
Abstract
The function of the fibrinolytic system was first identified to dissolve fibrin to maintain vascular patency. Connections between the fibrinolytic system and many other physiological and pathological processes have been well established. Dysregulation of the fibrinolytic system is closely associated with multiple pathological conditions, including thrombosis, inflammation, cancer progression, and neuropathies. Thus, molecules in the fibrinolytic system are potent therapeutic and diagnostic targets. This review summarizes the currently used agents targeting this system and the development of novel therapeutic strategies in experimental studies. Future directions for the development of modulators of the fibrinolytic system are also discussed.
Similar content being viewed by others


Introduction
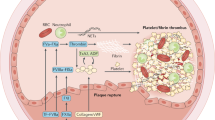
Fibrinolysis is the process of dissolving blood clots, thereby preventing the obstruction of blood vessels. Triggered by the activation of the fibrinolytic system, fibrinolysis is mainly regulated by proteases and protease inhibitors. In addition to the originally identified thrombolytic function, the fibrinolytic system has also been found to play pivotal roles in various physiological and pathological processes, e.g., tissue remodeling, immune responses, and cancer progression. The key enzyme of this system, plasmin, has two main physiological functions: (1) to degrade the deposited fibrin into soluble fibrin degradation products (FDP) in blood vessels and (2) to degrade base membranes (BM) or extracellular matrices (ECM) to facilitate tissue remodeling or cell migration (Fig. 1a). Plasmin is physiologically modulated by a specific inhibitor, α2-antiplasmin, and a nonspecific protease inactivator, α2-macroglobulin. Plasmin is mainly derived from inactive plasminogen by tissue- or urokinase-type plasminogen activators (tPA or uPA). In addition to tPA and uPA, plasminogen can also be activated by proteases in the contact activation system: plasma kallkrein (PK) or coagulation factor XIIa (fXIIa), albeit with lower activation efficacies than those of the plasminogen activators. The activities of plasminogen activators are modulated by plasminogen activator inhibitor-1 and −2 (PAI-1 and PAI-2). As PK serves as an alternative plasminogen activator, its physiological inhibitor, C1 esterase inhibitor, is also named plasminogen activator inhibitor-3 (PAI-3).

a Scheme of the fibrinolytic system. Plasmin (Plm) mainly performs two physiological functions: 1) to degrade fibrin (Fn) to maintain vascular fluidity and 2) to degrade the ECM and BM, facilitating tissue remodeling. Plasmin is physiologically regulated by α2-antiplasmin (α2-AP) or α2-macroglobulin (α2-MGB). Plasmin is activated via the cleavage of the inactive zymogen plasminogen (Plg) by plasminogen activators (tPA/uPA) or other enzymes, e.g., plasma kallikrein (PK) and coagulation factor XIIa (fXIIa). TPA and uPA are physiologically regulated by plasminogen activator inhibitor-1 and −2 (PAI-1 and PAI-2). FG and FDP indicate fibrinogen and fibrin degradation products, respectively. b Kallikrein-dependent activation of the pericellular fibrinolytic system. Cells normally attach to the ECM through the binding of the D2 domain of uPA receptor (uPAR) and vitronectin (Vn). The zymogen of uPA (pro-uPA) binds to the D1 domain of uPAR. Activated high molecular weight kininogen (HKa) competitively binds to the D2 domain of uPAR to replace the uPAR-Vn interaction. HKa then recruits PK to activate pro-uPA to generate active uPA, which triggers the fibrinolytic system and facilitates the migration of cells. The generated plasmin also activates pro-uPA to amplify the activity of the fibrinolytic system.
The Fibrinolytic System
Plasmin and plasminogen
In blood, plasmin normally exists as plasminogen, an inactive zymogen originally synthesized in the liver and circulates at a concentration of 200 µg/mL1. Plasminogen is a 7-domain protein comprising an N-terminus PAN/apple domain, five tandem kringle domains and a C-terminus catalytic domain. The PAN/apple domain is proteolytically removable, and the kringle domains are responsible for fibrin binding1,2. The PAN/apple domain-free plasmin can be further degraded by phosphoglycerate kinase to remove the catalytic domain. The remaining five kringle domains are collectively referred to as angiostatin, an endogenous angiogenesis inhibitor3. Plasminogen activation by tPA is normally considered to be responsible for thrombolysis in blood vessels. In contrast, uPA-triggered plasmin generation primarily occurs in tissues and is associated with tissue remodeling4. Plasmin is a versatile enzyme that has a broad spectrum of physiological substrates, including many blood proteins and extracellular proteins5. Moreover, plasmin is the activator of multiple functional proteins. For instance, plasmin activates the generation of over 7 matrix metalloproteinases (MMPs)6, which directly participate in the processes of tissue remodeling, cancer progression, pregnancy, etc7,8,9. Beyond its fibrinolytic property, plasmin also has anti-coagulative effects by degrading factors V and VIII, thus blocking coagulation cascades10.
UPA
UPA is a serine protease that consists of an N-terminus epidermal growth factor-like domain (GFD), a kringle domain, and a C-terminus catalytic domain. UPA is synthesized as an inactive single-chain (sc) zymogen. The activation of sc-uPA is proposed to require the binding of the membrane-bound uPA receptor (uPAR)11. PK, fXIIa, or plasmin sequentially approaches and activates sc-uPA11,12. In cancer progression, uPA is also reported to be activated by matriptase, a type II transmembrane serine protease13. Plasmin is the most efficient activator among these sc-uPA activators. Unlike plasmin and its myriad substrates, uPA is specific to plasminogen. However, uPA also exhibits several physiological functions independent of uPAR or plasminogen. For instance, uPAR deficiency did not affect uPA-related arterial neointima formation, neointimal cell accumulation, or smooth muscle cell migration14. Additionally, uPA was also reported to regulate the intravascular adherence of leukocytes independently of its proteolytic activity15.
TPA
TPA, the main plasminogen activator in blood, is a serine protease containing an N-terminus fibronectin type II domain, a growth factor-like domain, two kringle domains, and a C-terminus catalytic domain16. Similar to uPA, tPA has no other confirmed physiological substrate except plasminogen. However, unlike uPA, tPA binds fibrin through its fibronectin type II domain and the two kringle domains. Thus, tPA specifically binds to fibrin-bound plasminogen17. TPA is synthesized and stored in endothelial cells as a single-chain zymogen (sc-tPA). Endothelial cells release tPA into the blood upon stimulation by thrombin, histamine, bradykinin, or other molecules. Sc-tPA is then transformed into its two-chain form (tc-tPA) by proteolytic cleavage18. Unlike inactive sc-uPA, sc-tPA, and tc-tPA have comparable proteolytic activity19. TPA is also expressed on many cells in the central and peripheral nervous systems20,21,22. Dysregulated tPA activity has been associated with multiple neurological diseases, including demyelination23, Alzheimer’s disease24, and seizures25.
PAI-1
PAI-1 is the main suppressor of the fibrinolytic system and regulates tPA and uPA. PAI-1 covalently inactivates plasminogen activators and facilitates their metabolic clearance26. PAI-1 is synthesized in endothelial cells and stored in platelets. Once activated, platelets release PAI-1 to suppress fibrinolysis and thus promote coagulation27. Inflammation provokes the generation of thrombin, which also triggers the release of PAI-1 from platelets28. PAI-1 circulates in two forms, the active form and the latent form29. Active PAI-1 contains a solvent-exposed reactive central loop (RCL), which covalently blocks the active sites of plasminogen activators after proteolytic cleavage. Latent PAI-1 has an RCL that is embedded and thus loses its inhibitory activity. Active PAI-1 is unstable in vitro and irreversibly converts into the latent form within hours30. PAI-1 also exhibits physiological or pathological functions independent of the inhibition of plasminogen activators. For instance, as the main inhibitor of uPA, PAI-1 is expected to perform anticancer and anti-metastatic effects. However, PAI-1 was reported to manifest dual effects on the invasion of cancer cells. In different cancer cell lines, PAI-1 exhibited either suppressive31,32,33 or enhancing effects on invasion. The pro-cancer effects of PAI-1 work via the upregulation of the expression of oncogenes through other signaling pathways34,35,36.
uPAR
uPAR is a highly glycosylated glycosylphosphatidyl-inositol-anchored receptor with three homologous domains (D1, D2, and D3)37. The three domains establish a pocket via an intramolecular disulfide bridge for the accommodation of the GFD domain of uPA38. uPAR also accommodates other ligands, including vitronectin or cleaved high molecular weight kininogen (HKa), both of which bind to the D2 and D3 domains39,40. Notably, uPAR has no transmembrane domain. The signal transduction caused by extracellular uPAR binding occurs via interactions between uPAR and other membrane proteins41, including integrins42, G-protein-coupled receptors (GPCR)43,44, and epidermal growth factor receptor (EGFR)45. In addition to the membrane-bound full-length form, uPAR also exists in a soluble form (suPAR) in biological fluids or as a cleaved form (cuPAR), which lacks the D1 domain46. After proteolytic cleavage by plasmin or uPA, cuPAR (D2-D3) loses its binding affinity for uPA or vitronectin47,48 and thus constitutes a negative feedback component of the fibrinolytic system. SuPAR is released from cells or plasma membranes by the cleavage of phospholipases and can exist as either intact suPAR (D1-D2-D3) or cleaved suPAR (C-suPAR, D2-D3)49. Intact suPAR is able to accept all extracellular proteins bound by membrane-anchored uPAR. Hence, suPAR competes with membrane-anchored uPAR in its physiological functions, such as the reduction of pericellular uPA activity and ECM binding50. However, the functions of uPA- and vitronectin-independent uPAR are unaffected by suPAR51.
