
Abstract
Adult-type granulosa cell tumor (aGCT) is a rare malignant ovarian sex cord-stromal tumor, harboring recurrent FOXL2 c.C402G/p.C134W hotspot mutations in 97% of cases. These tumors are considered to have a favorable prognosis, however aGCTs have a tendency for local spread and late recurrences, which are associated with poor survival rates. We sought to determine the genetic alterations associated with aGCT disease progression. We subjected primary non-recurrent aGCTs (n = 7), primary aGCTs that subsequently recurred (n = 9) and their matched recurrences (n = 9), and aGCT recurrences without matched primary tumors (n = 10) to targeted massively parallel sequencing of ≥410 cancer-related genes. In addition, three primary non-recurrent aGCTs and nine aGCT recurrences were subjected to FOXL2 and TERT promoter Sanger sequencing analysis. All aGCTs harbored the FOXL2 C134W hotspot mutation. TERT promoter mutations were found to be significantly more frequent in recurrent (18/28, 64%) than primary aGCTs (5/19, 26%, p = 0.017). In addition, mutations affecting TP53, MED12, and TET2 were restricted to aGCT recurrences. Pathway annotation of altered genes demonstrated that aGCT recurrences displayed an enrichment for genetic alterations affecting cell cycle pathway-related genes. Analysis of paired primary and recurrent aGCTs revealed that TERT promoter mutations were either present in both primary tumors and matched recurrences or were restricted to the recurrence and absent in the respective primary aGCT. Clonal composition analysis of these paired samples further revealed that aGCTs display intra-tumor genetic heterogeneity and harbor multiple clones at diagnosis and relapse. We observed that in a subset of cases, recurrences acquired additional genetic alterations not present in primary aGCTs, including TERT, MED12, and TP53 mutations and CDKN2A/B homozygous deletions. Albeit harboring relatively simple genomes, our data provide evidence to suggest that aGCTs are genetically heterogeneous tumors and that TERT promoter mutations and/or genetic alterations affecting other cell cycle-related genes may be associated with disease progression and recurrences.
Similar content being viewed by others
Introduction
Adult-type granulosa cell tumors (aGCTs) of the ovary are a rare form of ovarian cancer (<5%) characterized by rather simple genomes and by the presence of recurrent FOXL2 p.C134W somatic missense mutations in ≥97% of cases [1,2,3]. Despite their indolent growth and overall good prognosis, recurrences occur in 10–30% of aGCTs [4,5,6,7]. These rare tumors exhibit long latency periods, with a median time to first recurrence of 4–7 years [4,5,6], with late recurrences reported up to 20–30 years following initial diagnosis [8]. Hence, the natural history of aGCTs poses therapeutic challenges, requiring long-term follow-up [5, 9, 10].
Somatic TERT promoter mutations (C228T and C250T), associated with telomerase activation, have been reported at high frequency in cancers (12% overall) [11], including gynecologic malignancies such as ovarian clear cell carcinomas (16%) [12, 13]. In addition, recent studies have reported a significantly higher frequency of the TERT C228T promoter hotspot mutations in recurrent (41–67%) than in primary aGCTs (22–29%) [14, 15]. Patients with primary aGCTs harboring TERT promoter hotspot mutations were also found to have a significantly worse overall survival than those with wild-type TERT [14]. Furthermore, KMT2D inactivating mutations have been reported to be associated with recurrences in aGCTs [16].
Although TERT promoter and KMT2D mutations appear to provide the basis for recurrences in a subset of aGCTs, the genetic basis of the clinical behavior in a substantial proportion of recurrent aGCTs has not been defined to date. Here, we sought to compare the repertoire of somatic genetic alterations of (1) primary aGCTs that did not recur within at least four years of follow-up, (2) primary aGCTs that recurred and (3) aGCT recurrences. Furthermore, given that samples from primary aGCTs and their respective relapses were available, we have also compared the TERT mutation status between paired primary and recurrent aGCTs, and investigated whether aGCTs would display intra-tumor genetic heterogeneity and if specific genetic alterations would be selected during progression from primary tumor to recurrence.
Materials and methods
Subjects and samples
Following approval by the Institutional Review Board (IRB) of the authors’ institutions, we retrieved representative hematoxylin and eosin and unstained tissue sections from formalin-fixed paraffin embedded aGCTs from Memorial Sloan Kettering Cancer Center (MSKCC, NY, USA), Fudan University Cancer Center (Shanghai, China), Hospital Universitario de Bellvitge (Barcelona, Spain), Hospital Universitario Arnau de Vilanova (Lleida, Spain), and Cleveland Clinic (OH, USA). Patient consents were obtained according to the protocols approved by the local IRBs of the authors’ institutions. Samples were anonymized prior to analysis. Samples from 40 cases were reviewed by eight pathologists (RB, SES, MV, CGP, XM-G, BPR, JSR-F, and DFD) following the criteria put forward by the World Health Organization [17]. Only cases where a consensus diagnosis of aGCT was achieved were included in this study (n = 38), and two cases were excluded. Patients were defined as having primary non-recurrent aGCTs if no recurrence was detected within at least 48 months of follow-up, based on the median and range of time-to-recurrence of aGCTs previously reported [4,5,6]. Our final series included 47 samples from 38 patients: (1) primary non-recurrent aGCTs (n = 10), (2) primary aGCTs that subsequently recurred (n = 9) and their matched recurrences (n = 9 from nine patients), and aGCT recurrences without matched primary tumors (n = 19; Table 1 and Supplementary Table S1). Surgical staging was performed according to the 2014 International Federation of Gynecology and Obstetrics system [18].
