
Abstract
The appearance of topologically protected spin-momentum locked surface states in topological insulators gives rise to robust room temperature spin currents making them ideal candidates for the realization of spintronic devices. New methods are needed to access and manipulate such currents with timescales that are compatible with modern electronics. Here we reveal that an optically induced long-lived (~10 ns), spin-polarized surface state excitation in topological insulators can be easily tuned in both magnitude and duration. Time-resolved angle-resolved photoemission spectroscopy, together with a quantitative model, reveals the ideal conditions for a surface photovoltage in two different topological insulators. Our model predicts that the reported effects are an intrinsic property of topological insulators, as long as the chemical potential falls within the band gap. This work demonstrates that persistent excited topological surface states are photon-accessible and easily tuned in both magnitude and duration, merging photonics- and spintronics-based devices in the same material.
Similar content being viewed by others
Introduction
The surface photovoltage effect (SPV) in conventional semiconductors is a well-known phenomenon used extensibly for semiconductor characterization across a wide range of material properties1,2. The SPV describes an illumination-induced change in the surface potential as a result of photoexcited carriers canceling intrinsic band bending. In conventional semiconductors, broken translational symmetry at the surface results in the formation of in-gap states. As a result, charge redistributes at the surface inducing an internal electric field and subsequent band bending. Upon illumination, charge that was locked in the in-gap states can migrate back to the bulk and cancel the intrinsic field producing a nonequilibrium potential1,2,3. The in-gap surface states of a topological insulator are symmetry-protected and spin-momentum locked inhibiting backward scattering, in contrast to the semiconductor SPV4,5. This translates into a highly conductive surface and generation of radial spin currents weakly affected by disorder.
Utilization of the topological SPV is constrained by the ability to perform operations at speeds comparable to present-day processors (10 GHz), which will require excitation lifetimes on the order of nanoseconds. Despite previous predictions6 and observations of the SPV effect7,8,9,10 in topological insulators, it is still not clear whether this is an intrinsic property of topological materials and under which conditions it can be realized. Moreover, previous reports rely on narrow temporal ranges and an indirect determination of the time constant, which has led to the misinterpretation of the SPV time profile11,12,13. Full characterization of the effect over a larger temporal range is needed in order to directly determine the time constant of the effect.
In this study we reveal that when the prototypical topological insulators Bi2Se3 and Bi2Te3 are doped into the bulk band gap and are pumped with 800 nm light, a strong, long-lived (~10 ns) and tunable SPV effect develops. We show that both the magnitude and timescale of the SPV can be easily manipulated by changing the fluence, making it very appealing for device applications. These findings were made possible by virtue of time-resolved angle-resolved photoemission spectroscopy (TR-ARPES) enabling direct measurement of the chemical potential as a function of pump–probe delay. Furthermore, our study employs a much larger delay range in both negative and positive delay values than any previous SPV study on topological insulators. We argue that the ideal situation to realize such an effect is when the chemical potential falls within the band gap, due to the intricate relation between the existence of a topological surface state, the band bending of the insulating bulk and photoexcited hole trapping in the bulk.
Results
Figure 1 presents transient ARPES spectra showing the distribution of electrons as a function of energy and momentum along a Γ−K cut through the Brillouin zone for the Bi2Se3 bulk-conductive and p-type (bulk Mg-doped) topological insulator. The delay of the probe with respect to the pump is represented by τ. Spectra were taken with ~30 μJ/cm2 incident fluence. As schematically shown in Fig. 1a, d, the Fermi level, EF, of the electron-doped sample intersects the bottom of the bulk conduction band. For the p-type sample (Fig. 1d), EF lies in the bulk gap near the bulk valence (bulk bands in light gray) and the surface bands are shifted down due to band bending. Band bending does not occur in the bulk-conductive samples, since the metallic bulk forbids a nonuniform potential. For the bulk-conductive samples at τ = −100 ps (Fig. 1b), photoemission intensity fills the upper Dirac cone up to the Fermi level. After optical excitation with 1.5 eV photons, states above EF are populated including the bulk conduction band and surface resonance states14. We observe a continuation of the topological surface state linearly dispersing >150 meV above EF. At long enough delay time (50 and 800 ps) the excited states have decayed back to equilibrium and states are populated only up to EF.