Pathological roles of the fibrinolytic system
The fibrinolytic system was first identified as a counterpart of the coagulation system. However, unlike the widely observed thrombotic diseases caused by dysregulation of the coagulation system, abnormal activation of the fibrinolytic system is clinically rare. In contrast to intravascular fibrinolysis, accumulating evidence demonstrates that extravascular fibrinolysis plays critical roles in multiple pathological processes.
Cancer
The association between uPA-triggered fibrinolysis and cancer progression has been known for over 50 years52. Similar to MMPs, plasmin was originally considered to only facilitate cancer metastasis by degrading the ECM11. However, as its pro-cancer mechanism was revealed, scientists found that the fibrinolytic system has impacts on multiple processes involved in cancer progression. Furthermore, crosstalk with other systems has also been identified. Recent studies demonstrated that PK and HKa of the contact activation system are involved in fibrinolytic processes on the surfaces of cancer cells or immune cells (Fig. 1b). Normally, cells connect with the ECM partially through the binding of the D2 domain of uPAR on cells to the somatomedin B (SMB) domain of vitronectin in the ECM. The fibrinolytic system facilitates cancer migration through a two-step process. First, HKa binds to the D2 domain of uPAR and outcompetes uPAR-vitronectin binding. The loss of uPAR-vitronectin binding facilitates the detachment of tumor cells from the ECM. Second, HKa recruits PK or fXIIa to activate pro-uPA, which binds to the D1 domain. Active uPA sequentially activates plasminogen to generate plasmin, which further activates pro-uPA and thereby amplifies plasmin generation. The known pro-cancer functions of plasmin are (1) to release cancer-related growth factors53, (2) to degrade pro-apoptotic factors54, and (3) to promote angiogenesis55 either by itself or by activating pro-cancer MMPs6. UPA-induced fibrinolysis exhibits pro-cancer effects synergistically with transforming growth factor-β (TGF-β) and MMPs56. TGF-β regulates the expression of pro-MMPs and pro-uPA. As part of a feedback loop, plasmin and active MMPs activate the precursor of TGF-β, which sequentially triggers the activation of multiple pro-cancer signaling pathways in the advanced stage of cancer57.
Inflammation
The fibrinolytic system exhibits critical functions involved in multiple aspects of inflammation progression. First, uPA and uPAR play pivotal roles in the extravasation of immune cells, including macrophages, leukocytes, B cells, and T cells. UPA and uPAR on the intravascular endothelial surface regulate the adherence of leukocytes to the endothelial walls15. Downstream plasmin and MMPs then disintegrate the BM and ECM proteins, facilitating the epithelial to mesenchymal transition (EMT) of immune cells58. In addition, after infection, bacteria invade and disseminate through recruitment of the host fibrinolytic system by surface-exposed plasminogen receptors to degrade the ECM or BM59. In addition, the fibrinolytic system has also been reported to modulate inflammation levels through the complement system. For instance, the administration of streptokinase (SK) or recombinant tPA (r-tPA) both caused activation of the complement pathway in patients with acute myocardial infarction (AMI)60. Similarly, both r-tPA and uPA administration caused a 2- to 3-fold increase in the plasma concentration of C3a61. In a septic mouse model, neutralizing uPAR suppressed the C5a signaling pathway in macrophages62. In glomerular mesangial cells, uPA upregulated the expression of +complement C5a receptor63. However, uPA-triggered plasmin generation was also reported to inactivate C5a in human fibrosarcoma cells64. In addition to its role in inflammation progression, plasminogen/plasmin was also reported to regulate some key steps involved in the resolution of inflammation, such as neutrophil apoptosis and macrophage reprogramming65.
Although no clinical data have shown that the inhibition of uPA leads to the amelioration of inflammation, numerous pieces of experimental evidence have suggested that uPA serves as a highly potent anti-inflammatory target. UPA-triggered fibrinolysis promoted the recruitment of inflammatory cells and induced organ fibrosis and dysfunction after myocardial infarction66. In rheumatoid arthritis (RA), fibrin deposition in synovial tissue and fluid correlates with the progression of arthritis67. In addition, the levels of D-dimer (a degradation product of fibrin generated by plasmin) in synovial tissue are suggested to predict the therapeutic outcomes of RA67,68. Plasmin facilitates the infiltration of immune cells into the synovial joints, which is impaired by knocking out plasminogen69. In addition, the suppression of the fibrinolytic system reduced cartilage degradation in SCID mice suffering from RA70. Interestingly, tPA and uPA were reported to play opposing roles in a collagen-induced arthritis mouse model; uPA−/− mice showed ameliorated symptoms, while tPA−/− mice showed aggravated symptoms71.
TPA is also upregulated in many inflammatory conditions72. The upregulation of tPA can be attributed to two reasons: (1) Inflammation is normally accompanied by the excessive generation of thrombin73, which promotes the release of tPA from storage granules in endothelial cells74. (2) Inflammatory cytokines, e.g., tumor necrosis factor (TNF) or interleukins (ILs), have also been reported to mediate the expression of tPA either positively or negatively75. For instance, TNF and IL-6 increased tPA expression in human peritoneal mesothelial cells and dental pulp cells, respectively76,77. In contrast, TNF and IL-1 decreased tPA levels in human umbilical vein endothelial cells (HUVEC)75,78. Recent evidence suggests that PAI-1 is tightly associated with inflammation, which appears irrelevant to its inhibitory effects on plasminogen activators. PAI-1 levels are locally enhanced at inflammatory sites79,80, which can have two causes. (1) PAI-1 stored in platelets is released upon platelet activation by excessive thrombin28. (2) Multiple inflammatory cytokines upregulate the expression of PAI-181,82,83. PAI-1 deficiency significantly downregulated the acute phase levels in endotoxin-induced septic models84,85. In a lipopolysaccharide (LPS)-induced septic mouse model, alcohol significantly enhanced pulmonary fibrin deposition and lung injury, which was apparently ameliorated by PAI-1 deficiency86.
The fibrinolytic system as therapeutic or diagnostic targets
One clinical use of plasmin inhibitor was to stop severe hemorrhage. Aprotinin, a proteinaceous plasmin inhibitor, was clinically used to prevent postsurgical or labor-induced hemorrhage87. However, aprotinin was withdrawn from the market in 2008 due to problems pertaining to its metabolism and specificity88. Although plasmin plays the most direct role in the dysregulation of the fibrinolytic system, plasmin itself is seldom considered as a therapeutic target. One likely reason is that plasmin, as a downstream factor, is involved in multiple critical biological processes, including thrombolysis and tissue remodeling. The off-target inhibition of plasmin might lead to systemic disorders. Evidence showed that plasminogen-deficient mice suffered from retarded growth, reduced fertility, and low survival89. In contrast, mice deficient in either tPA or uPA demonstrated slightly impaired thrombolytic properties but showed generally normal growth90. However, mice with both tPA and uPA deficiency demonstrated similar pathological symptoms as mice deficient in plasminogen91. Thus, the upstream factors, e.g., uPAR, uPA, tPA, and PAI-1, are more specific and safer targets and have several successful applications in clinical treatments.
Therapeutics targeting uPAR
UPAR has been certified to be pivotal in multiple pathological processes and is considered to be a diagnostic and therapeutic target. Due to the overexpression of uPAR on tumor cells, pericellular uPAR is a prognostic marker for multiple cancers. In addition, as elevated uPAR levels have been connected with multiple pathological processes, uPAR agonists have also been developed for the treatment of many other diseases.
UPAR antagonists as anticancer or anti-inflammatory agents
UPA binds to uPAR with high affinity (KD = 0.3 nM). Thus, peptides derived from the uPAR-binding regions of uPA have been studied as uPAR antagonists to interrupt uPA-uPAR interactions. Å6, an 8-mer peptide derived from uPA (residues 136–143), demonstrated excellent anticancer and anti-metastatic effects in experimental models and preclinical studies92,93,94. Å6 also entered phase I and II clinical trials for cancer treatment95,96,97. In addition, Å6 demonstrated promising therapeutic effects in other pathological experimental models. For instance, Å6 inhibited the degradation of vascular endothelial cadherin and elevated the alteration of the blood retinal barrier in diabetic rats98. In a laser-induced choroidal neovascularization mouse model, Å6 suppressed 95% of vascular formation99. In another study, Å6 inhibited retinal angiogenesis and neovascularization100. Moreover, Å6 suppressed LPS-induced inflammatory osteoclastogenesis and bone loss101. Similar to Å6, a peptide derived from residues 21–30 of uPA has also been re-engineered (21C and 29C mutants) as a cyclic peptide to inhibit uPAR. This peptide, WX-360, and its derivative, WX-360-NIe, suppressed tumor growth and metastasis in animal models102. Notably, the recombinant growth factor domain of uPA fused with the Fc domain of IgG suppressed tumor growth in a breast cancer–xenografted mouse model103 and a melanoma mouse model104.