Targeted capture massively parallel sequencing using the Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) [11, 19] was performed on tumor-normal pairs from 26 patients, including seven primary non-recurrent aGCTs, nine primary recurrent aGCTs and their matched recurrences, and ten aGCT recurrences. The remaining three primary non-recurrent aGCTs and nine aGCT recurrences from 12 additional patients did not yield sufficient DNA for MSK-IMPACT sequencing, and, therefore, were subjected to Sanger sequencing analyses to assess the presence of FOXL2 and TERT promoter hotspot mutations (see below; Supplementary Table S1).
Microdissection and DNA extraction
Representative sections of tumor tissue samples were microdissected under a stereomicroscope (Olympus SZ61) to ensure a tumor cell content >80%, as previously described [20, 21]. DNA from tumor and matched normal tissues was extracted using the DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturers’ instructions. DNA of sufficient quantity/ quality was obtained for Sanger sequencing for all 38 cases (47 samples) and for targeted massively parallel sequencing for 26 cases (35 samples; see below).
Assessment of FOXL2 and TERT promoter hotspot mutations by Sanger sequencing
PCR amplification of FOXL2 and TERT promoter hotspot loci was performed using the AmpliTaq Gold 360 Master Mix kit (Life Technologies, ThermoFisher Scientific) using previously described primers [22, 23]. PCR fragments were cleaned using ExoSAP It (ThermoFisher Scientific) and Sanger sequenced as previously described [22].
Targeted massively parallel sequencing
Microdissected tumor and matched normal DNA samples from primary non-recurrent aGCTs (n = 7), primary recurrent aGCTs (n = 9), and matched recurrences (n = 9), and aGCT recurrences (n = 10) were subjected to MSK-IMPACT sequencing of 410–468 cancer-related genes, as previously described [19, 24]. Sequencing data were processed and analyzed as previously described [21, 24]. In brief, reads were aligned to the reference human genome GRCh37 using the Burrows–Wheeler Aligner (v0.7.15) [25]. Local realignment, duplicate removal, and base quality recalibration were performed using the Genome Analysis Toolkit (v3.7) [26]. Somatic single-nucleotide variants (SNVs) were detected by MuTect (v1.0) [27], and small insertions and deletions (indels) were detected using a combination of Strelka (v2.0.15) [28], VarScan2 (v2.3.7) [29], Lancet (v1.0.0) [30], Scalpel (v0.5.3) [31], and Platypus [32]. Pathogenic mutations were defined as variants that were deleterious and/or mutational hotspots. In addition, mutations that were identified in the primary or recurrent tumor from a given patient were subsequently interrogated in the matched respective primary or recurrent sample using mpileup from SAMtools mpileup (version 1.2 htslib 1.2.1) [33]. Allele-specific copy number alterations (CNAs) and loss of heterozygosity (LOH) were defined using FACETS [34], as previously described [20, 21]. The fraction of the genome altered was computed from the CNAs obtained from FACETS. The cancer cell fraction of each mutation was determined using ABSOLUTE (v1.0.6) [35], as previously described [20, 24]. A combination of mutation function predictors was employed to define the potential functional impact of each missense SNV, as previously described [20, 21, 36]. Mutational hotspots were assigned according to Chang et al. [37]. The median depth of coverage of tumor and normal samples was 518x (range 120x–1223x) and 366x (range 117x−510x), respectively (Supplementary Table S2).
Pathway analyses
A MSigDB and DAVID pathway analysis was performed based on genes affected by nonsynonymous pathogenic somatic mutations, amplifications, or homozygous deletions in primary non-recurrent aGCTs (n = 7), primary aGCTs (n = 9) and matched recurrences (n = 9) and aGCT recurrences (n = 10) [38]. Pathways found to be significantly enriched (p < 0.01) were selected as previously reported [39]. In addition, a mutual exclusivity analysis was performed using combinations of mutually exclusive alterations (CoMET) with the use of a pair-wise Fisher’s exact test to detect the presence of significant pairs of genes [40].
Mutation-based tree construction
The mutation-based trees of the primary and matched aGCT recurrences were constructed using Treeomics [41] based on all synonymous and nonsynonymous mutations identified, as previously described [24]. For these analyses, a given mutation was considered “shared” if it was present in both the primary and matched aGCT recurrences. We defined mutations present only in the primary tumor or only in the recurrence as “private to the primary aGCT” and “private to the aGCT recurrence”, respectively.