a–c presents electron-doped (bulk-conductive) Bi2Se3 data whereas d–f presents p-type data. a, d Cartoons illustrate the surface state (red and blue) and bulk bands (gray) of Bi2Se3 with the dashed box representing the detector energy-momentum window for the respective dopings. Light gray bands in p-type samples are bulk bands deep in the bulk, and surface bands (red, blue, dark gray) are shifted down due to band bending. b, e k-space slices at τ = −100, 0, 50, and 750 ps for bulk-conductive (raw data) and p-type (second derivative in momentum). c, f k-integrated energy spectra as a function of pump–probe delay. All dynamics from pump excitation of bulk-conductive Bi2Se3 (c) occur within one 10-ps pixel at τ = 0. As for p-type (f), the delay scan exhibits a long 300 ps rise before the presence of the pump, and a slow >10 ns decay after τ = 0. The long 1 ns delay scans that reach −200 ps into the negative delay demonstrate that the negative delay chemical potential shift is separate from the positive delay decay process.
To better extract the timescale associated with such a process, in Fig. 1c we show the momentum-integrated spectral intensity across the entire delay range (−200 to 800 ps). A sudden change of intensity following the pump pulse is observed instantly at τ = 0, followed by a decay process that takes less than 10 ps to restore equilibrium. This timescale is in agreement with previous reports14,15,16,17 and has been discussed in terms of a bottleneck of charge in the bulk state moving through the surface state. In contrast, the p-type samples exhibit a much different response to optical pump as can be clearly seen in Fig. 1f. Two main differences are observed: (a) persistence of excitation well after τ = 10 ps; (b) apparent presence of excitation even for negative delay. At τ = 0, we see population 200 meV above EF in both Fig. 1e, f. Strikingly, a nonequilibrium chemical potential persists over a much longer delay window (800 ps), as compared to the bulk-conductive samples (see both panels c and f).
For more than 200 ps preceding pumping, the chemical potential remains above equilibrium EF and rises to ~120 meV to approach the chemical potential at positive delays as shown by the blue dashed line in Fig. 1f. In this case, photoelectrons appear to be affected by a pump pulse that arrives after they have been photoemitted. Previous studies have assumed the chemical potential shift at negative delay is the continuation of the SPV decay from a previous pump pulse, leading to incorrect estimates of the lifetime of the SPV (>4 μs) in similar systems7,8,9,10. However, our work reveals over a larger decay range that the negative time decay process is separate from the relaxation of the SPV and therefore unrelated to the previous pulse. This is confirmed by the chemical potential returning to equilibrium towards more negative delays (as can be inferred from Fig. 1f). A 4 μs timescale cannot explain the reversed decay shape of the negative delay effect. This peculiar behavior is consistent with previous observations of SPV in GaAs11 in which the chemical potential shift at negative delays has been associated with an SPV generated electric field influencing the photoemitted electrons shortly after leaving the sample surface. Therefore, the negative delay signal is due to in-vacuum electromagnetic interactions with the photoelectron and thus the effect is extrinsic to the material. The timescale of this effect is set by the kinetic energy of photoelectrons propagating away from the sample in vacuum and by the pump spot geometry11,12,13.
By considering a much larger range in both negative and positive delay values we are able to correctly and precisely measure the SPV lifetime. We note that the shorter negative delay range used in previous work7,8,9,10 is insufficient for observing the reverse decay profile and the negative delay chemical potential shift cannot be attributed to the effect of previous pump pulses. Turning our attention to the positive delay regime (Fig. 1f), two decay mechanisms can be identified due to the wide delay range covered in this experiment. From a two-exponential fit, we extract a timescale of 159 ps and a remarkably long timescale of 16.5 ns at a fluence of ~30 μJ/cm2.
With the insight that the SPV timescale lasts on the order of 10 ns, we investigate the effects of fluence on the magnitude and decay of the SPV. Figure 2 shows the fluence dependence of the SPV effect. Figure 2a displays Fermi energy fits of the momentum-integrated ARPES spectra at each delay for all measured fluences. The Fermi distribution is well-defined adiabatically for all delays except τ = 0 due to the SPV dynamics being much slower than the typical electron relaxation timescale. The peak excitation at τ = 0 was removed and replaced by the average value of the neighboring points.

a Fermi-edge fits (blue) to delay scan spectra represent the chemical potential as a function of delay at multiple fluences from 0.92 to 27.5 μJ/cm2. The decay scans are fitted best by the sum of two positive amplitude exponentials and one negative exponential term (red dashed). The SPV peak (yellow dots) occur at later delays with decreasing fluence. b The build-up time of the SPV peak log-log plotted as a function of fluence. The data fits best to a 3D diffusion model. Due to discrete delay steps, fluences above 10 μJ/cm2 have peak SPV values at τ = 0 and are not plotted. c SPV magnitude versus fluence as determined by the amplitude of the long ~10 ns timescale in the three-exponential model. The teal dashed line depicts the expected trend of the SPV magnitude from the theory discussed in the text. The inset figure demonstrates the same data on a log scale of fluence. d SPV decay constant versus fluence as determined by the time constant in the long timescale in the three-exponential model. The error bars are conservative estimates determined by reapplying the fits in the worst case scenarios for variation in pump fluence added in quadrature with the respective covariance from the fits. The teal-shaded region represents two standard deviations of slope and y-intercept of the weighted best fit trend line.
