
Abstract
Background
Saccharopolyspora erythraea (S. erythraea) is a Gram-positive bacterium widely used for the production of erythromycin, a potent macrolide antibiotic. However, the mechanism behind erythromycin production is poorly understood. In the high erythromycin-producer strain S. erythraea HL3168 E3, the level of copper ions positively correlates with erythromycin production. To explain this correlation, we performed a genome-based comparison between the wild-type strain NRRL23338 and the mutant strain HL3168 E3, and further characterized the identified gene(s) by targeted genome editing, mRNA transcript analysis, and functional analysis.
Results
The response regulator of the two-component system (TCS) encoded by the gene SACE_0101 in S. erythraea showed high similarity with CopR of TCS CopRS in Streptomyces coelicolor, which is involved in the regulation of copper metabolism. The deletion of SACE_0101 was beneficial for erythromycin synthesis most likely by causing changes in the intracellular copper homeostasis, leading to enhanced erythromycin production. In addition, Cu2+ supplementation and gene expression analysis suggested that SACE_0101 may be involved in the regulation of copper homeostasis and erythromycin production.
Conclusions
The mutation of SACE_0101 gene increased the yield of erythromycin, especially upon the addition of copper ions. Therefore, the two-component system gene SACE_0101 plays a crucial role in regulating copper homeostasis and erythromycin synthesis in S. erythraea.

Similar content being viewed by others


Introduction
Saccharopolyspora erythraea is used mainly at an industrial scale to produce erythromycin, an important broad-spectrum macrolide antibiotic used for industrial and clinical applications. The mechanism on how S. erythraea produces high-yield erythromycin is poorly understood, hindering its rational optimization. In 2007, the genome of S. erythraea NRRL23338 was published providing the necessary resources to fully understand the mechanism of erythromycin synthesis. This development may also provide the basis for the analytical research related to the directional modification of erythromycin-producing strains (Oliynyk et al. 2007).
The development of the next-generation sequencing method facilitates the rational optimization of industrial strains through pathway engineering or signal transduction analysis, such as reverse engineering. Through genomic alignment, we can easily identify point mutations, gene insertions or deletions, as well as other genetic recombination events that directly lead to genomic diversity (Couronne et al. 2003). Extensive genetic studies on high erythromycin producers are beneficial to elucidate the mechanism of high yield production and to provide potential molecular targets. Previous genome comparison between NRRL23338 and erythromycin-overexpressing strain Px revealed several mutations in the coding regions of the latter strain, especially in key enzyme genes that are involved in central carbon metabolism, nitrogen metabolism, and biosynthesis of secondary metabolites. Notably, several mutations were also found in genes with transcriptional and translational (global) regulatory functions that might explain the overproduction of erythromycin in Px strain (Peano et al. 2012).
Essentially, most genes involved in global regulations belong to the family of two-component system (TCS). In prokaryotes, TCS is the most important signal transduction system that functions as sensing machinery to environmental stimuli. TCS plays a significant role in the growth and regulation of antibiotic synthesis in Streptomyces (Bijlsma and Groisman 2003). Normally, TCS consisted mainly of histidine kinase (HK) and response regulator (RR). While HK senses specific environmental stimuli, the RR mediates the response through transcriptional regulation of the corresponding target genes (Mascher et al. 2016). For instance, TCS CopRS is closely related to the regulation of heavy metal copper ions in several bacteria. González-Quiñónez et al. (2019) proposed a model describing the relationship between cytosolic copper and Streptomyces morphogenesis. The model suggests that copper is restricted to the differentiation (aerial mycelium and sporulation) and secondary metabolism while the vegetative growth proceeds even under a strongly limited cytosolic copper concentration. When it comes to erythromycin fermentation, the addition of copper ions to the culture of a high erythromycin-producing strain at the beginning of the fermentation process produces a 24% increase in erythromycin yield (Liu et al. 2005).