Statistical analysis
The frequencies of somatic mutations affecting cancer genes in primary and recurrent aGCTs were compared using two-tailed Fisher’s exact test. Mutual exclusivity was tested using two-tailed Fisher’s exact test. The fraction of genome affected by CNAs in primary and recurrent aGCTs was evaluated using a Mann-Whitney U test. All p values were two-tailed, and 95% confidence intervals were adopted for all analyses.
Results
Clinico-pathologic features of primary and recurrent aGCTs
Our series encompassed aGCTs from 38 patients, including 10 patients with non-recurrent aGCTs (primary non-recurrent aGCTs) and 28 patients with recurrent disease. Of the 28 patients with recurrent disease, we analyzed samples from the primary tumor (primary recurrent aGCT) and matched recurrences from nine patients, and from the recurrent tumors only from 19 patients (aGCT recurrences; Supplementary Table S1).
The median age of patients at aGCT diagnosis was 52.5 years (range 34–87 years) in primary non-recurrent aGCTs (n = 10), 62 years (range 41–83 years) for patients with primary aGCT that developed recurrences (n = 9) and 56 years (range 34–89 years) for patients with aGCT recurrences without available matched primary tumors (n = 19). No significant differences in age at diagnosis were observed between primary non-recurrent aGCTs and primary aGCTs that recurred (p = 0.74, Student’s t test, Table 1, Supplementary Table S1). All patients (10/10, 100%) with primary non-recurrent aGCTs had early stage disease (IA and IC) at the time of diagnosis, whereas 1 (11%) and two patients (22%) with primary aGCTs that recurred had stage II and stage III disease at the time of diagnosis, respectively. No significant differences in stage were observed between primary non-recurrent aGCTs and primary aGCTs that recurred (p = 0.087, Fisher’s exact test), or other clinic-pathologic factors assessed (Table 1, Supplementary Table S1).
aGCT recurrences display distinct genomic profiles from primary aGCTs
Consistent with previous reports [2, 14, 15], all aGCTs analyzed in this study harbored FOXL2 p.C134W missense mutations as defined by MSK-IMPACT and/or Sanger sequencing (Fig. 1a). In addition, we identified recurrent TERT promoter mutations by Sanger and/or MSK-IMPACT sequencing, affecting not only the previously described C228T hotspot locus but also the C250T locus (Fig. 1a). In these 38 aGCTs, a significantly higher frequency of TERT promoter mutations was observed in aGCT recurrences (18/28, 64%) than in primary non-recurrent and primary recurrent aGCTs (5/19, 26.3%, p = 0.017, Fisher’s exact test, Fig. 1a, Table 2). While there was a stepwise increase in the frequency of TERT promoter mutations from primary non-recurrent aGCTs (2/7; 29%) to primary recurrent aGCTs (3/ 9; 33%) and aGCT recurrences (10/19; 52%; Fig. 1b), no significant differences in the TERT hotspot mutation frequency between primary aGCTs with (n = 9) and without (n = 10) recurrences were found (33 vs. 20% p = 0.434, Fisher’s exact test; Fig. 1a, Table 2).

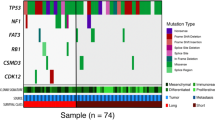
a FOXL2 and TERT promoter hotspot mutations in 38 adult-type granulosa cell tumors of the ovary (aGCTs) subjected to MSK-IMPACT and/or Sanger sequencing. Statistical significance was evaluated by Fisher’s exact test. b Nonsynonymous somatic mutations, amplifications, and homozygous deletions identified in primary adult-type granulosa cell tumors (aGCT) using MSK-IMPACT sequencing, including those without (non-recurrent, n = 7, left) and with (recurrent, n = 9, middle) subsequent recurrences, and in aGCT recurrences (n = 19, right). Cases are shown in columns and genes in rows. Genetic alterations are color-coded according to the legend. Indel small insertion and deletion, SNV single-nucleotide variant.