Besides the obvious change in magnitude, the most apparent fluence-dependent feature from this dataset is the shift of the SPV peak to more positive delays as fluence decreases (yellow circles), indicating that the nonequilibrium population requires a finite time to build-up at lower fluences. In order to account for the SPV build-up time at low fluences, the chemical potential versus delay curves were best fitted to a summed three-exponential decay model with one decaying exponential exhibiting a negative amplitude capturing the effect of the SPV build-up, as given by eq. (1).
The SPV build-up exponential term takes on a negative amplitude capturing the reduction in a SPV at short delays that would otherwise take on the form of two decaying exponentials alone. It is important to recognize that the time-dependent electric field resulting from the SPV formation affects the positive delay in a similar manner to the negative delay effect. More precisely, the measured chemical potential shift at a given delay is affected by the true chemical potential time profile of the SPV at more positive delays as the photoemitted electron is propagating through a decaying (or growing) electric field. As such, the decay profiles are most affected in delay regions with large time derivatives of the chemical potential shift12,13. Accordingly, the delay regions in which the SPV build-up is dominant are most affected by this phenomenon. This contributes to the nonzero chemical potential shift for the decay curves exhibiting delayed SPV onset. We note that our fit model does not take into account the positive delay effects of the induced time-dependent field but captures the total effect of the SPV build-up, decay, and in-flight time dependence phenomenologically.
We determine the peak SPV onset time (τbuild-up) by the maximum value of the resulting fit function for each fluence. The peak positions are represented by yellow circles in Fig. 2a and are summarized in Fig. 2b. As the pump fluence is decreased below saturation (~10 μJ/cm2), the onset of the SPV peak occurs at later delays reaching as large as ~150 ps near a fluence of 0.2 μJ/cm2. The long timescale associated with the build-up of this surface field is evidence for diffusive dynamics18,19,20. Within a diffusive model, the timescale for diffusion scales as τbuild-up ∝ λ2 21,22 leading to \({\tau }_{{\rm{build}\hbox{-}{up}}}\propto {f}^{\frac{-2}{d}}\) where λ is the mean free path, f is the pump fluence, and d is the degrees of freedom of travel (see Supplementary Fig. 6). The resulting fit for the diffusive models in one, two, and three dimensions is shown as dashed lines in Fig. 2b. By letting d be a free parameter in the fitting, the best fit is obtained for d = 3.18 ± 0.17. This is evidence that the delayed build-up of the SPV is due to slow diffusion in the bulk as opposed to a two-dimensional surface effect.
At high fluences, the negative-amplitude decay is nearly absent and the fit is dominated by a two-exponential model with timescales on the order of 100 ps and 1–10 ns, respectively. The amplitude corresponding to the nanosecond timescale represents the SPV amplitude and is presented in Fig. 2c. The fluence dependence of the SPV magnitude clearly shows a nonlinear trend followed by a quick saturation for fluences above 10 μJ/cm2 up to a maximum of ~125 meV. The energy error bars in Fig. 2c were determined by combining the energy variance of the chemical potential fit at a single delay and the variance of the majority component of the SPV amplitude from the exponential fit.
Figure 2d exhibits the SPV decay time as a function of fluence as measured by the longest timescale in the three-exponential fit. As fluence is decreased the SPV timescale trends to 0, being <1 ns below 0.1 μJ/cm2 and >10 ns above 20 μJ/cm2. The error bars were derived from a conservative consideration of the effect of pump fluctuations on the resulting delay curves added in quadrature to the variances of the long decay fit parameter (see Supplementary Notes for more detail). The teal region represents the best fit trend line using the fluence variation error bars as weights and accounting for all possible trend lines within two standard deviations of the slope and y-intercept. From Fig. 2d, we can conclude that the SPV timescale increases with fluence, reaching 100 ps timescales at low fluences near 0.1 μJ/cm2 and exceeding 10 ns at fluences above 10 μJ/cm2. The approximately linear trend in the semi-log plot implies a logarithm-like dependence of the SPV time constant with fluence.
