
Abstract
The real-time dissolution of the single-phase compositionally complex alloy (CCA), Al1.5TiVCr, was studied using an inline inductively coupled plasma method. Compositionally complex alloys (CCAs), a term encompassing high entropy alloys (HEAs) or multi-principal element alloys (MPEAs), are—in general—noted for their inherently high corrosion resistance. In order to gain an insight into the dissolution of Al1.5TiVCr alloy, atomic emission spectroelectrochemistry was utilised in order to measure the ion dissolution of the alloy during anodic polarisation. It was revealed that incongruent dissolution occurred, with preferential dissolution of Al, and essentially no dissolution of Ti, until the point of alloy breakdown. Results were correlated with X-ray photoelectron spectroscopy, which revealed a complex surface oxide inclusive of unoxidised metal, and metal oxides in disproportion to the bulk alloying element ratio.
Similar content being viewed by others

Introduction
The recent interest in high entropy alloys (HEAs)1,2,3 has generated a large number of studies regarding alloys that are derived from the design concepts behind high entropy alloys. Notionally, HEAs are alloys with five or more alloy components in near equi-atomic proportions, and which present a high entropy of mixing, resulting in a single-phase structure. However, as is nowadays well documented, such compositionally complex alloys (CCAs) which are also termed multi-principal element alloys (MPEAs), are a much broader category of alloys that may or may not possess a high entropy of mixing, may not be single-phase, and may also have less than five principle alloying elements4,5,6,7.
Of the works reported to date that have focused on the corrosion of HEAs, CCAs and MPEAs, there is, in the general case (notwithstanding exceptions) significant evidence that such alloys are highly corrosion resistant3,8,9,10,11,12. The high corrosion resistance of such alloys is an unintended, but welcome, consequence that is also coupled with alloy properties that include unprecedented hardness and high strength13,14. In order to mechanistically study the corrosion of complex and corrosion resistant alloys, exposure testing and subsequent microscopy is not compatible with slow rates of alloy dissolution, therefore electrochemical testing is required in order to measure and quantify such low levels of dissolution. Electrochemical testing, whilst extremely valuable (both critical and necessary) does not however offer any physical insight into the extent of corrosion or alloy dissolution, and requires supplementation with ancillary testing.
Of the most powerful analysis techniques now available to corrosion scientists and electrochemists, is the method developed by Ogle15,16,17, termed atomic emission spectroelectrochemistry (AESEC). The AESEC method employs the use of an electrochemical flow cell, which permits standard electrochemical testing, however the flow cell is outlet into an inductively coupled plasma (ICP) instrument - which may either be ICP optical emission spectroscopy (ICP-OES)15,16,17,18,19 or ICP mass spectroscopy (ICP-MS)20,21. This technique has been responsible for delineating the dissolution of numerous alloy systems, by providing an element-selective insight whilst providing the precise rates of alloy dissolution, including for highly corrosion resistant alloys21.
To the best of authors’ knowledge, the AESEC method has not yet been applied to understanding the dissolution of CCAs. Of the analysis to date regarding corrosion of CCAs, including recent X-ray photoelectron spectroscopy analysis, it is posited that corrosion of CCAs involves incongruent dissolution—whereby there is selective oxidation of alloy species in proportions that are unique to the bulk alloy composition (and alloying element ratio)11,22. In fact, the complexity of the dissolution associated with CCAs was also recently posited as being so unique, that it challenges the long-held views of the thermodynamic nature of metal alloy passivity23. A first-order requirement of validating and quantifying incongruent dissolution associated with CCAs would further elucidate the process of alloy dissolution; but then also serve as a basis for illuminating second-order effects such as dissolution mechanisms and the nature of what passivity means in the cases of CCAs (or at least, for the alloy and its associated CCA family studied herein). The present study applies AESEC to the Al1.5TiVCr alloy, which is a close variant to the equi-atomic AlTiVCr alloy studied and reported in detail previously by Qiu and co-workers4,11,23. The alloy notation for Al1.5TiVCr indicates that the alloy is nominally comprised of equi-atomic proportions of Ti, V and Cr, but the atomic concentration of Al is 1.5 times that of Ti, V and Cr. This alloy is being studied because it represents an alloy that is single-phase and of appreciably low density (of 4.65 g/cm3) in comparison to other CCAs reported to date; whilst also presenting high aqueous corrosion resistance relative to typical corrosion resistant alloys (CRAs) such as stainless steels (which nominally possess a density of >7.48 g/cm3) or Ni alloys (which nominally possess a density of >8.09 g/cm3).