Analysis of the somatic mutation data obtained from MSK-IMPACT further revealed that aGCTs overall displayed a relatively low mutation burden, with a median of 3 (range 1–7) somatic mutations in the genes analyzed, of which two (range 1–6) were nonsynonymous (Supplementary Table S3). Despite the numerically higher number of somatic mutations identified in aGCT recurrences (median 3, range 2–7), no statistically significant differences in the mutational burden were observed when compared with primary non-recurrent aGCTs (median 2, range 1–5; p = 0.47, Fisher’s exact test) or primary recurrent aGCTs (median 2, 1–5; p = 0.86, Fisher’s exact test). Recurrent mutations affecting known cancer-related genes such as GNAQ and KMT2C were identified, however none of these was statistically different between the groups (Fig. 1b). Also, inactivating KMT2D mutations, which have been reported to be associated with recurrence in aGCTs [16], were only found in a single sample in our study and affected a primary non-recurrent aGCT (aGCT35-P; Fig. 1b, Supplementary Table S3). In contrast, we identified TP53 pathogenic mutations only in aGCT recurrences (16%). Of note, the TP53-mutant aGCT recurrences did not harbor TERT promoter mutations (Fig. 1b); however formal mutual exclusivity analysis using CoMET showed that TERT promoter and TP53 mutations were not significantly mutually exclusive in aGCT recurrences (p = 0.062, Fisher’s exact test, Fig. 2a), likely due to the low number of samples/TP53 mutations. We further found a subclonal pathogenic mutation affecting TET2 (5%) that was restricted to an aGCT recurrence lacking a TERT promoter mutation (aGCT82-R; Fig. 1b, Supplementary Fig. S1), and STAG2 and IDH1 pathogenic mutations in primary recurrent aGCTs (11%) and their matched aGCT recurrences (5%) but not in primary non-recurrent aGCTs (0%) (Fig. 1b). None of these differences reached statistical significance, however, likely due to the small sample size.

a Mutual exclusivity analysis between TERT promoter hotspot mutations and TP53 mutations in adult-type granulosa cell tumor (aGCT) recurrences. The type of mutations is color-coded according to the legend. Mutual exclusivity analysis was performed using combinations of mutually exclusive alterations (CoMET) and Fisher’s exact test. b Fraction of the genome altered in primary non-recurrent aGCTs, primary recurrent aGCTs, and aGCT recurrences. c Frequency of loss-of-function somatic genetic alterations affecting genes in the canonical cell cycle pathway. Genes are depicted in blue rectangles, and the percentage of primary non-recurrent aGCTs (Prim), primary recurrent aGCTs (Prim Rec), and aGCT recurrences (Rec) altered is shown below each gene.
When assessing the CNAs in the aGCTs subjected to MSK-IMPACT sequencing, we found primary aGCTs and aGCT recurrences to display overall similar copy number profiles with similar fractions of the genome altered (primary non-recurrent aGCTs, median 11%, range 5–48%; primary recurrent aGCTs, median 9%, range 0–91%; aGCT recurrences, median 8%, range 0–52%, Fig. 2b). Nevertheless, a numerically higher frequency of CDKN2A/B homozygous deletions was observed in aGCT recurrences (3/19, 16%) and in primary recurrent aGCTs (1/9, 11%) as compared with primary non-recurrent aGCTs (0%). Similarly, homozygous deletions of BCL2L11 were also identified in primary recurrent aGCTs (2/9, 22%) and in aGCT recurrences (2/19, 11%) but not in primary non-recurrent aGCTs (Fig. 1b).
Recurrent aGCTs harbor somatic genetic alterations affecting the cell cycle pathway
Given the distinct genetic alterations observed in primary non-recurrent aGCTs and aGCT recurrences, we sought to determine the signaling pathways that are enriched in aGCT recurrences. For this, we performed a pathway analysis using the genes that were either affected by nonsynonymous pathogenic somatic mutations, amplifications, or homozygous deletions. This analysis revealed that there was an enrichment in cell cycle pathway genes that were altered in aGCT recurrences but not in primary aGCTs (Supplementary Table S4). As mentioned above, pathogenic TP53 alterations were solely found in aGCT recurrences (Fig. 2c, top), whereas CDKN2A/B homozygous deletions were identified in both aGCT recurrences and primary recurrent aGCTs but not in primary non-recurrent aGCTs (Fig. 2c, bottom left). In contrast, CDKN1B homozygous deletions were identified at different frequencies in primary non-recurrent aGCTs (14%), primary aGCTs that recurred (11%) and in aGCT recurrences (5%; Fig. 2c, bottom right). These findings suggest that in addition to the described TERT promoter mutations [14, 15] alterations in cell cycle-related genes and apoptosis might also play a role in the progression of aGCTs.
TERT promoter mutations in primary aGCTs and matched recurrences
To investigate the role of TERT promoter mutations in the progression of aGCTs, we next assessed the TERT promoter mutation status in primary aGCTs and their matched recurrences using MSK-IMPACT and Sanger sequencing. Of the nine pairs of primary and recurrent aGCTs included in this study, five harbored TERT mutations in at least one of the samples of a given patient based on MSK-IMPACT sequencing (Fig. 3). We found that in two cases (aGCT77 and aGCT80), the primary lesion lacked TERT promoter mutations, but that the matched aGCT recurrences harbored a clonal C250T mutation (aGCT77) or a subclonal C228T mutation (aGCT80). This finding provides evidence to suggest that, in these cases, TERT mutations were either selected from a minor subclone not detected in the sequencing of the primary tumor or were acquired during disease progression. In contrast, the primary aGCT18 and aGCT43 both harbored clonal C228T TERT promoter hotspot mutations, which were preserved in the respective recurrences. Finally, a subclonal C250T was found in both the primary and matched recurrence of case aGCT76 (Fig. 3).