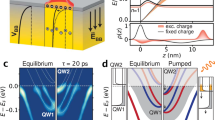
Figure 3 provides a schematic representation of a possible mechanism for the long-lived SPV in p-type Bi2Se3 similar to the reported mechanism from Papalazarou et al.10. Before pumping, the equilibrium conduction and valence band are expected to bend downwards due to surface state population and redistribution of charge from bulk to surface as seen in Fig. 3a. This downward band bending has been confirmed via X-ray photoelectron spectroscopy23 and is consistent with a p-type semiconductor2,24. After excitation, the top portion of the Dirac cone is populated while holes are generated in the bottom cone. Photoexcited electrons above the bulk conduction band and holes below the bulk valence band can quickly disperse into the bulk and recombine until the Fermi level is within the bulk band gap near the crystal surface as shown in Fig. 3b. Once the excited occupied states of the Dirac cone lie within the bulk band gap, the excited electrons can only recombine with the holes in the bottom cone. This process is already unfavorable due to the spin-momentum locking in the topological surface state. Within this time period, the downward band bending encourages electrons from the bulk to migrate to the upper Dirac cone and holes from the bottom Dirac cone to migrate into the bulk. A dipole field due to this charge migration keeps the holes locked a finite distance from the surface. This reduces the spatial overlap of the electron and hole wavefunctions resulting in an even greater enhancement of the photoexcited carrier lifetime. Figure 3c displays how such a dipole localizes holes far from the surface.

a Equilibrium occupation of the bands with intrinsic band bending due to charge in surface states. Upper image demonstrates density of states as a function of distance into the bulk from the surface. Bottom image represents ARPES spectra (mostly surface sensitive). b Immediately after photoexcitation, surface state recombination as well as migration into the bulk of photoexcited carriers results in a fast decay of less than 10 ps15. c However, a small portion of photoexcited holes migrate into bulk due to initial band bending and remain trapped due to their own dipole. Photoexcited holes and electrons are spatially separated and recombination is dramatically reduced. d Temperature-dependent data taken at a delay of 10 ps confirms that Fermi-edge broadening diminishes the SPV effect at high temperatures. The dashed curve is the model fit of the data considering temperature-dependence of the Fermi distribution for the migrating holes. Vertical dashed line is an approximate saturation temperature for the SPV. e Cartoon representation of diminished band bending as a result of thermal broadening of the Fermi-Dirac distribution. The Fermi-Dirac distributions are illustrated to the left of each cartoon. As the temperature increases, thermal carriers populate the conduction and valence bands. The additional carriers allow partial re-equilibration of the band bending field and therefore a smaller allowable surface-bulk dipole.
It is apparent within this description that bulk-conductive samples in which EF intersects the conduction band are incapable of producing an intrinsic SPV. In the metallic regime, the density of states is high both in the bulk and surface inhibiting large changes in chemical potential due to the surface state population. Furthermore, recombination of excited carriers is enhanced since the bulk conduction band and surface state are accessible for all energies above EF. In contrast, the reported band bending in bulk-conductive Bi2Se3 is an extrinsic process attributed to adatoms preferentially doping the surface25,26,27,28 as well as XUV ionization of the adsorbed species29,30,31 explained by the work of Frantzeskakis et al.32.
The lower limit on the length scale of the photoinduced dipole is determined by the penetration depth of the surface state wavefunction. In fact, the c-axis penetration of the surface state wavefunction can be approximated by the minimum thickness in which Bi2Se3 preserves a bulk boundary correspondence. Previous studies have reported the surface state penetration depth from a single Bi2Se3 surface to be two quintuple layers (~2 nm)33,34,35. This is consistent with theoretical models that predict a penetration depth of 1–2 nm36,37. Therefore, in order for the hole-dipole distance to play a large role in the long carrier lifetime, this distance would need to be on the order of 2 nm or greater.
To model the fluence dependence of the SPV magnitude, we study photocarrier dynamics at the surface of a topological insulator by utilizing the rate equation. We consider the recombination of the photoexcited carriers in the surface Dirac band and diffusion of holes into the bulk which leads to surface dipole formation and stabilizes long-lived photocarriers. The rate equations are given by
where Ne, Nh are the electron and hole concentrations in the surface Dirac band, respectively. Nh is the hole concentration a t the top of band bending formed by the excess holes (Nh = Ne − Nh). \({N}_{{\rm{d}}}^{\max }\) is the maximum hole concentration that the band bending can accommodate. αrec is the rate of surface electron and hole recombination and αtun is the rate of hole tunneling into the bulk. The pump pulse creates photoexcited electrons and holes in the surface Dirac bands that evolve in time according to the rate equations. Eventually, Nh(t) goes to zero due to recombination, while Ne(t) and Nh(t) converge into nonzero values which determines the density of the long-lived surface dipole.
