust. N. Z. J. Obstet. Gynaecol. 45, 207–210 (2005).
Westgate, J. A., Gunn, A. J. & Gunn, T. R. Antecedents of neonatal encephalopathy with fetal acidaemia at term. Br. J. Obstet. Gynaecol. 106, 774–782 (1999).
Heida, J. E. et al. Comparison of ex vivo stability of copeptin and vasopressin. Clin. Chem. Lab. Med. 55, 984–992 (2017).
Ruth, V., Fyhrquist, F., Clemons, G. & Raivio, K. O. Cord plasma vasopressin, erythropoietin, and hypoxanthine as indices of asphyxia at birth. Pediatr. Res. 24, 490–494 (1988).
Schlapbach, L. J. et al. Copeptin concentration in cord blood in infants with early-onset sepsis, chorioamnionitis and perinatal asphyxia. BMC Pediatr. 11, 38 (2011).
Kelen, D. et al. Serum copeptin and neuron specific enolase are markers of neonatal distress and long-term neurodevelopmental outcome. PLOS ONE 12, e0184593 (2017).
Acknowledgements
The present study was funded by grants from the Health Research Council of New Zealand (grant number 17/601), the Auckland Medical Research Foundation (grant number 1108004) and New Zealand Lottery Grants Board (grant numbers 209214 and 340855). The funding sources had no role in the design or undertaking of the study, the interpretation of results, writing of the article nor the decision to submit this article for publication.
Author information
Authors and Affiliations
Contributions
These experiments were conducted in the Fetal Physiology and Neuroscience Group laboratory at the University of Auckland. A.J.G., P.P.D. and C.A.L. conceived the hypotheses, experimental design and analysis protocols for the study. C.A.L., P.P.D., J.O.D., and A.J.G. were responsible for data collection. C.A.L., M.K. and P.P.D. performed the analysis. C.A.L. drafted the manuscript. All authors were involved in data interpretation, in the editing and revision of the manuscript, approved the final version of the manuscript and agreed to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lear, C.A., Kasai, M., Drury, P.P. et al. Plasma vasopressin levels are closely associated with fetal hypotension and neuronal injury after hypoxia-ischemia in near-term fetal sheep. Pediatr Res 88, 857–864 (2020). https://doi.org/10.1038/s41390-020-0845-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-020-0845-2