Chiron Corporation screened a phage-display library to identify uPAR antagonists105, but no further studies were reported on the antitumor effects of these peptides. Small molecules that interrupted the interaction between uPAR and vitronectin inhibited the invasion of cancer cells in vitro106. The Meroueh group synthesized a series of pyrrolidinone and piperidinone derivatives that bind to the uPA-binding pocket of uPAR107. They also reported a series of small-molecule uPAR antagonists that orthosterically inhibited uPA-uPAR binding and simultaneously allosterically inhibited uPAR-vitronectin binding108. Eefting et al. reported a hybrid protein containing ATF (uPAR antagonist), TIMP-1 (MMP inhibitor), and BPTI (plasmin inhibitor) to inhibit ECM degradation in smooth muscle cells and preventvein graft diseases109. In addition, anti-uPAR antibodies that block the interactions of uPAR and its ligands exhibited the suppression of tumor growth and metastasis in animal models110,111.
UPAR as an antitumor target for diagnosis and therapy
Ploug et al. reported a high-affinity 10-mer linear uPAR-binding peptide, AE105 (KD = 0.4 nM), which was developed by combinatorial chemistry112. In later studies, AE105 was conjugated with 64Cu-labeled DOTA, a positron emission tomography (PET) agent, for in vivo tumor imaging through binding to uPAR113,114. This probe showed encouraging results in its Phase I clinical trial in Denmark for the imaging of breast, prostate, and bladder cancers115,116. UPAR-binding ligands have also been used to promote the accumulation of antitumor agents at tumor sites. Rajagopal et al. developed a uPAR-targeting toxin by fusing a truncated Pseudomonas exotoxin with ATF, the amino terminal fragment of uPA, and evaluated the antitumor effects in vitro and in vivo117. In another study, full-length pro-uPA was fused with saporin to enhance its specificity for tumor cell lines118. In addition, ATF-conjugated iron oxide nanoparticles were developed for magnetic resonance imaging (MRI) of cancers119,120.
Based on our understanding of the structural features of uPAR, we developed a series of uPAR-targeting anticancer agents (Fig. 2). First, we reported a method of uPAR-targeted photodynamic therapy (PDT) that involved the conjugation of a photosensitizer (zinc phthalocyanine, ZnPc) with ATF (Fig. 2a)121. Conjugation with ATF remarkably enhanced the antitumor specificity and efficacy of ZnPc in vitro and in vivo. In the following study, we developed a uPAR-targeting drug carrier, ATF-HSA, a fusion protein of ATF and human serum albumin (HSA)122. The fusion protein not only has the receptor binding capability of ATF but also has a long circulating time and drug carrier properties, which are derived from HSA123. We used ATF-HSA to deliver a ZnPc photosensitizer122 or doxorubicin124 to tumor sites in cancer-engrafted mouse models (Fig. 2b, c). ATF-HSA-loaded ZnPc was a specific tumor imaging agent and illuminated the tumor site in mice when excited at a wavelength of 630 nm. In addition to its excellent tumor-targeting capability, it also demonstrated strong antitumor effects in vitro and in vivo122. We also observed that ATF-HSA-loaded DOX demonstrated reduced cardiotoxicity accompanied by an enhanced antitumor effect, in contrast to free DOX124. In our recent work, we formulated ATF-HSA into a nanoparticle to encapsulate a larger amount of ZnPc photosensitizer (Fig. 2d)125. This ATF-HSA nanoparticle is able to disintegrate by contacting uPAR on the tumor surface and release ZnPc, leading to a potent photodynamic effect. Such high sensitivity to pericellular uPAR bestows the ATF-HSA nanoparticle with the property of uPAR-responsive drug release.

UPAR has been certified to be overexpressed on tumor cell surfaces. We developed a series of uPAR-targeting therapeutics. a ATF-ZnPc conjugate: a ZnPc-based photosensitizer was covalently conjugated with the uPAR-binding fragment of uPA (ATF). b, c Recombinant ATF-HSA, which integrates the uPAR-targeting property and the drug-loading property of HSA, delivered ZnPc (b) or doxorubicin (DOX, c) to tumor sites. d A uPAR-targeting drug carrier, developed by using ATF-HSA nanoparticles, encapsulated and released loaded ZnPc on the tumor surface.
Detection of suPAR levels in plasma
Elevated suPAR levels in body fluids have been observed in multiple pathological conditions. Hayek et al. reported that elevated plasma suPAR levels were associated with decreased glomerular filtration rates and renal dysfunction in patients with chronic kidney diseases126. In addition, increased plasma suPAR levels were also found in patients with lung, breast, ovary, and colon cancers127. In addition, the plasma suPAR level was suggested as a nonspecific marker of sepsis128, and the determination of both the suPAR level and the procalcitonin level was able to enhance the accuracy of sepsis diagnosis129. Moreover, high plasma suPAR levels were also observed in patients with HIV infection130 or diabetes131. Thus, suPAR is a potent indicator of health crises132.
SuPAR circulates in two forms: full-length suPAR (D1-D2-D3, also named active suPAR) and cleaved suPAR (C-suPAR, D2-D3). C-suPAR lacks the capability to bind most ligands except formyl peptide receptor-like 1 and 2 (FPRL 1 and 2)133. Thus, active suPAR, rather than C-suPAR, is more ideal for use as a pathological prognostic marker. Although ELISA kits have been commercially available for determining the plasma level of suPAR134, the detected epitope is not well defined. In addition, none of these ELISA methods can distinguish active suPAR from C-suPAR. To precisely determine the plasma level of active suPAR, we developed a new ELISA-based approach based on our recombinant ATF-HSA molecule (Fig. 3a)135. ATF-HSA specifically binds to the central pocket of uPAR, which accommodates uPA via its ATF fragment. The binding of uPAR to uPA requires the participation of all three D1-D2-D3 domains. Thus, C-suPAR, which lacks the D1 domain, cannot be captured by ATF-HSA. Using this approach, we found that the plasma levels of active suPAR in 20 pregnant women were significantly higher than those in healthy donors, which was consistent with the effects of the overactivation of the fibrinolytic system required for uterine remodeling136,137.

Mechanistic schemes of ELISA kits for the detection of active suPAR (a) and active PAI-1 (b).
UPA inhibitors as anticancer agents
In the fibrinolytic system, uPA was the first recognized target for cancer treatment. Studies of the inhibition of uPA can be dated back to the 1960s52. Upamostat, a prodrug of a small-molecule uPA inhibitor, was FDA-approved as an orphan drug for the treatment of pancreatic cancer in 2017. Upamostat was originally developed by WILEX AG and was licensed to RedHill Biopharma Ltd. and the Link Health Group in 2014. In its phase II clinical trial, Upamostat demonstrated only mild toxicity but enhanced the survival rates of volunteer patients138. Other uPA inhibitors have also been developed for antitumor uses. CJ-463 (benzylsulfonyl-D-Ser-Ser-4-amidinobenzylamide) is a 4-mer peptide-like uPA inhibitor (Ki = 20 nM)139. CJ-463 significantly suppressed cancer metastasis in a lung cancer mouse model140. A peptidic irreversible uPA inhibitor with an IC50 of 57 nM141 was used for the single-photon emission computed tomography of breast cancer142. Heinis et al. reported a series of bicyclic peptides by screening with phage display or affinity maturation143,144,145. Amiloride, an oral potassium-sparing diuretic, was found to inhibit the proteolytic function of uPA with an IC50 of 7 µM146. However, amiloride is not suitable for clinical anticancer treatments because of its low daily dose ceiling147. Buckley et al. reported an amiloride derivative with enhanced uPA affinity (Ki = 53 nM) and thus enhanced its antitumor effects. Meanwhile, this compound loses the diuretic effect of amiloride, suggesting that it can be used at a much higher maximum daily dose148. Professor Peter Andreasen of Aarhus University, in collaboration with us, reported a series of peptide-based inhibitors of human and murine uPA with high potency and specificity. The relevant studies have been thoroughly reviewed before149. After Professor Andreasen passed away in late 2016, we continued this project. In our recent work, we reported a highly potent and specific peptide inhibitor of murine uPA, IG-2 (KD = 6.7 nM). However, in contrast to its high potency in vitro, IG-2 demonstrated moderate anticancer and anti-metastatic effects in animal models150, which is likely largely due to the unfavorable pharmacokinetic profile of the peptides.
Recombinant tPA variants for the treatment of acute thrombosis
Thrombosis is caused by an imbalance in the coagulation system and the fibrinolytic system, leading to excessive fibrin deposition in blood vessels. In addition to suppressing the coagulation system, promoting the fibrinolytic system is an alternative therapeutic strategy for thrombotic diseases. Recombinant tPA (r-tPA) and purified uPA were both approved by the US FDA in 1987 and 1978, respectively, as clinical thrombolytic drugs151. R-tPA is more favorable than uPA as a thrombolytic agent because tPA specifically recognizes fibrin-bound plasminogen, which leads to the specific degradation of solid fibrin, while uPA does not have a plasminogen- or fibrin-binding domain and activates both circulating and fibrin-bound plasminogen, which can increase the risk of fibrinogen lysis in the blood17,152. R-tPA is used for the treatment of severe thrombotic diseases (e.g., acute ischemic stroke) under extremely strict administration criteria, as the administration of r-tPA confers a high risk of mass hemorrhage153. Several types of r-tPA have been commercially available, including Alteplase, Retaplase, or Tenecteplase, but only one uPA product is clinically used for deep venous thrombosis or pulmonary embolism154.