From the final value of Ne(t), we determine the chemical potential after photoexcitation, by treating the potential difference induced by the spatially separated charge as a capacitor, such that V = eNe(t)ldip∕ε. We assume an approximate dipole separation length (ldip) of 10 nm, consistent with previous observations of the band bending depth23. The dipole creating the long-lived SPV (in Fig. 3c) is formed by ~1% of the initial photoexcited electrons as represented by the maximum dipole occupancy \({N}_{{\rm{d}}}^{\max }\). The model suggests that the timescale of hole transport into the bulk and recombination at the surface are comparable. Furthermore, the comparison of the values of the rates and the initial excited carrier concentrations affirms that the majority of excited charge has recombined or has been transported to the bulk within a few picoseconds, consistent with the quick relaxation process between τ = 0 and 10 ps in both p-type and bulk-conductive samples.
Despite the rapid recombination process, a finite number of holes still diffuse into the bulk to sustain a prolonged dipole. Applying this model and assuming the rates are fixed, the SPV magnitude as a function of fluence closely matches the behavior of the data as seen by the teal dashed line in Fig. 2c.
We can further check this model by characterizing the temperature dependence of the SPV. As temperature increases, thermal carriers start to occupy the top of the band bending making tunneling to the bulk less efficient. Additionally, increasing temperature mitigates band bending due to the appearance of extra thermal carriers at the surface24,38.
In order to incorporate these effects minimally in our model, we introduced temperature dependence in \({N}_{{\rm{d}}}^{\max }\) as \({N}_{{\rm{d}}}^{\max }\propto f({\epsilon }_{{\rm{d}}}(T))\), where f is the Fermi distribution function and ϵd is the dipole state binding energy. We adopt a phenomenological form for the binding energy, ϵd(T) = 86[meV] − bkBT. The maximal energy of 86 meV is a tenable value for the dipole state binding energy, being smaller than the Bi2Se3 gap. It is reasonable to expect that increased temperature weakens the band bending reducing the binding energy of the dipole state. This temperature dependence of binding energy is phenomenologically incorporated with linear term bkBT. In fact, we can confirm this dependence with experimental data featured in Fig. 3d. The temperature-dependence from our model captures the temperature-induced change of band bending profile and thus the SPV magnitude as shown by the black dashed line in Fig. 3d. The graph displays the chemical potential shift due to the SPV above the saturation fluence as of 10 ps after the pump as a function of temperature. The SPV weakens at higher temperatures and saturates at temperatures below 125 K. Figure 3e illustrates the mitigation of band bending at higher temperature as a result of additional thermal carriers due to Fermi-edge broadening. The additional holes in the bulk valence partially cancel the band bending field. Our model describes an equivalent picture in that more holes in the bulk valence band at equilibrium result in less room for migrating holes after photoexcitation.
The model described above suggests that the observed SPV might represent a more general property of TIs and provides a general recipe for the effect: chemical potential within the gap and strong in-gap surface states that enable a charge redistribution at the surface. All appropriately doped topological insulators satisfy this condition by definition39,40,41. To test this idea we have studied Sn-doped Bi2Te3 when the Fermi energy lies in the gap nearest the bulk conduction band (n-type) as presented in Fig. 4. Upon pumping an SPV effect, lasting beyond the delay range (>400 ps) is observed (see Fig. 4b, c). In contrast to the p-type samples (shown in Fig. 1), the surface bands in n-type samples are expected to bend upward, resulting in an SPV with the chemical potential shifting downward upon illumination2,3,24. This can be observed clearly in Fig. 4b, where the Dirac node location is marked in black and the Fermi level is represented in white. The bands rigid shift downward due to the 800 nm excitation. Near τ = 0, the hot-carrier recombination is visible as a peak above the Fermi level, lasting ~20 ps, comparable to the bulk-conductive case (see Supplementary Fig. 3). As for the p-type sample, the decay profile for negative delays points to an extrinsic in-vacuum effect on the photoelectrons. The presence of the SPV in a second topological insulator with a different chemical dopant demonstrates the attainability of this effect and the possible universality among the topological insulators.

a Cartoon depiction of Bi2Te3 band structure and momentum-energy detection window. Bands at the surface (dark gray, red, and blue) are shifted upward by band bending. b k-integrated energy spectrum as a function of pump-probe delay, Dirac node position and chemical potential are indicated by the black dotted line and white line, respectively. This time, the chemical potential dips downward towards τ = 0, returning to equilibrium beyond 400 ps. c k-space slices at τ = −100, 0, 50, and 400 ps illustrating the downward chemical potential shift in both the positive and negative delays.