Results and discussion
Alloy microstructure
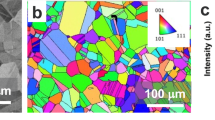
The Al1.5TiVCr alloy studied herein was investigated in the as-arc melted condition. In the as-arc melted condition, the Al1.5TiVCr alloy is single-phase, with the general microstructure typified by the electron backscatter diffraction inverse pole figure map presented in Fig. 1a. Transmission electron microscopy (TEM) of Al1.5TiVCr in dark field mode, as shown in Fig. 1b, reveals fine nanoscale domain-like structure (indicated by arrows) when imaged using the (100) superlattice reflection along the [001] zone axis. Accompanying selected area diffraction patterns collected from the [001] and [011] zone axis, reveal the characteristics of a uniform B2 structure (Fig. 1c, d).

a Microstructure of as-cast single-phase Al1.5TiVCr as represented from an EBSD inverse pole figure map, b Dark field TEM image obtained using the (100) superlattice reflection as determined from the selected area electron diffraction (SAED) pattern along the [001] zone axis (revealing the extremely fine domains (indicated by arrows)). c, d SAED patterns of the alloy along [001] and [011] zone axis, revealing characteristics of a uniform B2 structure.
Atomic emission spectroelectrochemistry
The ion dissolution rate, represented in the form of an equivalent current density, for Al1.5TiVCr exposed to quiescent 0.1 M NaCl under open circuit conditions is presented in Fig. 2a. It is observed that the method employed herein is not only capable of providing an equivalent current density, but the unique ion dissolution current density for individual species, namely, \(i_{{\mathrm{Al}}}^{3 + }\), \(i_{{\mathrm{Ti}}}^{4 + }\), \(i_{\mathrm{V}}^{3 + }\), and \(i_{{\mathrm{Cr}}}^{3 + }\) as determined in real time by inline ICP-MS analysis. Therefore, the method employed is capable of providing accurate dissolution rates on an elemental basis. The results in Fig. 2a indicate that upon immersion, the dissolution of Al is greatest, particularly in the first ~250 s of immersion, but then remaining as the principle dissolving element of the alloy. The dissolution of species at open circuit was observed to occur in the order of: \(i_{{\mathrm{Al}}}^{3 + }\) » \(i_{\mathrm{V}}^{3 + }\) » \(i_{{\mathrm{Cr}}}^{3 + }\) > \(i_{{\mathrm{Ti}}}^{4 + }\), providing direct evidence for preferential dissolution of Al and V, and incongruent alloy dissolution in real time (and in situ).

a Open circuit potential (OCP) exposure for 15 mins, b open circuit condition and subsequent potentiodynamic polarisation exposure for the alloy produced herein in 0.1 M NaCl. The sum of ICP determined dissolution current itot and the potentiostat applied current density ipstat during PDP exposure are also included for comparison purpose. c Potentiodynamic polarisation data presented in the format of applied potential vs. current density \(i_{{\mathrm{Al}}}^{3 + }\), \(i_{{\mathrm{Ti}}}^{4 + }\), \(i_{\mathrm{V}}^{3 + }\), \(i_{{\mathrm{Cr}}}^{3 + }\), itot, and ipstat.
The application of a polarising signal that is afforded by the atomic emission spectroelectrochemistry method, permits for the collection of a significant insight from the data presented in Fig. 2b. The data shown in Fig. 2b, includes the ion dissolution current for three distinct regimes within the test. The ion dissolution is shown for ~500 s of open circuit conditioning, followed by the ion dissolution shown for the period of potentiodynamic polarisation (PDP), which also lasted ~500 s, covering a potential range of ~ 1.5 V, at a potential scan rate 3 mV/s. It should be noted that the polarising signal commenced at a potential 100 mV below the open circuit potential and was scanned upwards through the corrosion potential and to increasing anodic potentials (and thus, the first ~100 mV of polarisation was cathodic). Finally, upon cessation of the applied polarisation signal, the ion dissolution rate is also shown for a period of open circuit (~200 s).