Clinically used r-tPAs require a total dose of up to 90 mg per patient to achieve sufficient thrombolytic effects. This dose is equivalent to an initial plasma concentration of ~320 nM (assuming a total blood volume of 4.5 l), which is significantly higher than the endogenous tPA concentration of ~0.1 nM. This high dose requirement is mainly due to inhibition by endogenous PAI-1 (~0.4 nM), which is further upregulated in thrombotic conditions155. Bennett et al. reported a full-length r-tPA variant (KtPA, with mutations of KHRR 296–299 to AAAA), which showed 90-fold increased resistance to PAI-1 and similar fibrinolytic activity as wild-type r-tPA156,157. In a rabbit thrombotic model, KtPA prevented fibrin deposition at lower doses than wild-type r-tPA did158. We also discovered an r-tPA variant with 5-fold increased fibrinolytic activity and 30-fold increased PAI-1 resistance compared to clinically used r-tPA (Fig. 4a)159. This new r-tPA variant was rationally designed based on our crystal structure of the tPA:PAI-1 complex160. In contrast to full-length KtPA with mutations in kringle domain 2, our r-tPA variant, tPA-SPD (A146Y), has only a single catalytic domain (also called the serine protease domain, SPD) with a single mutation. More importantly, our tPA-SPD variant (A146Y) enhanced fibrinolytic activity, while KtPA produced no increase in fibrinolytic activity compared to R-tPA. In a subsequent study, we used a pulmonary embolism mouse model to confirm the enhanced thrombolytic effects of tPA-SPD (A146Y) in vivo.

a The high-efficiency R-tPA-SPD-A146Y agent demonstrated 5-fold increased fibrinolytic activity and 30-fold increased PAI-1 resistance compared to clinical r-tPA. b A recombinant PAI-1 inhibitor, PAItrap, derived from the inactive catalytic domain of uPA. c PAItrap fused with HSA demonstrated prolonged circulation time and enhanced thrombolytic effects. d A small-molecule natural product, embelin, demonstrated high inhibitory potency against PAI-1 and suppressed inflammation and thrombosis in a sepsis-induced DIC model. Blood flow is indicated by the yellow arrows.
PAI-1 inhibitors as thrombolytic agents
PAI-1 is the most important physiological modulator of tPA and the fibrinolytic system. Upregulated PAI-1 levels inactivate plasma tPA and suppress fibrinolysis in thrombotic or inflammatory conditions. Monoclonal antibodies against PAI-1 decreased fibrin deposition in thrombotic animal models, indicating that PAI-1 inhibition is an effective antithrombotic strategy161,162. Thus far, a large number of small-molecule or peptidic PAI-1 inhibitors have been developed163,164. Tiplasinin, also named tiplaxtinin or PAI-039, is a small-molecule PAI-1 inhibitor (IC50 = 2.7 µM) that has been widely studied in multiple experimental thrombotic models and preclinical studies165,166,167. PAI-039 docked in the vitronectin-binding region of PAI-1 in a computational model and thus inhibited vitronectin-bound PAI-1 less efficiently than free PAI-1168. By screening a natural product library, we identified a compound, embelin, as a potent PAI-1 inhibitor (IC50 = 1.6 µM) (Fig. 4d)169. Interestingly, although embelin and PAI-039 are not similar in structure, the crystal structure of PAI-1:embelin complex demonstrated that embelin also bind to the vitronectin-binding region of PAI-1. Embelin demonstrated thrombolytic effects in three thrombotic mouse models induced by electric current, FeCl3, and laser irradiation. By combining the fibrinolytic property we identified with the previously reported anti-inflammatory property170, we demonstrated that embelin is a potent therapeutic agent for sepsis-induced disseminated intravascular coagulation [to be published]. In the subsequent work, we designed and synthesized a series of embelin derivatives and found that the best compound showed a 10-fold enhanced inhibitory potency against PAI-1 (IC50 = 0.18 µM)171.
We also reported two proteinaceous PAI-1 inhibitors (PAItrap) derived from the inactive recombinant catalytic domain of uPA (Fig. 4b, c)172,173. UPA is known to bind to PAI-1 through the catalytic domain with very high affinity. We used inactive uPA (S195A) to avoid inducing proteolytic activity. Depletion of the kringle and GFD domains was to prevent off-target uPAR-binding. Based on our previously reported crystal structure of the uPA-PAI-1 complex174, we optimized the uPA-PAI-1 interaction by mutating some critical residues in uPA and generated a version of the catalytic domain of the uPA variant with five mutations (PAItrap), which demonstrated high affinity to PAI-1 (KD = 0.15 nM). PAItrap also demonstrated high specificity among other homogenous serpin proteins, e.g., PAI-2, protease nexin 1, antithrombin, or α2-AP (IC50 > 10 µM). In a mouse thrombotic model, PAItrap significantly reduced fibrin deposition and platelet aggregation. However, as a small protein (~25 kDa), PAItrap showed a very short half-life in vivo (<5 min in mouse vessels). To enhance the circulation time, we fused PAItrap with HSA and optimized PAI-1 binding by using computational chemistry, leading to the second generation PAItrap, PAItrap(H37R)-HSA173. PAItrap(H37R)-HSA demonstrated an ~300-fold prolonged circulating time, a 7-fold enhanced efficacy in preventing platelet accumulation, and a 3-fold higher efficacy in reducing fibrin deposition.
Determination of PAI-1 concentrations in plasma
PAI-1 and uPA have been validated as biomarkers for breast or prostate cancers in the clinical uses of level of evidence 1 studies by the American Cancer Society175. In addition, elevated plasma PAI-1 levels have also been associated with multiple pathological conditions, e.g., septic disease course176, hypertension in American Indians177, obesity adiposis178, and type II diabetes mellitus179. Thus, the determination of the plasma PAI-1 concentration is of potential diagnostic and prognostic importance. Several ELISA kits for determining plasma PAI-1 levels are commercially available. However, the results of these kits showed poor consistency, as the results determined by seven different kits varied by 4- to 6-fold180. One likely reason is that PAI-1 exists in four different forms, and the different kits determined the concentration of different forms of PAI-1. In ELISA assays, the target proteins are normally captured by monoclonal antibodies that typically recognize the exosites rather than the active site (RCL) of PAI-1 and thus fail to distinguish the active form of PAI-1 from other forms. We reported an ELISA assay for the precise detection of active-form PAI-1 based on PAItrap(H37R)-HSA (Fig. 3b)181. PAItrap(H37R)-HSA specifically recognized active PAI-1 via exposed RCL but did not bind other forms of PAI-1 in which the RCL loop was either cleaved or embedded.
Conclusion
As a well-studied system, the fibrinolytic system has been shown to play versatile roles in many essential biological processes and is of considerable interest for the discovery of therapeutic targets. Medicinal molecules targeting the fibrinolytic system have been successfully used in clinical treatments or have been subjected to clinical trials, including the uPA inhibitor Upamostat for cancer treatment, r-tPA for acute thrombotic diseases, the radioactive uPAR-binding peptide for PET cancer diagnosis, and the plasmin inhibitor aprotinin for postsurgical hemostasis. Although these agents have achieved success in clinical treatments, their drawbacks are still obvious. For instance, Upamostat shows poor specificity, as shown by its Ki values that demonstrate its near-inhibition of homologous proteases, which function as coagulation factors, although no severe coagulative disorders were observed in clinical trials182. R-tPA is the only FDA-approved and widely used treatment for acute ischemic stroke, but it has only been used in a very small number of patients due to its various limitations. Thus, studies on the development of novel strategies with higher efficiency and specificity have shown a robust increase. This review summarizes the efforts to target the fibrinolytic system via novel diagnostic and therapeutic strategies by us and others. Accumulating evidence verifies the presence of crosstalk between the fibrinolytic system and inflammatory processes. Depletion or inhibition of uPA, PAI-1, or uPAR decreased inflammatory levels, indicating that the fibrinolytic system is a potent anti-inflammatory target. In addition, multiple studies demonstrated that the plasma levels of suPAR or PAI-1 were adequate indicators of the degree of inflammation. Medicinal modulators of the fibrinolytic system also demonstrated promising results in inflammatory disease models. Thus, modulation of the fibrinolytic system is an attractive strategy for anti-inflammatory therapies. In addition, the roles of tPA and plasmin in neuronal diseases have been increasingly recognized, strengthening the therapeutic importance of tPA. In addition, the dysregulation of PAI-1 has been associated with obesity or diabetes. Hence, the development of therapeutics targeting the fibrinolytic system is still attractive to academia and the pharmaceutical industry.
References
Castellino, F. J. & Ploplis, V. A. Structure and function of the plasminogen/plasmin system. Thromb. Haemost. 93, 647–654 (2005).
Law, R. H. et al. The X-ray crystal structure of full-length human plasminogen. Cell Rep. 1, 185–190 (2012).
Cao, Y. & Xue, L. Angiostatin. Semin Thromb. Hemost. 30, 83–93 (2004).
Urano, T., Castellino, F. J. & Suzuki, Y. Regulation of plasminogen activation on cell surfaces and fibrin. J. Thromb. Haemost. https://doi.org/10.1111/jth.14157 (2018).
Hervio, L. S. et al. Negative selectivity and the evolution of protease cascades: the specificity of plasmin for peptide and protein substrates. Chem. Biol. 7, 443–453 (2000).