Discussion
Our proposed model describing photoexcited hole migration and subsequent dipole trapping matches the SPV response in magnitude versus fluence. Furthermore, the introduction of Fermi-edge broadening to the model captures the temperature response and the consideration of bulk diffusion dynamics describes the delayed onset of the SPV. However, the model does not take into account decay mechanisms for the nonequilibrium dipole corresponding to the two timescales of the long SPV lifetime. Diffusive spreading of the dipole on the surface is expected and could explain one of these timescales. Further time-resolved measurements with spatial resolution may elucidate the diffusion rates of surface and bulk excited carriers. Understanding the dipole decay mechanisms will be crucial to engineering a topological SPV system with even longer lifetimes.
The presence of a topological SPV in TIs opens up new avenues for applications of the SPV given its unique traits from a conventional SPV such as a spin-momentum locked diffusion current at the surface that inherits forbidden backscattering, i.e. ballistic transport4,5. Moreover, the robustness of the topological surface state to contamination ensures an environmentally stable SPV42. As the catalog of known topological insulators expands, time-resolved measurements will determine whether the SPV is truly an inherent property of topological insulators and expound on universal behaviors of the topological SPV.
In order to exploit the topological SPV for spintronics applications, long-range spin transport as well as spin manipulation need to be confirmed and developed. We suggest further observation that surface charge is indeed transported far from the region of photoexcitation. This can be resolved via a pump−probe experiment with a spatially resolved probe rastering outside the pumped region. Furthermore, studies are needed to investigate whether optical techniques can be used to manipulate the spin of these long-lived carriers. By weakly breaking time-reversal symmetry such that the system is still topologically nontrivial, one may be able to selectively create decay channels for one spin branch and not the other paving way for long-lived spin current manipulation in topological materials.
Conclusion
A decade of academic excitement in topological insulators has resulted in few devices and applications. This is partly due to a lack of techniques capable of manipulating the behavior of topological insulators. In this study, we have revealed a large amplitude SPV with a long lifetime on the order of 10 ns. Furthermore, we demonstrate wide range control of the amplitude, timescale, and location of the SPV. The amplitude and lifetime of the SPV in p-type Bi2Se3 exhibit logarithm-like tunability via fluence. We can manipulate the SPV lifetime over a wide range, from approximately 100 ps to 10 ns. Not only can we control the amplitude and lifetime of the SPV, but we can selectively excite the spin-momentum locked bands spatially with localized illumination. In practice, diffusion of the surface charge from a pump geometry of choice can result in complex spatially dependent spin transport varying both in magnitude, lifetime, and spin direction. Such extensive manipulation opens up the phase space for new and exciting spintronic devices.
Methods
The Bi2Se3 single crystals were grown via directional slow solidification with ≈1 at. % Mg substitution with bismuth to achieve a Fermi energy in the bulk gap. The Bi2Te3 crystals were grown through the same process with Sn-doping of ≈0.3 at. % with respect to bismuth. The crystals were cleaved before measurement along the (111) plane at a pressure of 5 × 10−11 Torr to ensure a clean surface for ARPES. Fluence-dependent measurements were acquired at the base temperature of our system with liquid nitrogen, ~80 K. Temperature-dependent measurements were taken using a resistive heater and thermal probe close to the sample stage.
The Bi2Se3 sample is pumped with 1.5 eV photons generated by a cavity-dumped Ti:sapphire oscillator operating with a repetition rate of 500 kHz. It is then probed with 6 eV photons generated by frequency quadrupling in two stages of BBO crystals. The delay between pump and probe pulses is controlled by a mechanical translation stage that varies the pump path length, with τ = 0 referring to temporal overlap of the two pulses and positive delays referring to the probe pulse arriving after the pump. The pump and probe beam spot full-width-half-maxes on the sample were 127 and 80 μm respectively. The pump beam drifted no more than 20 μm across the entire range of the delay stage determined via CCD camera43. In order to achieve fluence-dependent measurements, the pump power was filtered to achieve fluences in the range of 0.09−27.5 μJ/cm2. All reported fluence values are for incident fluence on the Bi2Se3 surface.
Both the pump and probe were p-polarized incident on the sample. The equilibrium electronic population is imaged with the pump blocked in order to establish the equilibrium Fermi level, when only the bulk valence band and the bottom of the upper topological surface state Dirac cone are populated. With the pump exposed, pump-induced excited states above EF are imaged at controllable delay times before and after excitation.
Data availability
The datasets collected and analysis performed by this study are available from the corresponding author upon reasonable request.
References
Schroder, D. K. Surface voltage and surface photovoltage: history, theory and applications. Meas. Sci. Technol. 12, R16 (2001).