From Fig. 2b, the differentiation between the ion dissolution of alloying elements during open circuit is difficult to discern owing to the scale of the plotted data—however, this is covered further below. The application of a polarisation is indicated by the signal denoted as ipstat. It is noted that during the application of a polarising signal, the ion dissolution rate remains low, until about ~850 s (corresponding to a potential of ~ +0.4 VSCE). At this stage, a corresponding increase can also be seen in the signal denoted as itot. The itot signal corresponds to the sum of all of \(i_{{\mathrm{Al}}}^{3 + }\) + \(i_{{\mathrm{Ti}}}^{4 + }\) + \(i_{\mathrm{V}}^{3 + }\) + \(i_{{\mathrm{Cr}}}^{3 + }\). It is noted that at all times, the itot signal remains lower than the ipstat signal, which is indicative that not all of the polarising signal is converted to ion dissolution; meaning that a surface film is developing during the anodic polarisation scan. One advantage of AESEC technique is the ability to demonstrate the real-time elemental current density vs. the applied polarising signal during polarisation. The results in Fig. 2b indicate that the principal s ource of the itot signal is from \(i_{{\mathrm{Al}}}^{3 + }\), and to a lesser extent, \(i_{\mathrm{V}}^{3 + }\), \(i_{{\mathrm{Cr}}}^{3 + }\) and \(i_{{\mathrm{Ti}}}^{4 + }\). The relative proportions are however best discerned from a logarithmic plot, as discussed below. Finally, upon cessation of the polarising signal, it is noted that ion dissolution persists for some ~200 s (which corresponds to the remaining period of assessment for the testing conducted herein), albeit with a diminishing ion dissolution signal. However, it is important to bear in mind the difficulty in interpreting such decay as the time constant associated with the ion transport within the flow cell was not quantitatively defined. The diminishing of the ion dissolution signal indicates that ‘repassivation’ of the Al1.5TiVCr occurred, following anodic polarisation. It is noteworthy that Ti4+ establishes a relatively lower ion dissolution current in the most rapid time, without a secondary peak, suggesting the highest repassivation ability of Ti among the alloying elements and a greater contribution from Ti in repassivation. It seems that the repassivation of Ti also contributes to lower ion dissolution current of other metal cations including V3+, Cr3+ and Al3+ after 1100 s.
Figure 2c reveals a portion of the AESEC data (as reported in Fig. 2b corresponding to the period of potentiodynamic polarisation) presented in the familiar format of applied potential vs. current density, with the unique current density provided for \(i_{{\mathrm{Al}}}^{3 + }\), \(i_{{\mathrm{Ti}}}^{4 + }\), \(i_{\mathrm{V}}^{3 + }\), \(i_{{\mathrm{Cr}}}^{3 + }\), itot, and ipstat. The representation in Fig. 2c provides an additional insight into the dissolution of Al1.5TiVCr. Whilst it was difficult from Fig. 2b to discern the differences in ion dissolution between unique alloying elements, particularly at the early stages of polarisation, Fig. 2c permits ready differentiation (owing to the logarithmic axis). The results in Fig. 2c reveal that the dissolution is incongruent, not only with the principal ion dissolution associated with \(i_{{\mathrm{Al}}}^{3 + }\), but there are definitive differences in the ion dissolution rates from \(i_{\mathrm{V}}^{3 + }\), \(i_{{\mathrm{Cr}}}^{3 + }\) and \(i_{{\mathrm{Ti}}}^{4 + }\) (all of which are present in the alloy in equal proportions). The assessment of Fig. 2 visually reveals the following: There are relatively uniform ion dissolution rates for all elements until ~ +0.3 VSCE; The ion dissolution rates are unique depending on the element being dissolved, indicating incongruent dissolution and unique dissolution rates for all of the four elements in the alloy; At ~ +0.3 VSCE, there is an increase in the \(i_{{\mathrm{Cr}}}^{3 + }\) signal, and a corresponding decrease in the \(i_{{\mathrm{Al}}}^{3 + }\) signal, At ~ +0.45 VSCE, there is a sharp increase in the \(i_{{\mathrm{Al}}}^{3 + }\) and the \(i_{\mathrm{V}}^{3 + }\) signals, which when added to the increasing \(i_{{\mathrm{Cr}}}^{3 + }\) signal, results in a marked increase in the itot signal detected; Uniquely, the \(i_{{\mathrm{Ti}}}^{4 + }\) signal begins to increase (by a marked ‘breakdown’) at ~ +0.7 VSCE.