TERT promoter hotspot mutations and their clonality identified in primary aGCTs (left) and their matched recurrences (right) using targeted MSK-IMPACT sequencing. The TERT mutations were validated by Sanger sequencing and the electropherograms of all samples are shown. Cancer cell fractions are color-coded according to the legends and clonal mutations are depicted by a yellow box. Arrows in electropherograms indicate TERT promoter mutations.
Clonal composition analysis of paired primary and aGCT recurrences
To interrogate the genetic alterations in addition to TERT promoter mutations that might be associated with clinical progression of aGCTs, we performed a clonal composition analysis of the nine primary aGCTs and their matched recurrences. Our analyses revealed that both the primary aGCTs and their matched recurrences shared clonal mutations in FOXL2 (p.C134W) in all nine cases analyzed (Fig. 4, Supplementary Fig. S2). Furthermore, primary tumors and their matched recurrences also shared somatic mutations affecting KMT2C (p.A1685S), MYOD1 (p.S260F), KMT2D (p.C5481F), PIK3R1 (p.W624R), and the TERT promoter (Fig. 4, Supplementary Fig. S2). We observed, however, that in a subset of cases, the recurrences acquired additional somatic mutations or CNAs not present in the primary aGCT: we identified clonal MED12 (aGCT77, p.Q2076dup), clonal SH2D1A (aGCT78, p.P97S), and subclonal TET2 (aGCT82, p.C1281Vfs*82) mutations restricted to the recurrences. Furthermore, two aGCT recurrences acquired alterations in cell cycle-related genes such as TP53 mutations (aGCT78, p.F338Lfs*7; aGCT82, p.Y236H) or CDKN2A/B homozygous deletions (aGCT79), which were not detected in their respective primary tumors (Fig. 4).

Representative hematoxylin and eosin micrographs (magnification, 200×; left), cancer cell fractions (clonal frequency) of mutations (top right) and mutation-based trees (bottom right) depicting the clonal evolution of matched primary and recurrent adult-type granulosa cell tumors (aGCTs) of the ovary for (a) aGCT77, (b) aGCT80, (c) aGCT78, (d) aGCT82, and (e) aGCT79. Cancer cell fractions are color-coded according to the legend. Clonal mutations are depicted by a yellow box. The length of the trunk and branches of the phylogenetic trees is proportional to the number of shared and private mutations identified in primary and recurrent aGCTs. Scale bars, 200 µM. P primary tumor, R recurrence, T truncal.
Discussion
Despite their relatively indolent behavior, management of a subset of aGCTs remains challenging due to their unpredictable behavior and late relapses. Identification of markers predictive of disease recurrences/metastases has been the subject of considerable interest. Here we not only confirm the presence of TERT promoter hotspot mutations in aGCTs but also find that somatic genetic alterations affecting cell cycle progression and apoptosis-related genes may play a role in the progression from primary aGCTs to recurrences.
Recent studies have reported a highly recurrent somatic mutation (C228T) in the promoter region of TERT in aGCTs [14, 15]. In our study, in addition to the C228T mutation (80%), we also identified recurrent C250T TERT hotspot mutations in 20% of the aGCTs harboring TERT promoter mutations. Whilst TERT promoter mutations have been reported to be an early genetic event in several cancer types [42,43,44], in aGCTs one study has suggested that these mutations are a late event [14]. The overall frequency of TERT promoter hotspot mutations in our series was significantly higher in aGCT recurrences than in primary aGCTs. TERT promoter mutations have been reported to lead to increased TERT expression and telomerase activation, to overcome the proliferative barrier imposed by telomere shortening, and to promote both immortalization and tumorigenesis [45]. Of note, the analysis of paired primary and recurrent aGCTs revealed that TERT promoter mutations were likely acquired during disease progression in two cases (aGCT77 and aGCT80), whereas in three cases (aGCT18, aGCT43 and aGCT76) these mutations were present in both primary tumors and matched recurrences. These findings suggest that depending on the context and activation of other signaling pathways, TERT promoter mutations may either be an early event and play a role in the development of more aggressive primary disease, that has the potential to spread and recur; or, in other contexts, TERT promoter mutations may be a late event and acquired during progression, disease spread or recurrence [14, 15]. Further studies are required to understand the interplay between TERT promoter mutations and other genetic or epigenetic alterations, and TERT’s activation and role in aGCT maintenance and progression. It is unlikely, however, that TERT promoter mutation status alone would be the sole genetic predictor of recurrence. In our series, only one third of primary aGCTs with recurrences harbored a TERT hotspot mutation, as did two primary aGCTs (20%) without subsequent recurrences after more than 8 years of follow-up.