Zhang, Z. & Yates, J. T. Band bending in semiconductors: chemical and physical consequences at surfaces and interfaces. Chem. Rev. 112, 5520–5551 (2012).
Kronik, L. & Shapira, Y. Surface photovoltage phenomena: theory, experiment, and applications. Surf. Sci. Rep. 37, 1–206 (1999).
Hsieh, D. et al. A tunable topological insulator in the spin helical dirac transport regime. Nature 460, 1101–1105 (2009).
Hasan, M. Z. & Kane, C. L. Colloquium: topological insulators. Rev. Mod. Phys. 82, 3045–3067 (2010).
Fregoso, B. M. & Coh, S. Intrinsic surface dipole in topological insulators. J. Phys.: Condens. Matter 27, 422001 (2015).
Neupane, M. et al. Gigantic surface lifetime of an intrinsic topological insulator. Phys. Rev. Lett. 115, 116801 (2015).
Sánchez-Barriga, J. et al. Laser-induced persistent photovoltage on the surface of a ternary topological insulator at room temperature. Appl. Phys. Lett. 110, 141605 (2017).
Yoshikawa, T. et al. Enhanced photovoltage on the surface of topological insulator via optical aging. Appl. Phys. Lett. 112, 192104 (2018).
Papalazarou, E. et al. Unraveling the dirac fermion dynamics of the bulk-insulating topological system Bi2Te2Se. Phys. Rev. Mater. 2, 104202 (2018).
Yang, S.-L., Sobota, J. A., Kirchmann, P. S. & Shen, Z.-X. Electron propagation from a photo-excited surface: implications for time-resolved photoemission. Appl. Phys. A 116, 85–90 (2013).
Ulstrup, S. et al. Ramifications of optical pumping on the interpretation of time-resolved photoemission experiments on graphene. J. Electron Spectrosc. Relat. Phenomena 200, 340–346 (2015).
Tanaka, S.-I. Utility and constraint on the use of pump-probe photoelectron spectroscopy for detecting time-resolved surface photovoltage. J. Electron Spectrosc. Relat. Phenomena 185, 152–158 (2012).
Jozwiak, C. et al. Spin-polarized surface resonances accompanying topological surface state formation. Nat. Commun. 7, 13143 (2016).
Sobota, J. A. et al. Ultrafast optical excitation of a persistent surface-state population in the topological insulator Bi2Se3. Phys. Rev. Lett. 108, 117403 (2012).
Cacho, C. et al. Momentum-resolved spin dynamics of bulk and surface excited states in the topological insulator Bi2Se3. Phys. Rev. Lett. 114, 097401 (2015).
Hajlaoui, M. et al. Ultrafast surface carrier dynamics in the topological insulator Bi2Te3. Nano Lett. 12, 3532–3536 (2012).
Tian, W., Zhao, C., Leng, J., Cui, R. & Jin, S. Visualizing carrier diffusion in individual single-crystal organolead halide perovskite nanowires and nanoplates. J. Am. Chem. Soc. 137, 12458–12461 (2015).
Wang, Y., Zhi, M. & Chan, Y. Delayed exciton formation involving energetically shallow trap states in colloidal CsPbBr3 quantum dots. J. Phys. Chem. C 121, 28498–28505 (2017).
Dittrich, T., Mora-Seró, I., García-Belmonte, G. & Bisquert, J. Temperature dependent normal and anomalous electron diffusion in porous TiO2 studied by transient surface photovoltage. Phys. Rev. B 73, 045407 (2006).
Bazant, M. Z., Thornton, K. & Ajdari, A. Diffuse-charge dynamics in electrochemical systems. Phys. Rev. E 70, 021506 (2004).
Biesheuvel, P. M., Fu, Y. & Bazant, M. Z. Diffuse charge and faradaic reactions in porous electrodes. Phys. Rev. E 83, 061507 (2011).
ViolBarbosa, C. E. et al. Direct observation of band bending in the topological insulator Bi2Se3. Phys. Rev. B 88, 195128 (2013).
Zhang, H. M., Sakamoto, K., Hansson, G. V. & Uhrberg, R. I. G. High-temperature annealing and surface photovoltage shifts on Si(111)7 × 7. Phys. Rev. B 78, 035318 (2008).
Bianchi, M., Hatch, R. C., Mi, J., Iversen, B. B. & Hofmann, P. Simultaneous quantization of bulk conduction and valence states through adsorption of nonmagnetic impurities on Bi2Se3. Phys. Rev. Lett. 107, 086802 (2011).
Benia, H. M., Lin, C., Kern, K. & Ast, C. R. Reactive chemical doping of the Bi2Se3 topological insulator. Phys. Rev. Lett. 107, 177602 (2011).