X-ray photoelectron spectroscopy surface analysis of Al1.5TiVCr
The abridged results from X-ray photoelectron spectroscopy (XPS) characterisation of Al1.5TiVCr are presented in Fig. 3. The XPS results from native surface oxide upon Al1.5TiVCr (naturally developed in laboratory air) are presented in Fig. 3a, b.

a native surface oxide upon Al1.5TiCrV, b data accompanying the results in a for the native surface oxide upon Al1.5TiCrV, however processed to provide the relative fraction of oxide to unoxidised metal (M0), c the surface film upon Al1.5TiCrV following an anodic potentiodynamic polarisation scan (as per Fig. 2c), d data accompanying the results in c for the surface oxide following anodic polarisation upon Al1.5TiCrV, however processed to provide the relative fraction of oxide to unoxidised metal (M0). Note that etching stopped at 60 min. The final data point (plotted at just above 60 min. for comparison) was measured after the samples had either been exposed very briefly to air (3a and b or left under vacuum for three days (3c and d). See text for details.
The results in Fig. 3a indicate the measured atomic concentrations as a function of surface depth, defined by etch time. As has been noted in prior works regarding XPS of a like alloy23, the sputtering rates for CCAs are not (accurately) known and therefore sputtering time is not converted to depth. In the present study we do not report the detailed spectra, but the analysis of spectra based on the analysis method previously reported in ref. 23. The results in Fig. 3a indicate that for 0 min etching time (which corresponds to the outer surface of the oxide), there is the highest concentration of oxygen (O) and the principal alloying element detected is Al (followed by Cr, V and Ti). It is noted that during etching, the expectation of realising a metal-only surface was not observed, with the O concentration decreasing (to ~20 mins etching) and then remaining relatively consistent thereafter. This inability to etch to a wholly metal substrate, even under ultra-high vacuum, was previously discussed23 and was attributed to the surface being highly reactive, immediately oxidising upon etching – even with the very minimal oxygen present in the XPS chamber (see Methods). Such a finding is characteristic of CCAs, and suggests that the origins of corrosion resistance of CCAs may not be in their low reactivity, but in high reactivity that leads to rapid oxidation and the development of protective surface films. It is noted, that XPS analysis was repeated numerous times for the samples discussed in this work, with highly consistent results (even when left in the XPS chamber for a conditioning for 3 days, a testing that is not reported herein). Based on the data format presented, it is notionally considered that the first few minutes of etching corresponds to the surface film propagation, with a transition through the film being completed within ~5 min of etching, which is better evidenced by Fig. 3b.
Figure 3b further analyses the data from the native surface film, differentiating the data in Fig. 3a according to whether the species is an oxide (MX+) or unoxidised metal (M0). It is seen that there is an inhomogeneous surface oxide as a function of etching time. The XPS data from the outer surface of the alloy indicates that the highest proportion of metal species is present as an oxide (~60%). However, this proportion of oxide-to-unoxidised metal changes rapidly (within ~5 min of etching) with depth into the outer surface. In particular, for V and Cr, they are principally present as unoxidised metal (for the low proportion of their overall presence) in the surface film following ~5 mins of etching. At all times however, the proportion of Al oxide to Al metal remains >1, such that (also when interpreted with Al having the highest surface concentration), the principle surface oxide is Al oxide with XPS confirms that the cation is predominantly Al3+, doped with unoxidised metal from the alloying elements. Overall, all four metals exist at the surface (top few nm) in metallic (unoxidised metal, M0) as well as oxidised form, and based on the experiments herein, the prevalent state is the most stable form (Al3+, Ti4+, Cr3+ and V3+). It is believed that an important feature of the data presented herein, and similarly in previous studies23, is the nontrivial proportion of unoxidised metal (M0) incorporated into the surface film—which is postulated as being a key contributor to the corrosion resistance of CCAs, although the mechanism is hitherto unexplored11.
Figure 3c, d present the XPS results corresponding to the Al1.5TiVCr surface following anodic polarisation (as per Fig. 2). Of note is that, following polarisation, the total atomic concentration of Al (Fig. 3c) is lower at the outer surface of the alloy, and remains ~5–7 at. % lower through the surface. However, of principle difference between the XPS results for the native surface and the previously anodically polarised is the determination that outer surface film (Fig. 3d) is principally comprised of oxides of the alloying elements (with negligible unoxidised metal) until ~5 min of etching, which is consistent with abovementioned development of a surface film during AESEC testing. Following ~5 min of etching the proportion of oxide-to-unoxidised metal was similar to that of the native alloy surface film.