Deryugina, E. I. & Quigley, J. P. Cell surface remodeling by plasmin: a new function for an old enzyme. J. Biomed. Biotechnol. 2012, 564259 (2012).
Nissinen, L. & Kahari, V. M. Matrix metalloproteinases in inflammation. Biochimica et. biophysica acta 1840, 2571–2580 (2014).
Chen, J. & Khalil, R. A. Matrix metalloproteinases in normal pregnancy and preeclampsia. Prog. Mol. Biol. Transl. Sci. 148, 87–165 (2017).
Cui, N., Hu, M. & Khalil, R. A. Biochemical and biological attributes of matrix metalloproteinases. Prog. Mol. Biol. Transl. Sci. 147, 1–73 (2017).
Nogami, K. et al. Mechanisms of plasmin-catalyzed inactivation of factor VIII: a crucial role for proteolytic cleavage at Arg336 responsible for plasmin-catalyzed factor VIII inactivation. J. Biol. Chem. 282, 5287–5295 (2007).
Santibanez, J. F. UrokinaseType Plasminogen activator and the molecular mechanisms of its regulation in cancer. Protein Pept. Lett. 24, 936–946 (2017).
Ichinose, A., Fujikawa, K. & Suyama, T. The activation of pro-urokinase by plasma kallikrein and its inactivation by thrombin. J. Biol. Chem. 261, 3486–3489 (1986).
Lee, S. L., Dickson, R. B. & Lin, C. Y. Activation of hepatocyte growth factor and urokinase/plasminogen activator by matriptase, an epithelial membrane serine protease. J. Biol. Chem. 275, 36720–36725 (2000).
Carmeliet, P. et al. Receptor-independent role of urokinase-type plasminogen activator in pericellular plasmin and matrix metalloproteinase proteolysis during vascular wound healing in mice. J. Cell Biol. 140, 233–245 (1998).
Reichel, C. A. et al. Urokinase-type plasminogen activator promotes paracellular transmigration of neutrophils via Mac-1, but independently of urokinase-type plasminogen activator receptor. Circulation 124, 1848–1859 (2011).
Kruithof, E. K. & Dunoyer-Geindre, S. Human tissue-type plasminogen activator. Thromb. Haemost. 112, 243–254 (2014).
Collen, D. Fibrin-specific thrombolytic therapy. Thromb. Res Suppl. 8, 3–14 (1988).
Stricker, R. B., Wong, D., Shiu, D. T., Reyes, P. T. & Shuman, M. A. Activation of plasminogen by tissue plasminogen activator on normal and thrombasthenic platelets: effects on surface proteins and platelet aggregation. Blood 68, 275–280 (1986).
Boose, J. A., Kuismanen, E., Gerard, R., Sambrook, J. & Gething, M. J. The single-chain form of tissue-type plasminogen activator has catalytic activity: studies with a mutant enzyme that lacks the cleavage site. Biochem.-Us 28, 635–643 (1989).
Teesalu, T., Kulla, A., Asser, T., Koskiniemi, M. & Vaheri, A. Tissue plasminogen activator as a key effector in neurobiology and neuropathology. Biochem Soc. Trans. 30, 183–189 (2002).
Melchor, J. P. & Strickland, S. Tissue plasminogen activator in central nervous system physiology and pathology. Thromb. Haemost. 93, 655–660 (2005).
Medcalf, R. L. Fibrinolysis: from blood to the brain. J. Thromb. Haemost. 15, 2089–2098 (2017).
Cuzner, M. L. & Opdenakker, G. Plasminogen activators and matrix metalloproteases, mediators of extracellular proteolysis in inflammatory demyelination of the central nervous system. J. Neuroimmunol. 94, 1–14 (1999).
Melchor, J. P., Pawlak, R. & Strickland, S. The tissue plasminogen activator-plasminogen proteolytic cascade accelerates amyloid-beta (Abeta) degradation and inhibits Abeta-induced neurodegeneration. J. Neurosci. 23, 8867–8871 (2003).
Wu, Y. P. et al. The tissue plasminogen activator (tPA)/plasmin extracellular proteolytic system regulates seizure-induced hippocampal mossy fiber outgrowth through a proteoglycan substrate. J. Cell Biol. 148, 1295–1304 (2000).
Gettins, P. G. Serpin structure, mechanism, and function. Chem. Rev. 102, 4751–4804 (2002).
Brogren, H. et al. Platelets synthesize large amounts of active plasminogen activator inhibitor 1. Blood 104, 3943–3948 (2004).
Huebner, B. R. et al. Thrombin provokes degranulation of platelet alpha-granules leading to the release of active plasminogen activator inhibitor-1 (PAI-1). Shock 50, 671–676 (2018).
Lang, I. M., Marsh, J. J., Moser, K. M. & Schleef, R. R. Presence of active and latent type 1 plasminogen activator inhibitor associated with porcine platelets. Blood 80, 2269–2274 (1992).
Kindell, D. G., Keck, R. W. & Jankun, J. Comparison between the clot-protecting activity of a mutant plasminogen activator inhibitor-1 with a very long half-life and 6-aminocaproic acid. Exp. Ther. Med. 9, 2339–2343 (2015).
Hjortland, G. O. et al. Modulation of glioma cell invasion and motility by adenoviral gene transfer of PAI-1. Clin. Exp. Metastasis 20, 301–309 (2003).
Inoue, M. et al. Plasminogen activator inhibitor-1 (PAI-1) gene transfection inhibits the liver metastasis of pancreatic cancer by preventing angiogenesis. Oncol. Rep. 14, 1445–1451 (2005).
Rubina, K. A. et al. Increased expression of uPA, uPAR, and PAI-1 in psoriatic skin and in basal cell carcinomas. Arch. Dermatol. Res. 309, 433–442 (2017).
Pavon, M. A. et al. Enhanced cell migration and apoptosis resistance may underlie the association between high SERPINE1 expression and poor outcome in head and neck carcinoma patients. Oncotarget 6, 29016–29033 (2015).
Hirahata, M. et al. PAI-1, a target gene of miR-143, regulates invasion and metastasis by upregulating MMP-13 expression of human osteosarcoma. Cancer Med. 5, 892–902 (2016).
Zhang, W. et al. Endothelial cells promote triple-negative breast cancer cell metastasis via PAI-1 and CCL5 signaling. FASEB J. 32, 276–288 (2018).
Ngo, J. C. et al. Structural basis for therapeutic intervention of uPA/uPAR system. Curr. Drug Targets 12, 1729–1743 (2011).
Huai, Q. et al. Structure of human urokinase plasminogen activator in complex with its receptor. Science 311, 656–659 (2006).
Colman, R. W. et al. Binding of high molecular weight kininogen to human endothelial cells is mediated via a site within domains 2 and 3 of the urokinase receptor. J. Clin. Invest. 100, 1481–1487 (1997).
Huai, Q. et al. Crystal structures of two human vitronectin, urokinase and urokinase receptor complexes. Nat. Struct. Mol. Biol. 15, 422–423 (2008).
Blasi, F. & Carmeliet, P. uPAR: a versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol. 3, 932–943 (2002).
Tang, C. H. & Wei, Y. The urokinase receptor and integrins in cancer progression. Cell Mol. Life Sci. 65, 1916–1932 (2008).
Resnati, M. et al. The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proc. Natl Acad. Sci. USA 99, 1359–1364 (2002).
de Paulis, A. et al. Urokinase induces basophil chemotaxis through a urokinase receptor epitope that is an endogenous ligand for formyl peptide receptor-like 1 and -like 2. J. Immunol. 173, 5739–5748 (2004).
Liu, D., Aguirre Ghiso, J., Estrada, Y. & Ossowski, L. EGFR is a transducer of the urokinase receptor initiated signal that is required for in vivo growth of a human carcinoma. Cancer Cell 1, 445–457 (2002).
Montuori, N. & Ragno, P. Multiple activities of a multifaceted receptor: roles of cleaved and soluble uPAR. Front. Biosci. 14, 2494–2503 (2009).
Sidenius, N. & Blasi, F. Domain 1 of the urokinase receptor (uPAR) is required for uPAR-mediated cell binding to vitronectin. FEBS Lett. 470, 40–46 (2000).
Montuori, N., Carriero, M. V., Salzano, S., Rossi, G. & Ragno, P. The cleavage of the urokinase receptor regulates its multiple functions. J. Biol. Chem. 277, 46932–46939 (2002).
Wilhelm, O. G. et al. Cellular glycosylphosphatidylinositol-specific phospholipase D regulates urokinase receptor shedding and cell surface expression. J. Cell Physiol. 180, 225–235 (1999).
Wilhelm, O. et al. Recombinant soluble urokinase receptor as a scavenger for urokinase-type plasminogen activator (uPA). Inhibition of proliferation and invasion of human ovarian cancer cells. FEBS Lett. 337, 131–134 (1994).
Montuori, N., Visconte, V., Rossi, G. & Ragno, P. Soluble and cleaved forms of the urokinase-receptor: degradation products or active molecules? Thromb. Haemost. 93, 192–198 (2005).
Misheneva, V. S. [Effect of some inhibitors on the urokinase activity of the liver of normal and tumor-bearing. animals]. Ukrains’kyi biokhimichnyi zhurnal 35, 566–572 (1963).