Zhu, Z.-H. et al. Rashba spin-splitting control at the surface of the topological insulator Bi2Se3. Phys. Rev. Lett. 107, 186405 (2011).
Lin, C.-Y. et al. Orbital-dependent spin textures in Bi2Se3 quantum well states. Phys. Rev. B 98, 075149 (2018).
Kordyuk, A. A. et al. Photoemission-induced gating of topological insulators. Phys. Rev. B 83, 081303 (2011).
Jiang, R. et al. Reversible tuning of the surface state in a pseudobinary Bi2(Te-Se)3 topological insulator. Phys. Rev. B 86, 085112 (2012).
Frantzeskakis, E. et al. Dirac states with knobs on: interplay of external parameters and the surface electronic properties of three-dimensional topological insulators. Phys. Rev. B 91, 205134 (2015).
Frantzeskakis, E. et al. Trigger of the ubiquitous surface band bending in 3D topological insulators. Phys. Rev. X 7, 041041 (2017).
Wang, Z. et al. Dimensional crossover and topological nature of the thin films of a three-dimensional topological insulator by band gap engineering. Nano Lett. 19, 4627–4633 (2019).
Wu, L. et al. A sudden collapse in the transport lifetime across the topological phase transition in (Bi1−xInx)2Se3. Nat. Phys. 9, 410–414 (2013).
Zhang, Y. et al. Crossover of the three-dimensional topological insulator Bi2Se3 to the two-dimensional limit. Nat. Phys. 6, 584–588 (2010).
Zhang, W., Yu, R., Zhang, H.-J., Dai, X. & Fang, Z. First-principles studies of the three-dimensional strong topological insulators Bi2Te3, Bi2Se3 and Sb2Te3. N. J. Phys. 12, 065013 (2010).
Pertsova, A. & Canali, C. M. Probing the wavefunction of the surface states in Bi2Se3 topological insulator: a realistic tight-binding approach. N. J. Phys. 16, 063022 (2014).
Kronik, L. & Shapira, Y. Surface photovoltage spectroscopy of semiconductor structures: at the crossroads of physics, chemistry and electrical engineering. Surf. Interface Anal. 31, 954–965 (2001).
Fu, L., Kane, C. L. & Mele, E. J. Topological insulators in three dimensions. Phys. Rev. Lett. 98, 106803 (2007).
Moore, J. E. & Balents, L. Topological invariants of time-reversal-invariant band structures. Phys. Rev. B 75, 121306 (2007).
Chen, Y. L. et al. Experimental realization of a three-dimensional topological insulator, Bi2Te3. Science 325, 178–181 (2009).
Liu, Z. et al. Robust topological surface state against direct surface contamination. Phys. E 44, 891–894 (2012).
Smallwood, C. L., Jozwiak, C., Zhang, W. & Lanzara, A. An ultrafast angle-resolved photoemission apparatus for measuring complex materials. Rev. Sci. Instrum. 83, 123904 (2012).
Acknowledgements
We thank C.H. Stansbury for developing the ARPES analysis software. Both the experimental and theoretical part of this work were primarily supported by the Director, Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division, of the U.S. Department of Energy, under Contract No. DE-AC02-05CH11231, as part of the Ultrafast Materials Science Program (KC2203). S.C. was supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE1852814 and DGE1106400. T.M. was supported by the EPiQS Initiative of the Gordon and Betty Moore Foundation through Grant No. GBMF4537. J.G.A. acknowledges support from the National Science Foundation under Grant No. 1607753. A.L. acknowledges support from the Gordon and Betty Moore Foundation’s EPiQS grant GBMF4838 that has enabled this research.
Author information
Authors and Affiliations
Contributions
S.C., R.M., and K.G. devised the experiment and carried out the data collection. S.C. analyzed the experimental data. Theory and calculations were performed by T.M. and J.E.M. Samples were prepared by J.G.A. Experimental planning and infrastructure were provided by A.L. and Z.H. All authors contributed to the interpretation and writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ciocys, S., Morimoto, T., Mori, R. et al. Manipulating long-lived topological surface photovoltage in bulk-insulating topological insulators Bi2Se3 and Bi2Te3. npj Quantum Mater. 5, 16 (2020). https://doi.org/10.1038/s41535-020-0218-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41535-020-0218-4
This article is cited by
-
Ultrafast enhancement of electron-phonon coupling via dynamic quantum well states
Communications Materials (2023)
-
Driving ultrafast spin and energy modulation in quantum well states via photo-induced electric fields
npj Quantum Materials (2022)
-
Optical manipulation of Rashba-split 2-dimensional electron gas
Nature Communications (2022)