In our series, TP53 mutations were identified in 16% of aGCT recurrences but were not present in primary aGCTs. Importantly, we observed that these TP53 mutations were acquired during disease progression in cases that did not harbor TERT promoter mutations. Alexiadis et al. also reported a TP53 mutation in a recurrent aGCT that also lacked TERT promoter mutations but they had a lower frequency of TP53 mutations overall in their series (4%, 1/22) [15]. Our findings suggest that TP53 may play a role in the progression of aGCTs, in particular in those lacking TERT promoter mutations.
Apart from TP53, we observed an enrichment of alterations in cell cycle genes in aGCT recurrences, including CDKN2A/B homozygous deletions. Given the small numbers assessed, further studies are warranted to define the frequency and role of cell cycle-related genes in the progression of aGCTs. Whilst homozygous deletions or loss-of-function mutations of CDKN2A/B are frequent events in various human primary solid and hematopoietic neoplasias [46, 47], these have not been previously reported in aGCTs. Gene expression analyses comparing wild-type FOXL2 and mutant FOXL2 C402G (p.C134W) transfected COV434 aGCT cells in vitro revealed the presence of differentially expressed genes associated with cell death and cell proliferation in mutant FOXL2 cells, such as CDKN1A, CDKN2A, and CDK6 [48]. Inactivating mutations in the chromatin remodeling gene KMT2D have been recently reported to be strongly associated with aGCT recurrences [16]. The frequency of KMT2D loss-of-function mutations was very low in the aGCT analyzed here (one truncating mutation identified in one primary non-recurrent aGCT35). Furthermore, even when taking all nonsynonymous KMT2D mutations into account, no difference in frequency between primary aGCTs with/without recurrences and aGCT recurrences could be identified.
Clonal composition analysis revealed that primary aGCTs and their matched recurrences, despite their generally simple genomes with few mutations and CNAs, display intra-lesion heterogeneity harboring clonal and subclonal mutations. In the progression from primary to metastatic disease, the acquisition of additional mutations, LOH of the wild-type allele of a given mutated gene as well as clonal shifts of genes affected by somatic mutations have been described [49,50,51]. Through the analysis of paired primary and matched recurrent aGCTs, we found that the acquisition of mutations, including those affecting TP53, TET2, MED12, and SH2D1A, was most commonly associated with disease progression.
Our study has important limitations. The sample size of the study is small, given the rarity of aGCTs, and larger validation studies are warranted. In addition, given the multi-institutional nature of our study, survival analyses could not be performed. Furthermore, our findings may not be applicable to the general population, given that the majority of the patients included in our cohort were treated at tertiary referral centers that tend to see higher risk populations. The recurrence rates observed here were similar to those reported in the literature, however [5, 7]. Given the limited amounts of DNA available from these lesions, we restricted our sequencing analysis to 410–468 cancer-related genes. We cannot rule out, however, that there are other genes which may play a role in the progression of aGCTs. Nonetheless, our findings provide support to the notion that TERT promoter hotspot mutations are the most recurrent genetic events affecting aGCTs and might be associated with disease progression in a subset of cases. We further identified genetic alterations affecting cell cycle-related genes, which may be associated with aGCT progression. Finally, our data suggest that whilst aGCTs harbor simple genomes, intra-tumor heterogeneity is present in this rare subtype of pure sex cord tumor.
References
Wang YK, Bashashati A, Anglesio MS, Cochrane DR, Grewal DS, Ha G, et al. Genomic consequences of aberrant DNA repair mechanisms stratify ovarian cancer histotypes. Nat Genet. 2017;49:856–65.
Shah SP, Kobel M, Senz J, Morin RD, Clarke BA, Wiegand KC, et al. Mutation of FOXL2 in granulosa-cell tumors of the ovary. N Engl J Med. 2009;360:2719–29.
Jamieson S, Butzow R, Andersson N, Alexiadis M, Unkila-Kallio L, Heikinheimo M, et al. The FOXL2 C134W mutation is characteristic of adult granulosa cell tumors of the ovary. Mod Pathol. 2010;23:1477–85.
Lee IH, Choi CH, Hong DG, Song JY, Kim YJ, Kim KT, et al. Clinicopathologic characteristics of granulosa cell tumors of the ovary: a multicenter retrospective study. J Gynecol Oncol. 2011;22:188–95.
Sun HD, Lin H, Jao MS, Wang KL, Liou WS, Hung YC, et al. A long-term follow-up study of 176 cases with adult-type ovarian granulosa cell tumors. Gynecol Oncol. 2012;124:244–9.
Bryk S, Farkkila A, Butzow R, Leminen A, Heikinheimo M, Anttonen M, et al. Clinical characteristics and survival of patients with an adult-type ovarian granulosa cell tumor: a 56-year single-center experience. Int J Gynecol Cancer. 2015;25:33–41.
Lee YK, Park NH, Kim JW, Song YS, Kang SB, Lee HP. Characteristics of recurrence in adult-type granulosa cell tumor. Int J Gynecol Cancer. 2008;18:642–7.