Mekkawy, A. H., Pourgholami, M. H. & Morris, D. L. Involvement of urokinase-type plasminogen activator system in cancer: an overview. Medicinal Res. Rev. 34, 918–956 (2014).
Prager, G. W. et al. Urokinase mediates endothelial cell survival via induction of the X-linked inhibitor of apoptosis protein. Blood 113, 1383–1390 (2009).
Poettler, M. et al. The urokinase receptor (CD87) represents a central mediator of growth factor-induced endothelial cell migration. Thromb. Haemost. 108, 357–366 (2012).
Santibanez, J. F., Obradovic, H., Kukolj, T. & Krstic, J. Transforming growth factor-beta, matrix metalloproteinases, and urokinase-type plasminogen activator interaction in the cancer epithelial to mesenchymal transition. Dev. Dyn. 247, 382–395 (2018).
Fabregat, I., Fernando, J., Mainez, J. & Sancho, P. TGF-beta signaling in cancer treatment. Curr. Pharm. Des. 20, 2934–2947 (2014).
Reichel, C. A., Kanse, S. M. & Krombach, F. At the interface of fibrinolysis and inflammation: the role of urokinase-type plasminogen activator in the leukocyte extravasation cascade. Trends Cardiovasc. Med. 22, 192–196 (2012).
Bhattacharya, S., Ploplis, V. A. & Castellino, F. J. Bacterial plasminogen receptors utilize host plasminogen system for effective invasion and dissemination. J. Biomed. Biotechnol. 2012, 482096 (2012).
Agostoni, A. et al. Activation of complement and kinin systems after thrombolytic therapy in patients with acute myocardial infarction. A comparison between streptokinase and recombinant tissue-type plasminogen activator. Circulation 90, 2666–2670 (1994).
Schaiff, W. T. & Eisenberg, P. R. Direct induction of complement activation by pharmacologic activation of plasminogen. Coron. Artery Dis. 8, 9–18 (1997).
Yang, X. S. et al. Protein kinase C-delta mediates sepsis-induced activation of complement 5a and urokinase-type plasminogen activator signaling in macrophages. Inflamm. Res. 63, 581–589 (2014).
Shushakova, N. et al. Urokinase-induced activation of the gp130/Tyk2/Stat3 pathway mediates a pro-inflammatory effect in human mesangial cells via expression of the anaphylatoxin C5a receptor. J. Cell Sci. 118, 2743–2753 (2005).
Higazi, A. A.-R. & Barghouti, I. I. Inactivation of human anaphylatoxin C5a and C5a des-Arg through cleavage by the plasminogen activator activity of a human fibrosarcoma cell line. J. Biol. Chem. 269, 25529–25533 (1994).
Sugimoto, M. A. et al. Plasmin and plasminogen induce macrophage reprogramming and regulate key steps of inflammation resolution via annexin A1. Blood 129, 2896–2907 (2017).
Minami, E. et al. The role of macrophage-derived urokinase plasminogen activator in myocardial infarct repair: urokinase attenuates ventricular remodeling. J. Mol. Cell Cardiol. 49, 516–524 (2010).
Carmassi, F., de Negri, F., Morale, M., Song, K. Y. & Chung, S. I. Fibrin degradation in the synovial fluid of rheumatoid arthritis patients: a model for extravascular fibrinolysis. Semin Thromb. Hemost. 22, 489–496 (1996).
Weinberg, J. B., Pippen, A. M. & Greenberg, C. S. Extravascular fibrin formation and dissolution in synovial tissue of patients with osteoarthritis and rheumatoid arthritis. Arthritis Rheum. 34, 996–1005 (1991).
Li, J. et al. The plasminogen activator/plasmin system is essential for development of the joint inflammatory phase of collagen type II-induced arthritis. Am. J. Pathol. 166, 783–792 (2005).
Serrati, S. et al. Reduction of in vitro invasion and in vivo cartilage degradation in a SCID mouse model by loss of function of the fibrinolytic system of rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 63, 2584–2594 (2011).
Cook, A. D., Braine, E. L., Campbell, I. K. & Hamilton, J. A. Differing roles for urokinase and tissue-type plasminogen activator in collagen-induced arthritis. Am. J. Pathol. 160, 917–926 (2002).
Lin, L. & Hu, K. Tissue plasminogen activator and inflammation: from phenotype to signaling mechanisms. Am. J. Clin. Exp. Immunol. 3, 30–36 (2014).
Levi, M. & van der Poll, T. Inflammation and coagulation. Crit. Care Med. 38, S26–S34 (2010).
Prosper, F. et al. Mobilization of peripheral blood progenitor cells with a combination of cyclophosphamide, r-metHuSCF and filgrastim in patients with breast cancer previously treated with chemotherapy. Leukemia 17, 437–441 (2003).
Larsson, P. et al. Effects of IL-1beta and IL-6 on tissue-type plasminogen activator expression in vascular endothelial cells. Thromb. Res. 123, 342–351 (2008).
Sitter, T. et al. Modulation of procoagulant and fibrinolytic system components of mesothelial cells by inflammatory mediators. Am. J. Physiol. 271, R1256–R1263 (1996).
Hosoya, S. et al. Stimulatory effect of interleukin-6 on plasminogen activator activity from human dental pulp cells. J. Endod. 24, 331–334 (1998).
Ulfhammer, E. et al. TNF-alpha mediated suppression of tissue type plasminogen activator expression in vascular endothelial cells is NF-kappaB- and p38 MAPK-dependent. J. Thromb. Haemost. 4, 1781–1789 (2006).
Juhan-Vague, I., Moerman, B., De Cock, F., Aillaud, M. F. & Collen, D. Plasma levels of a specific inhibitor of tissue-type plasminogen activator (and urokinase) in normal and pathological conditions. Thromb. Res. 33, 523–530 (1984).
Kluft, C. et al. The postoperative fibrinolytic shutdown: a rapidly reverting acute phase pattern for the fast-acting inhibitor of tissue-type plasminogen activator after trauma. Scand. J. Clin. Lab Invest. 45, 605–610 (1985).
Campbell, I. K., Wojta, J., Novak, U. & Hamilton, J. A. Cytokine modulation of plasminogen activator inhibitor-1 (PAI-1) production by human articular cartilage and chondrocytes. Down-regulation by tumor necrosis factor alpha and up-regulation by transforming growth factor-B basic fibroblast growth factor. Biochim. Et. Biophys. Acta 1226, 277–285 (1994).
Rega, G. et al. Inflammatory cytokines interleukin-6 and oncostatin m induce plasminogen activator inhibitor-1 in human adipose tissue. Circulation 111, 1938–1945 (2005).
Rawson, R. et al. TGF-beta1-induced PAI-1 contributes to a profibrotic network in patients with eosinophilic esophagitis. J. Allergy Clin. Immunol. 138, 791–800 (2016). e794.
Renckens, R. et al. The role of plasminogen activator inhibitor type 1 in the inflammatory response to local tissue injury. J. Thromb. Haemost. 3, 1018–1025 (2005).
Gupta, K. K., Xu, Z., Castellino, F. J. & Ploplis, V. A. Plasminogen activator inhibitor-1 stimulates macrophage activation through Toll-like Receptor-4. Biochem. Biophys. Res. Commun. 477, 503–508 (2016).
Poole, L. G. et al. Plasminogen Activator Inhibitor-1 Is Critical in Alcohol-Enhanced Acute Lung Injury in Mice. Am. J. Respir. Cell Mol. Biol. 57, 315–323 (2017).
Robert, S., Wagner, B. K., Boulanger, M. & Richer, M. Aprotinin. Ann. Pharmacother. 30, 372–380 (1996).
Reis, S. E. & Feldman, A. H. Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. N. Engl. J. Med. 336, 1453 (1997). author reply 1453–1454.
Ploplis, V. A. et al. Effects of disruption of the plasminogen gene on thrombosis, growth, and health in mice. Circulation 92, 2585–2593 (1995).
Carmeliet, P. et al. Physiological consequences of loss of plasminogen activator gene function in mice. Nature 368, 419–424 (1994).
Lijnen, H. R. Pathophysiology of the plasminogen/plasmin system. Int J. Clin. Lab Res 26, 1–6 (1996).
Guo, Y. et al. A peptide derived from the nonreceptor binding region of urokinase plasminogen activator (uPA) inhibits tumor progression and angiogenesis and induces tumor cell death in vivo. FASEB J. 14, 1400–1410 (2000).
Guo, Y., Mazar, A. P., Lebrun, J. J. & Rabbani, S. A. An antiangiogenic urokinase-derived peptide combined with tamoxifen decreases tumor growth and metastasis in a syngeneic model of breast cancer. Cancer Res. 62, 4678–4684 (2002).
Boyd, D. D., Kim, S. J., Wang, H., Jones, T. R. & Gallick, G. E. A urokinase-derived peptide (A6) increases survival of mice bearing orthotopically grown prostate cancer and reduces lymph node metastasis. Am. J. Pathol. 162, 619–626 (2003).
Berkenblit, A. et al. A6, a urokinase plasminogen activator (uPA)-derived peptide in patients with advanced gynecologic cancer: a phase I trial. Gynecol. Oncol. 99, 50–57 (2005).
Ghamande, S. A. et al. A phase 2, randomized, double-blind, placebo-controlled trial of clinical activity and safety of subcutaneous A6 in women with asymptomatic CA125 progression after first-line chemotherapy of epithelial ovarian cancer. Gynecol. Oncol. 111, 89–94 (2008).