To investigate the re-oxidation behaviour of both the native surface and anodically polarised one, a “repeat” test was carried out after 60 min of etching (see Fig. 3). The native surface was briefly exposed to air, while the anodically polarised one was kept under vacuum for three days. It can be seen from both Fig. 3b, d that there was a large increase in fraction of Ti oxide, V oxide and Cr oxide, however, a much lesser increase of Al oxide in both cases. It is interesting considering Al is the most reactive constitutional element to form oxides indicated by the most negative ΔG for metal oxidation (−ΔG0 = −RTlnpO2). It is likely due to the fact that Al was largely oxidised during sputtering (>50%). It is also possible related to the slow kinetics of Al oxide formation, such slow-growing of alumina (Al2O3) has been reported in Al-containing high temperature alloys and such slow-forming, dense oxide is beneficial for their oxidation property24.
General discussion
The data in Fig. 2, namely Fig. 2c indicates that the dissolution of Al1.5TiVCr is incongruent. It is noted that each element has a unique dissolution rate, and a unique potential at which the unique ion dissolution rate increases with potential. It was also revealed that the itot < ipstat at all times during the AESEC testing, indicating the development of a surface film upon the alloy. The precise quantification of ion dissolution rates indicates the ‘corrosion rate’ of the alloy tested herein by a rapid in-situ methodology, also indicating that the alloy is capable of repassivation. However, it is important to bear in mind that one inherent limitation of this AESEC technique as the oxidation state of each element cannot be determined by ICP-MS. The ion dissolution rate of each element is calculated based on the most stable oxidation state of each element. In this study, CCA was polarised to a high potential (~1.0 VSCE), there is a possibility of oxidation state change during polarisation, particularly for V and Cr. Despite the difficulty in empirical determination of actual oxide states, interpretation of the XPS analysis herein revealed that qualitatively, the fraction of V4+ and V5+ increased from around ~30 to ~60%, and the fraction of Cr5+ was also increased and proportionally higher following polarisation. To even more accurately determine the ion dissolution rate, it might be useful to explore pure metals of numerous oxidation states using AESEC method, which will be important future work (and not carried out herein). The work herein is finite in its remit, but it has been the first demonstration of AESEC upon a CCA. The accompanying XPS results indicate that the CCA tested has a surface film that is principally comprised of the Al oxide, with the less reactive (or more noble) elements entrapped in the surface film, including in the unoxidised metallic state. As noted by a review of the corrosion of HEAs3 and in works that have recently been studying the corrosion of HEAs (and CCAs)23,25,26,27,28,29,30,31,32,33,34,35,36,37 there is important—and significant—future work required to rationalise the corrosion and surface films developed upon such complex alloys.
The present study has indicated that the dissolution of the alloy Al1.5TiVCr was incongruent, both during open circuit exposure, and during potentiodynamic polarisation. The inline ICP-MS method employed (AESEC) was capable of providing significant insights into the alloy dissolution mechanism. It was revealed that Al3+ dissolution was 1–2 orders of magnitude greater than V, Cr and Ti, respectively. In that context, the significant majority of the total dissolution current (itot) measured was from Al dissolution. The measured itot was lower than the measured potentiostat current, ipstat, indicative of the development of a surface film upon the alloy during dissolution. Upon cessation of the applied anodic potential, the real-time repassivation of Al1.5TiVCr was readily observed. The aforementioned surface film developed on Al1.5TiVCr was studied using XPS. The XPS analysis of the native surface oxide, and that following polarisation were notionally consistent, revealing a low proportion of Ti and the dominance of Al in the film. The predominant oxide in the surface film was that of oxidised Al, and to a significantly lesser extent, Cr, V and then Ti. There was evidence that there was unoxidised metal in the surface film, previously posited in prior work, with evidence of unoxidised metal for all of Al, Ti, Cr and V.
Methods
Alloy production
The Al1.5TiVCr alloy was produced by melting pure (99.99 wt.%) metal pieces (chunks) as starting materials on a water-cooled copper hearth in an argon atmosphere, using an arc melting furnace. The ingot was remelted five times to ensure homogeneity.