Hines JF, Khalifa MA, Moore JL, Fine KP, Lage JM, Barnes WA. Recurrent granulosa cell tumor of the ovary 37 years after initial diagnosis: a case report and review of the literature. Gynecol Oncol. 1996;60:484–8.
Jamieson S, Fuller PJ. Molecular pathogenesis of granulosa cell tumors of the ovary. Endocr Rev. 2012;33:109–44.
Mangili G, Ottolina J, Gadducci A, Giorda G, Breda E, Savarese A, et al. Long-term follow-up is crucial after treatment for granulosa cell tumours of the ovary. Br J Cancer. 2013;109:29–34.
Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–13.
Wu RC, Ayhan A, Maeda D, Kim KR, Clarke BA, Shaw P, et al. Frequent somatic mutations of the telomerase reverse transcriptase promoter in ovarian clear cell carcinoma but not in other major types of gynaecological malignancy. J Pathol. 2014;232:473–81.
Nishikimi K, Nakagawa K, Tate S, Matsuoka A, Iwamoto M, Kiyokawa T, et al. Uncommon human telomerase reverse transcriptase promoter mutations are associated with poor survival in ovarian clear cell carcinoma. Am J Clin Pathol. 2018;149:352–61.
Pilsworth JA, Cochrane DR, Xia Z, Aubert G, Farkkila AEM, Horlings HM, et al. TERT promoter mutation in adult granulosa cell tumor of the ovary. Mod Pathol. 2018;31:1107–15.
Alexiadis M, Rowley SM, Chu S, Leung DTH, Stewart CJR, Amarasinghe KC, et al. Mutational landscape of ovarian adult granulosa cell tumors from whole exome and targeted TERT promoter sequencing. Mol Cancer Res. 2019;17:177–85.
Hillman RT, Celestino J, Terranova C, Beird HC, Gumbs C, Little L, et al. KMT2D/MLL2 inactivation is associated with recurrence in adult-type granulosa cell tumors of the ovary. Nat Commun. 2018;9:2496.
Zaloudek CJ, Mooney EE, Staats PN, Young RH. Sex cord-stromal tumours—pure sex cord tumours, In: Kurman RJ, Carcangiu ML, Herrington CS, Young RH, editors. WHO classification of tumours of female reproductive organs. 4th edn. vol. 6. Lyon: IARC; 2014. p. 50–3.
Prat J. Oncology FCoG. FIGO’s staging classification for cancer of the ovary, fallopian tube, and peritoneum: abridged republication. J Gynecol Oncol. 2015;26:87–9.
Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–64.
Pareja F, Lee JY, Brown DN, Piscuoglio S, Gularte-Merida R, Selenica P, et al. The genomic landscape of mucinous breast cancer. J Natl Cancer Inst. 2019. https://doi.org/10.1093/jnci/djy216. [Epub ahead of print].
Geyer FC, Li A, Papanastasiou AD, Smith A, Selenica P, Burke KA, et al. Recurrent hotspot mutations in HRAS Q61 and PI3K-AKT pathway genes as drivers of breast adenomyoepitheliomas. Nat Commun. 2018;9:1816.
Piscuoglio S, Ng CK, Murray M, Burke KA, Edelweiss M, Geyer FC, et al. Massively parallel sequencing of phyllodes tumours of the breast reveals actionable mutations, and TERT promoter hotspot mutations and TERT gene amplification as likely drivers of progression. J Pathol. 2016;238:508–18.
Conlon N, Schultheis AM, Piscuoglio S, Silva A, Guerra E, Tornos C, et al. A survey of DICER1 hotspot mutations in ovarian and testicular sex cord-stromal tumors. Mod Pathol. 2015;28:1603–12.
Weigelt B, Bi R, Kumar R, Blecua P, Mandelker DL, Geyer FC, et al. The landscape of somatic genetic alterations in breast cancers from ATM germline mutation carriers. J Natl Cancer Inst. 2018;110:1030–4.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8.
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–9.
Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–7.
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76.
Narzisi G, Corvelo A, Arora K, Bergmann EA, Shah M, Musunuri R, et al. Genome-wide somatic variant calling using localized colored de Bruijn graphs. Commun Biol. 2018;1:20.
Narzisi G, O’Rawe JA, Iossifov I, Fang H, Lee YH, Wang Z, et al. Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nat Methods. 2014;11:1033–6.
Rimmer A, Phan H, Mathieson I, Iqbal Z, Twigg SRF, Consortium WGS, et al. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet. 2014;46:912–8.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131.
Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–21.
Martelotto LG, Ng CK, De Filippo MR, Zhang Y, Piscuoglio S, Lim RS, et al. Benchmarking mutation effect prediction algorithms using functionally validated cancer-related missense mutations. Genome Biol. 2014;15:484.
Chang MT, Bhattarai TS, Schram AM, Bielski CM, Donoghue MTA, Jonsson P, et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discov. 2018;8:174–83.
Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic signaling pathways in the cancer genome atlas. Cell. 2018;173:321–37. e10.
Leiserson MD, Wu HT, Vandin F, Raphael BJ. CoMEt: a statistical approach to identify combinations of mutually exclusive alterations in cancer. Genome Biol. 2015;16:160.
Reiter JG, Makohon-Moore AP, Gerold JM, Bozic I, Chatterjee K, Iacobuzio-Donahue CA, et al. Reconstructing metastatic seeding patterns of human cancers. Nat Commun. 2017;8:14114.
Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, et al. The genetic evolution of melanoma from precursor lesions. N Engl J Med. 2015;373:1926–36.
Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac-Sage P, Laurent C, et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun. 2013;4:2218.
Cheng L, Davidson DD, Wang M, Lopez-Beltran A, Montironi R, Wang L, et al. Telomerase reverse transcriptase (TERT) promoter mutation analysis of benign, malignant and reactive urothelial lesions reveals a subpopulation of inverted papilloma with immortalizing genetic change. Histopathology. 2016;69:107–13.
Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM, Hockemeyer D. Cancer-associated TERT promoter mutations abrogate telomerase silencing. Elife. 2015;4. https://doi.org/10.7554/eLife.07918.
Pinyol M, Cobo F, Bea S, Jares P, Nayach I, Fernandez PL, et al. p16(INK4a) gene inactivation by deletions, mutations, and hypermethylation is associated with transformed and aggressive variants of non-Hodgkin’s lymphomas. Blood. 1998;91:2977–84.
Roussel MF. The INK4 family of cell cycle inhibitors in cancer. Oncogene. 1999;18:5311–7.
Rosario R, Araki H, Print CG, Shelling AN. The transcriptional targets of mutant FOXL2 in granulosa cell tumours. PLoS ONE. 2012;7:e46270.
Murali R, Selenica P, Brown DN, Cheetham RK, Chandramohan R, Claros NL, et al. Somatic genetic alterations in synchronous and metachronous low-grade serous tumours and high-grade carcinomas of the adnexa. Histopathology. 2019;74:638–50.
Ng CKY, Bidard FC, Piscuoglio S, Geyer FC, Lim RS, de Bruijn I, et al. Genetic heterogeneity in therapy-naive synchronous primary breast cancers and their metastases. Clin Cancer Res. 2017;23:4402–15.
Schrijver W, Selenica P, Lee JY, Ng CKY, Burke KA, Piscuoglio S, et al. Mutation profiling of key cancer genes in primary breast cancers and their distant metastases. Cancer Res. 2018;78:3112–21.
Acknowledgements
Some tissue samples were obtained with the support of IRBLLEIDA Biobank (PT 17/0015/0027) and IDIBELL Biobank (PT 17/0015/0024), each of them members of the ISCIII Platform of Spanish Biobanks.
Funding
Research reported in this publication was funded in part by a Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute (grant no. P30CA008748). JSR-F is partially funded by a Breast Cancer Research Foundation grant. BW is partially funded by Breast Cancer Research Foundation, Cycle for Survival, and Stand Up To Cancer grants.
Author information
Authors and Affiliations
Contributions
SES, BW, and DFD conceived the study. RB, AV, SG, XM-G, BPR, and DFD provided tissue samples. RB, SES, MV, CGP, XM-G, BPR, JSR-F, and DFD conducted pathology review. RB, ADCP, and EMdS performed sample processing. ADCP, LF, and PS performed bioinformatics analyses. Data acquisition, interpretation, and analysis was performed by RB, ADCP, EMdS, FP, PS, LF, RAS, NRA-R, JSR-F, DFD, and BW. ADCP, EMdS, FP, SHK, JSR-F, and BW drafted the manuscript, and all authors edited and approved the final draft of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Co-principal investigators for this study: Deborah F. DeLair, Britta Weigelt
Supplementary information
Rights and permissions
About this article

Cite this article
Da Cruz Paula, A., da Silva, E.M., Segura, S.E. et al. Genomic profiling of primary and recurrent adult granulosa cell tumors of the ovary. Mod Pathol 33, 1606–1617 (2020). https://doi.org/10.1038/s41379-020-0514-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-0514-3
This article is cited by
-
Uterine PEComas: correlation between melanocytic marker expression and TSC alterations/TFE3 fusions
Modern Pathology (2022)
-
Mullerian adenosarcoma: clinicopathologic and molecular characterization highlighting recurrent BAP1 loss and distinctive features of high-grade tumors
Modern Pathology (2022)
-
ESR1 hotspot mutations in endometrial stromal sarcoma with high-grade transformation and endocrine treatment
Modern Pathology (2022)
-
T1 bladder carcinoma with variant histology: pathological features and clinical significance
Virchows Archiv (2022)
-
Genomic alterations in gynecological malignancies: histotype-associated driver mutations, molecular subtyping schemes, and tumorigenic mechanisms
Journal of Human Genetics (2021)