Gold, M. A. et al. A phase II study of a urokinase-derived peptide (A6) in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol. Oncol. 125, 635–639 (2012).
Navaratna, D. et al. A peptide inhibitor of the urokinase/urokinase receptor system inhibits alteration of the blood-retinal barrier in diabetes. FASEB J. 22, 3310–3317 (2008).
Das, A., Boyd, N., Jones, T. R., Talarico, N. & McGuire, P. G. Inhibition of choroidal neovascularization by a peptide inhibitor of the urokinase plasminogen activator and receptor system in a mouse model. Arch. Ophthalmol. 122, 1844–1849 (2004).
McGuire, P. G., Jones, T. R., Talarico, N., Warren, E. & Das, A. The urokinase/urokinase receptor system in retinal neovascularization: inhibition by A6 suggests a new therapeutic target. Invest Ophthalmol. Vis. Sci. 44, 2736–2742 (2003).
Kanno, Y., Maruyama, C., Matsuda, A. & Ishisaki, A. uPA-derived peptide, A6 is involved in the suppression of lipopolysaccaride-promoted inflammatory osteoclastogenesis and the resultant bone loss. Immun. Inflamm. Dis. 5, 289–299 (2017).
Sato, S. et al. High-affinity urokinase-derived cyclic peptides inhibiting urokinase/urokinase receptor-interaction: effects on tumor growth and spread. FEBS Lett. 528, 212–216 (2002).
Tressler, R. J. et al. Urokinase receptor antagonists: discovery and application to in vivo models of tumor growth. APMIS 107, 168–173 (1999).
min, H. Y. et al. Urokinase receptor antagonists inhibit angiogenesis and primary tumor growth in syngeneic mice. Cancer Res. 56, 2428–2433 (1996).
Goodson, R. J., Doyle, M. V., Kaufman, S. E. & Rosenberg, S. High-affinity urokinase receptor antagonists identified with bacteriophage peptide display. Proc. Natl Acad. Sci. USA 91, 7129–7133 (1994).
Rea, V. E. et al. Discovery of new small molecules targeting the vitronectin-binding site of the urokinase receptor that block cancer cell invasion. Mol. Cancer Ther. 12, 1402–1416 (2013).
Mani, T. et al. Probing binding and cellular activity of pyrrolidinone and piperidinone small molecules targeting the urokinase receptor. ChemMedChem 8, 1963–1977 (2013).
Liu, D., Zhou, D., Wang, B., Knabe, W. E. & Meroueh, S. O. A new class of orthosteric uPAR.uPA small-molecule antagonists are allosteric inhibitors of the uPAR.vitronectin interaction. Acs Chem. Biol. 10, 1521–1534 (2015).
Eefting, D. et al. A novel urokinase receptor-targeted inhibitor for plasmin and matrix metalloproteinases suppresses vein graft disease. Cardiovasc. Res. 88, 367–375 (2010).
Rabbani, S. A. & Gladu, J. Urokinase receptor antibody can reduce tumor volume and detect the presence of occult tumor metastases in vivo. Cancer Res. 62, 2390–2397 (2002).
Bauer, T. W. et al. Targeting of urokinase plasminogen activator receptor in human pancreatic carcinoma cells inhibits c-Met- and insulin-like growth factor-I receptor-mediated migration and invasion and orthotopic tumor growth in mice. Cancer Res. 65, 7775–7781 (2005).
Ploug, M. et al. Peptide-derived antagonists of the urokinase receptor. affinity maturation by combinatorial chemistry, identification of functional epitopes, and inhibitory effect on cancer cell intravasation. Biochemistry 40, 12157–12168 (2001).
Persson, M. et al. Quantitative PET of human urokinase-type plasminogen activator receptor with 64Cu-DOTA-AE105: implications for visualizing cancer invasion. J. Nucl. Med. 53, 138–145 (2012).
Persson, M. et al. Improved PET imaging of uPAR expression using new (64)Cu-labeled cross-bridged peptide ligands: comparative in vitro and in vivo studies. Theranostics 3, 618–632 (2013).
Persson, M. et al. First-in-human uPAR PET: imaging of cancer aggressiveness. Theranostics 5, 1303–1316 (2015).
Skovgaard, D., Persson, M. & Kjaer, A. Urokinase plasminogen activator receptor-PET with (68)Ga-NOTA-AE105: first clinical experience with a novel PET ligand. PET Clin. 12, 311–319 (2017).
Rajagopal, V. & Kreitman, R. J. Recombinant toxins that bind to the urokinase receptor are cytotoxic without requiring binding to the alpha(2)-macroglobulin receptor. J. Biol. Chem. 275, 7566–7573 (2000).
Cavallaro, U., del Vecchio, A., Lappi, D. A. & Soria, M. R. A conjugate between human urokinase and saporin, a type-1 ribosome-inactivating protein, is selectively cytotoxic to urokinase receptor-expressing cells. J. Biol. Chem. 268, 23186–23190 (1993).
Yang, L. et al. Receptor-targeted nanoparticles for in vivo imaging of breast cancer. Clin. Cancer Res. 15, 4722–4732 (2009).
Hansen, L. et al. Targeting of peptide conjugated magnetic nanoparticles to urokinase plasminogen activator receptor (uPAR) expressing cells. Nanoscale 5, 8192–8201 (2013).
Chen, Z. et al. Zinc phthalocyanine conjugated with the amino-terminal fragment of urokinase for tumor-targeting photodynamic therapy. Acta Biomater. 10, 4257–4268 (2014).
Li, R. et al. A novel tumor targeting drug carrier for optical imaging and therapy. Theranostics 4, 642–659 (2014).
Li, R., Zheng, K., Yuan, C., Chen, Z. & Huang, M. Be Active or Not: the Relative Contribution of Active and Passive Tumor Targeting of Nanomaterials. Nanotheranostics 1, 346–357 (2017).
Zheng, K. et al. Dual actions of albumin packaging and tumor targeting enhance the antitumor efficacy and reduce the cardiotoxicity of doxorubicin in vivo. Int. J. Nanomed. 10, 5327–5342 (2015).
Li, S. et al. Nanoparticle binding to urokinase receptor on cancer cell surface triggers nanoparticle disintegration and cargo release. Theranostics 9, 884–899 (2019).
Hayek, S. S. et al. Soluble Urokinase Receptor and Chronic Kidney Disease. N. Engl. J. Med. 373, 1916–1925 (2015).
Brunner, N. et al. The urokinase plasminogen activator receptor in blood from healthy individuals and patients with cancer. APMIS 107, 160–167 (1999).
Donadello, K., Scolletta, S., Covajes, C. & VincentJ. L. suPAR as a prognostic biomarker in sepsis. BMC Med. 10, 2 (2012).
Zeng, M. et al. Clinical value of soluble urokinase-type plasminogen activator receptor in the diagnosis, prognosis, and therapeutic guidance of sepsis. Am. J. Emerg. Med 34, 375–380 (2016).
Rasmussen, L. J. et al. Soluble urokinase plasminogen activator receptor (suPAR) is a novel, independent predictive marker of myocardial infarction in HIV-1-infected patients: a nested case-control study. HIV Med. 17, 350–357 (2016).
Drechsler, C. et al. Soluble urokinase plasminogen activator receptor and outcomes in patients with diabetes on hemodialysis. Clin. J. Am. Soc. Nephrol. 12, 1265–1273 (2017).
Hall, S. S. Omen in the blood. Science 360, 254–258 (2018).
Iribarren, P., Zhou, Y., Hu, J., Le, Y. & Wang, J. M. Role of formyl peptide receptor-like 1 (FPRL1/FPR2) in mononuclear phagocyte responses in Alzheimer disease. Immunol. Res. 31, 165–176 (2005).
Stephens, R. W. et al. ELISA determination of soluble urokinase receptor in blood from healthy donors and cancer patients. Clin. Chem. 43, 1868–1876 (1997).
Zhou, X. et al. An ELISA method detecting the active form of suPAR. Talanta 160, 205–210 (2016).
Basu, H. K. & Jeffcoate, N. Local fibrinolytic activity in the pregnant uterus. Am. J. Obstet. Gynecol. 107, 1188–1194 (1970).
Osime, O. E., Ese-Onakewhor, J. U. & Kolade, S. O. Fibrinolytic changes in pregnant women on highly active antiretroviral therapy. Saudi Med. J. 36, 200–203 (2015).
Mack, G. S. & Marshall, A. Lost in migration. Nat. Biotechnol. 28, 214–229 (2010).
Schweinitz, A. et al. Design of novel and selective inhibitors of urokinase-type plasminogen activator with improved pharmacokinetic properties for use as antimetastatic agents. J. Biol. Chem. 279, 33613–33622 (2004).
Henneke, I. et al. Inhibition of urokinase activity reduces primary tumor growth and metastasis formation in a murine lung carcinoma model. Am. J. Respir. Crit. Care Med. 181, 611–619 (2010).
Joossens, J., der Veken, Van, Lambeir, P., Augustyns, A. M. & Haemers, K. A. Development of irreversible diphenyl phosphonate inhibitors for urokinase plasminogen activator. J. Med Chem. 47, 2411–2413 (2004).
Vangestel, C. et al. Preclinical evaluation of [(111) In]MICA-401, an activity-based probe for SPECT imaging of in vivo uPA activity. Contrast Media Mol. Imaging 11, 448–458 (2016).