Microstructural characterisation
Electron backscatter diffraction (EBSD) of the metallographically prepared Al1.5TiVCr surface was carried out using an FEI Quanta 3D FEG, equipped with a Pegasus Hikari EBSD System and using TSL software. Specimen preparation involved gradually polishing to 1 µm finish using diamond paste following by final polishing with 0.05 µm oxide polishing suspension (OPS).
Transmission electron microscopy (TEM) was carried out using an FEI Tecnai T20 instrument. Specimen preparation for TEM was carried out using the focused ion beam (FIB) lift-out technique, in order to create an electron transparent lamella.
AESEC
Atomic emission spectroelectrochemical (AESEC) measurements involved the use of a scanning flow cell coupled with an inductively coupled plasma mass spectrometer (ICP-MS). A conventional three-electrode configuration was employed for electrochemical testing, with such a configuration realised using a commercially available electrochemical flow cell (012799, ALS, Tokyo, Japan). Unbuffered quiescent 0.1 M NaCl was used as the electrolyte, which was continuously pumped past the working electrode surface and injected onto the ICP-MS instrument using a flow rate of ~0.13 mL/min, allowing for real-time inline elemental analysis of the following elements 27Al, 52Cr, 48Ti, 51V. Electrochemical testing was carried out using a potential scan rate of 3 mV/s, and potentiodynamic polarisation was carried out in an upward scan direction from the open circuit potential (OCP) ̶ 100 mVSCE. A Bio-logic SP-50 potentiostat was used for the electrochemical control during AESEC. The ICP-MS instrument used in this study was a Perkin Elmer Optima 8000 ICP-MS Spectrometer using Syngistix software. The ICP-MS instrument detection limit (C2σ) for the following element Al, Cr, Ti, V was 3, 2, 0.5, and 0.5 µg/L.
The AESEC method has a high sensitivity to the detection of dissolved ion concentration, as well as micrometre-sized solids (precipitates or particles) during the process of open circuit exposure or polarisation of metallic alloys. The calculation of the concentration of released metal ions and particles as a function of time has been presented in numerous studies15,38,39 and as per prior works, the instantaneous metal dissolution rate was calculated (expressed in µg/s cm2) using the following equation:
With CM the volume adjusted ion concentration (µg/L), f the electrolyte flow rate (mL/s) and A the exposed working electrode surface area (0.6 cm2)39. The conversion of the instantaneous dissolution rate to the partial elemental current density (iMn+) was determined via Faraday’s law:
where n is the charge on the ion, F is the Faraday constant and MA is the atomic mass of the element M.
XPS analysis
X-ray photoelectron spectroscopy (XPS) was carried out using an AXIS Nova spectrometer (Kratos Analytical, UK) with a monochromated Al Kα source, and utilising the standard aperture (0.7 mm × 0.3 mm). Survey spectra were acquired at a pass energy of 160 eV. Higher resolution spectra were recorded from peaks at 40 eV pass energy, and binding energies were referenced to the Ti2p3/2 metal peak at 453.9 eV analysed at an emission angle of 0 degrees. The reported depth profiling experiments were conducted using an Ar Gas Cluster Ion Source with a cluster size of Ar1000+ and impact energy of 10 keV. The total vacuum chamber pressure was ~10−9 mbar, and the residual gas was analysed using an MKS Instruments e-Vision 2 quadrupole mass spectrometer.
Data availability
All relevant data are available from authors upon reasonable request.
References
Yeh, J. W. et al. Nanostructured high-entropy alloys with multiple principal elements: Novel alloy design concepts and outcomes. Adv. Eng. Mater. 6, 299–303 (2004).
Zhang, Y. et al. Microstructures and properties of high-entropy alloys. Prog. Mater. Sci. 61, 1–93 (2014).
Qiu, Y., Thomas, S., Gibson, M. A., Fraser, H. L. & Birbilis, N. Corrosion of high entropy alloys. npj Mater. Degrad. 1, 15 (2017).
Qiu, Y. et al. A lightweight single-phase AlTiVCr compositionally complex alloy. Acta Mater. 123, 115–124 (2017).
Jensen, J. K. et al. Characterization of the microstructure of the compositionally complex alloy Al1Mo0.5Nb1Ta0.5Ti1Zr1. Scr. Mater. 121, 1–4 (2016).
Senkov, O. N., Senkova, S. V., Miracle, D. B. & Woodward, C. Mechanical properties of low-density, refractory multi-principal element alloys of the Cr–Nb–Ti–V–Zr system. Mater. Sci. Eng. A 565, 51–62 (2013).
Soni, V. et al. Phase stability as a function of temperature in a refractory high-entropy alloy. J. Mater. Res. 33, 3235–3246 (2018).
Lu, P. et al. Computational materials design of a corrosion resistant high entropy alloy for harsh environments. Scr. Mater. 153, 19–22 (2018).
Quiambao, K. F. et al. Passivation of a corrosion resistant high entropy alloy in non-oxidizing sulfate solutions. Acta Mater. 164, 362–376 (2019).
Qiu, Y., Gibson, M. A., Fraser, H. L. & Birbilis, N. Corrosion characteristics of high entropy alloys. Mater. Sci. Technol. 31, 1235–1243 (2015).
Qiu, Y. et al. Microstructure and corrosion properties of the low-density single-phase compositionally complex alloy AlTiVCr. Corros. Sci. 133, 386–396 (2018).
Ayyagari, A. et al. Reciprocating sliding wear behavior of high entropy alloys in dry and marine environments. Mater. Chem. Phys. 210, 162–169 (2018).
Li, Z., Pradeep, K. G., Deng, Y., Raabe, D. & Tasan, C. C. Metastable high-entropy dual-phase alloys overcome the strength-ductility trade-off. Nature 534, 227–230 (2016).
Qiu, Y. et al. Microstructural evolution, electrochemical and corrosion properties of Al CoCrFeNiTi high entropy alloys. Mater. Des. 170, 107698 (2019).
Ogle, K. Atomic emission spectroelectrochemistry: a new look at the corrosion, dissolution and passivation of complex materials. Corros. Mater. 37, 60–67 (2012).
Lebouil, S., Gharbi, O., Volovitch, P. & Ogle, K. Mg dissolution in phosphate and chloride electrolytes: insight into the mechanism of the negative difference effect. Corrosion 71, 234–241 (2015).
Swiatowska, J., Volovitch, P. & Ogle, K. The anodic dissolution of Mg in NaCl and Na{sub 2}SO{sub 4} electrolytes by atomic emission spectroelectrochemistry. Corros. Sci. 52, 2372–2378 (2010).
Gharbi, O., Birbilis, N. & Ogle, K. In-situ monitoring of alloy dissolution and residual film formation during the pretreatment of Al-alloy AA2024-T3. J. Electrochem. Soc. 163, C240–C251 (2016).
Yan, Y. et al. Investigating ion release using inline ICP during in situ scratch testing of an Mg-Li(-Al-Y-Zr) alloy. Electrochem. Commun. 99, 46–50 (2019).
Klemm, S. O., Topalov, A. A., Laska, C. A. & Mayrhofer, K. J. Coupling of a high throughput microelectrochemical cell with online multielemental trace analysis by ICP-MS. Electrochem. Commun. 13, 1533–1535 (2011).
Lutton, K., Gusieva, K., Ott, N., Birbilis, N. & Scully, J. R. Understanding multi-element alloy passivation in acidic solutions using operando methods. Electrochem. Commun. 80, 44–47 (2017).
Ayyagari, A. V., Gwalani, B., Muskeri, S., Mukherjee, S. & Banerjee, R. Surface degradation mechanisms in precipitation-hardened highentropy alloys. NPJ Mater. Degrad. 2, 1–10 (2018).
Qiu, Y. et al. A surface study of the native oxide upon a compositionally complex alloy. Corrosion 74, 1312–1317 (2018).
Mortazavi, N. et al. Interplay of water and reactive elements in oxidation of alumina-forming alloys. Nat. Mater. 17, 610–617 (2018).
Raza, A., Abdulahad, S., Kang, B., Ryu, H. J. & Hong, S. H. Corrosion resistance of weight reduced AlxCrFeMoV high entropy alloys. Appl. Surf. Sci. 485, 368–374 (2019).
Wen, X. et al. Tribological and corrosion properties of Ni‐Cr‐Co‐Ti‐V multi‐principal element alloy prepared by vacuum hot‐pressing sintering. Adv. Eng. Mater. 21, 1801239 (2018).