Angelini, A. et al. Bicyclic peptide inhibitor reveals large contact interface with a protease target. Acs Chem. Biol. 7, 817–821 (2012).
Chen, S. et al. Bicyclic peptide ligands pulled out of cysteine-rich peptide libraries. J. Am. Chem. Soc. 135, 6562–6569 (2013).
Chen, S. et al. Dithiol amino acids can structurally shape and enhance the ligand-binding properties of polypeptides. Nat. Chem. 6, 1009–1016 (2014).
Vassalli, J. D. & Belin, D. Amiloride selectively inhibits the urokinase-type plasminogen activator. FEBS Lett. 214, 187–191 (1987).
Perazella, M. A. Drug-induced hyperkalemia: old culprits and new offenders. Am. J. Med. 109, 307–314 (2000).
Buckley, B. J. et al. 6-Substituted Hexamethylene Amiloride (HMA) Derivatives As Potent And Selective Inhibitors Of The Human Urokinase Plasminogen Activator For Use In Cancer. J. Med Chem. 61, 8299–8320 (2018).
Xu, P., Andreasen, P. A. & Huang, M. Structural Principles In The Development Of Cyclic Peptidic Enzyme Inhibitors. Int J. Biol. Sci. 13, 1222–1233 (2017).
Wang, D. et al. Suppression of Tumor Growth And Metastases By Targeted Intervention In Urokinase Activity With Cyclic Peptides. J. Med Chem. 62, 2172–2183 (2019).
Medcalf, R. L. & Davis, S. M. Plasminogen activation and thrombolysis for ischemic stroke. Int. J. Stroke.: Off. J. Int. Stroke. Soc. 7, 419–425 (2012).
Lippi, G., Mattiuzzi, C. & Favaloro, E. J. Novel and emerging therapies: thrombus-targeted fibrinolysis. Semin Thromb. Hemost. 39, 48–58 (2013).
Minematsu, K. et al. Guidelines for the intravenous application of recombinant tissue-type plasminogen activator (alteplase), the second edition, October 2012: a guideline from the Japan Stroke Society. J. Stroke Cerebrovasc. Dis. 22, 571–600 (2013).
Ouriel, K. & Kandarpa, K. Safety of thrombolytic therapy with urokinase or recombinant tissue plasminogen activator for peripheral arterial occlusion: a comprehensive compilation of published work. J. Endovasc. Ther. 11, 436–446 (2004).
Jang, I. K. et al. Differential sensitivity of erythrocyte-rich and platelet-rich arterial thrombi to lysis with recombinant tissue-type plasminogen activator. A possible explanation for resistance to coronary thrombolysis. Circulation 79, 920–928 (1989).
Bennett, W. F. et al. High resolution analysis of functional determinants on human tissue-type plasminogen activator. J. Biol. Chem. 266, 5191–5201 (1991).
Refino, C. J. et al. A variant of t-PA (T103N, KHRR 296-299 AAAA) that, by bolus, has increased potency and decreased systemic activation of plasminogen. Thromb. Haemost. 70, 313–319 (1993).
Krishnamurti, C., Keyt, B., Maglasang, P. & Alving, B. M. PAI-1-resistant t-PA: low doses prevent fibrin deposition in rabbits with increased PAI-1 activity. Blood 87, 14–19 (1996).
Peng, S. et al. tPA Point Mutation at Autolysis Loop Enhances Resistance to PAI-1 Inhibition and Catalytic Activity. Thromb. Haemost. 119, 77–86 (2019).
Gong, L. et al. Crystal Structure Of The Michaelis Complex Between Tissue-type Plasminogen Activator And Plasminogen Activators Inhibitor-1. J. Biol. Chem. 290, 25795–25804 (2015).
Biemond, B. J. et al. Thrombolysis and reocclusion in experimental jugular vein and coronary artery thrombosis. Effects of a plasminogen activator inhibitor type 1-neutralizing monoclonal antibody. Circulation 91, 1175–1181 (1995).
van Giezen, J. J. & Wahlund, G., Nerme, & Abrahamsson, T. The Fab-fragment of a PAI-1 inhibiting antibody reduces thrombus size and restores blood flow in a rat model of arterial thrombosis. Thromb. Haemost. 77, 964–969 (1997).
Brown, N. J. Therapeutic potential of plasminogen activator inhibitor-1 inhibitors. Therapeutic Adv. cardiovasc. Dis. 4, 315–324 (2010).
Li, S. H. & Lawrence, D. A. Development of inhibitors of plasminogen activator inhibitor-1. Methods Enzymol. 501, 177–207 (2011).
Elokdah, H. et al. Tiplaxtinin, a novel, orally efficacious inhibitor of plasminogen activator inhibitor-1: design, synthesis, and preclinical characterization. J. Med. Chem. 47, 3491–3494 (2004).
Baxi, S. et al. Dose-dependent thrombus resolution due to oral plaminogen activator inhibitor (PAI)-1 inhibition with tiplaxtinin in a rat stenosis model of venous thrombosis. Thromb. Haemost. 99, 749–758 (2008).
Hennan, J. K. et al. Effect of tiplaxtinin (PAI-039), an orally bioavailable PAI-1 antagonist, in a rat model of thrombosis. J. Thromb. Haemost. 6, 1558–1564 (2008).
Gorlatova, N. V. et al. Mechanism of inactivation of plasminogen activator inhibitor-1 by a small molecule inhibitor. J. Biol. Chem. 282, 9288–9296 (2007).
Lin, Z. et al. Structural insight into inactivation of plasminogen activator inhibitor-1 by a small-molecule antagonist. Chem. Biol. 20, 253–261 (2013).
Lu, H., Wang, J., Wang, Y., Qiao, L. & Zhou, Y. Embelin and Its Role in Chronic Diseases. Adv. Exp. Med. Biol. 928, 397–418 (2016).
Miyazaki, H. et al. Design, synthesis, and evaluation of orally active inhibitors of plasminogen activator inhibitor-1 (PAI-1) production. Bioorg. Med. Chem. Lett. 18, 6419–6422 (2008).
Gong, L. et al. A specific plasminogen activator inhibitor-1 antagonist derived from inactivated urokinase. J. Cell Mol. Med. 20, 1851–1860 (2016).
Peng, S. et al. A long-acting PAI-1 inhibitor reduces thrombus formation. Thromb. Haemost. 117, 1338–1347 (2017).
Lin, Z. et al. Structural basis for recognition of urokinase-type plasminogen activator by plasminogen activator inhibitor-1. J. Biol. Chem. 286, 7027–7032 (2011).
Duffy, M. J., McGowan, P. M., Harbeck, N., Thomssen, C. & Schmitt, M. uPA and PAI-1 as biomarkers in breast cancer: validated for clinical use in level-of-evidence-1 studies. Breast Cancer Res. 16, 428 (2014).
Chi, Y. F. et al. Association between PAI-1 polymorphisms and plasma PAI-1 level with sepsis in severely burned patients. Genet Mol. Res. 14, 10081–10086 (2015).
Peng, H. et al. Relationship between plasma plasminogen activator inhibitor-1 and hypertension in American Indians: findings from the Strong Heart Study. J. Hypertens. 35, 1787–1793 (2017).
Barnard, S. A., Pieters, M. & De Lange, Z. The contribution of different adipose tissue depots to plasma plasminogen activator inhibitor-1 (PAI-1) levels. Blood Rev. 30, 421–429 (2016).
Wang, J. et al. Association between Plasma Levels of PAI-1, tPA/PAI-1 Molar Ratio, and Mild Cognitive Impairment in Chinese Patients with Type 2 Diabetes Mellitus. J. Alzheimers Dis. 63, 835–845 (2018).
Longstaff, C. Measuring fibrinolysis: from research to routine diagnostic assays. J. Thromb. Haemost. 16, 652–662 (2018).
Shang, L. et al. A novel ELISA for the detection of active form of plasminogen activator inhibitor-1 based on a highly specific trapping agent. Anal. Chim. Acta 1053, 98–104 (2019).
Heinemann, V. et al. Phase II randomised proof-of-concept study of the urokinase inhibitor upamostat (WX-671) in combination with gemcitabine compared with gemcitabine alone in patients with non-resectable, locally advanced pancreatic cancer. Br. J. Cancer 108, 766–770 (2013).
Acknowledgements
This work was supported by the Natural Science Foundation of Fujian Province (2018J0105, 2018J05031), grants from the National Key R&D Program of China (2017YFE0103200), and the Natural Science Foundation of China (21708043, 31400637, 31670739). Natural Science Foundation of Fujian Province (2018J0105, 2018J05031). National Key R&D Program of China (2017YFE0103200). National Natural Science Foundation of China (21708043, 31400637, 31670739).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, H., Xu, L., Yu, S. et al. Therapeutics targeting the fibrinolytic system. Exp Mol Med 52, 367–379 (2020). https://doi.org/10.1038/s12276-020-0397-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-020-0397-x
This article is cited by
-
Identification of hub pathways and drug candidates in gastric cancer through systems biology
Scientific Reports (2022)
-
TRPM7 restrains plasmin activity and promotes transforming growth factor-β1 signaling in primary human lung fibroblasts
Archives of Toxicology (2022)
-
Platelets in aging and cancer—“double-edged sword”
Cancer and Metastasis Reviews (2020)