Yang, S. et al. Effect of Cr content on corrosion behavior of AlCr x FeNi2Cu1. 6 high entropy alloys. Mater. Res. Express 6, 076501 (2019).
Jiang, Y. Q., Li, J., Juan, Y. F., Lu, Z. J. & Jia, W. L. Evolution in microstructure and corrosion behavior of AlCoCrxFeNi high-entropy alloy coatings fabricated by laser cladding. J. Alloy. Compd. 775, 1–14 (2019).
Zhou, Q. et al. Corrosion behavior of Hf0.5Nb0.5Ta0.5Ti1.5Zr refractory high-entropy in aqueous chloride solutions. Electrochem. Commun. 98, 63–68 (2018).
Wei, L., Liu, Y., Li, Q. & Cheng, Y. F. Effect of roughness on general corrosion and pitting of (FeCoCrNi)0.89(WC)0.11 high-entropy alloy composite in 3.5wt.% NaCl solution. Corros. Sci. 146, 44–57 (2018).
Shi, Y. et al. Homogenization of Al x CoCrFeNi high-entropy alloys with improved corrosion resistance. Corros. Sci. 133, 120–131 (2018).
Cui, Z. et al. Microstructure and corrosion properties of FeCoNiCrMn high entropy alloy coatings prepared by high speed laser cladding and ultrasonic surface mechanical rolling treatment.Mater. Lett. 259, 126769 (2019).
Aliyu, A. & Srivastava, C. Microstructure-corrosion property correlation in electrodeposited AlCrFeCoNiCu high entropy alloys-graphene oxide composite coatings. Thin Solid Films 686, 137434 (2019).
Nene, S. S. et al. Corrosion-resistant high entropy alloy with high strength and ductility. Scr. Mater. 166, 168–172 (2019).
Torbati-Sarraf, H., Shabani, M., Jablonski, P. D., Pataky, G. J. & Poursaee, A. The influence of incorporation of Mn on the pitting corrosion performance of CrFeCoNi high entropy alloy at different temperatures. Mater. Des. 184, 108170 (2019).
Han, Z. et al. The corrosion behavior of ultra-fine grained CoNiFeCrMn high-entropy alloys. J. Alloy Compd. 816, 152583 (2019).
Xiang, C. et al. Microstructure and corrosion behavior of AlCoCrFeNiSi0.1 high-entropy alloy. Intermetallics 114, 106599 (2019).
Gharbi, O. & Birbilis, N. Clarifying the dissolution mechanisms and electrochemistry of Mg2Si as a function of solution pH. J. Electrochem. Soc. 165, C497–C501 (2018).
Liu, R., Scully, J., Williams, G. & Birbilis, N. Reducing the corrosion rate of magnesium via microalloying additions of group 14 and 15 elements. Electrochim. Acta 260, 184–195 (2018).
Acknowledgements
We thank the Monash Centre for Electron Microscopy (MCEM). Dr. Mark Gibson is also gratefully acknowledged for technical assistance with alloy production.
Author information
Authors and Affiliations
Contributions
Y.Q. designed the alloy studied, carried out electron microscopy and coordinated experiments. R.L. acquired and processed ICP-MS data, with technical support and oversight from O.G. T.G. was responsible for the acquisition, processing and evaluation of XPS data. O.G., S.C., S.T. and H.L.F. commented on the paper and discussed findings. N.B. supervised this study. N.B. and Y.Q. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qiu, Y., Liu, R., Gengenbach, T. et al. Real-time dissolution of a compositionally complex alloy using inline ICP and correlation with XPS. npj Mater Degrad 4, 7 (2020). https://doi.org/10.1038/s41529-020-0112-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-020-0112-3
This article is cited by
-
Recent research progress on the passivation and selective oxidation for the 3d-transition-metal and refractory multi-principal element alloys
npj Materials Degradation (2023)
-
High-Entropy Alloys: A Critical Review of Aqueous Corrosion Behavior and Mechanisms
High Entropy Alloys & Materials (2023)
-
Machine-learning-guided descriptor selection for predicting corrosion resistance in multi-principal element alloys
npj Materials Degradation (2022)
-
pyMPEALab Toolkit for Accelerating Phase Design in Multi-principal Element Alloys
Metals and Materials International (2022)
-
A Review on Corrosion Properties of High Entropy Alloys Fabricated by Additive Manufacturing
Transactions of the Indian Institute of Metals (2022)