The whole genome of S. erythraea HL3168 E3 (hereafter indicated as E3), a high erythromycin producer, was sequenced to potentially explain the genome-level mechanism behind the high yield production of erythromycin (unpublished work). E3 was previously obtained by performing multiple rounds of random mutagenesis and screening based on the wild-type strain. In contrast to the wild-type (WT) strain NRRL23338, the genome of E3 presents 255 single-nucleotide variations, 2 insertion sites, and 3 deletion sites. Among the non-synonymous mutations within the coding sequences (CDSs), the majority of mutations are located in the genes involved in central metabolism, followed by genes coding for transcriptional regulatory factors and translational components. In this study, we identified SACE_0101 as one of the genes with point mutation in the E3 strain (Additional file 1: Figure S1), and its DNA sequence showed high homology to CopR in S. coelicolor. We also found that SACE_0101 plays a role in the regulation of copper homeostasis and that intracellular copper homeostasis is crucial for high erythromycin yield in the E3 strain. To the best of our knowledge, this is the first report that demonstrates the role of a two-component system in copper regulation and erythromycin production in S. erythraea.
Materials and methods
Strains and media
Strains and plasmids used or constructed in this study are listed in Table 1. The wild-type strain S. erythraea NRRL23338 [purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ)] was used as the parental strain for mutant strain construction. YS medium was used for sporulation, and its preparation contained the following per liter: 3 g yeast extract, 5 g tryptone, 5 g sucrose, and 20 g agar. Escherichia coli DH5α was used for cloning. E. coli ET12567 was used for obtaining unmethylated DNA for transformation into S. erythraea. E. coli was grown at 37 °C on Luria–Bertani (LB) medium supplemented with the required antibiotics (50 μg/mL apramycin, 50 μg/mL kanamycin, 25 μg/mL chloramphenicol). Conjugal transfer of S. erythraea was performed on ISP4 solid medium (Hopwood 1985). Antibiotics (15 μg apramycin, 40 μg kanamycin, and 25 μg nalidixic acid) were overlaid on ISP4 plates for conjugators’ screening. The modified minimal liquid medium contained the following per liter: 20 g (NH4)2SO4, 50 g casamino acids (Difco™, BD), 0.6 g MgSO4·7H2O, 1 g ZnSO4·7H2O, 1 g FeSO4·7H2O, 1 g MnCl2·4H2O, 1 g CaCl2, and 15 mL NaH2PO4/K2HPO4 buffer (0.1 M, pH = 6.8) was added in 1 L liquid medium. After autoclaving, 20 g glucose was added into 1 L medium as a sole carbon source (Hodgson. 1982). Different concentrations of CuSO4 were also added in the medium as a growth factor.
Construction of mutant in S. erythraea
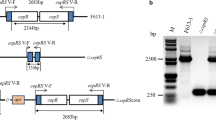
The schematic of the generated plasmid is shown in Fig. 1. The plasmid template pOJ260-Δ0101 was used to replace the target gene SACE_0101. The upstream (amplified with primers 0101UF and 0101UR) and downstream fragments (amplified with primers 0101DF and 0101DR) of the gene SACE_0101 were amplified starting from the 5′ homologous overlap sequences. The amplified products were inserted into the pOJ260 plasmid through HindIII and EcoRI restriction sites [Gibson Assembly® 1-Step Kit (NEB)]. Diagnostic PCR was performed to validate the correct insert (Additional file 1: Figure S2). The recombinant plasmid pOJ260-Δ0101 was transformed into E. coli ET12567 and then successfully introduced into S. erythraea by intergeneric conjugation (Hou et al. 2008). The pOJ260 is a suicide-type plasmid that could not replicate in S. erythraea. When the plasmid is integrated into the genome of the host, the kanamycin-resistant gene (neo gene) integrated in the plasmid is also expressed; hence, the successfully recombined strain could be selected by the resistance marker. SACE_0101-inactivation mutant strain (WT-Δ0101) was confirmed by PCR with 4 pairs of primers (Fig. 2a). All the primers used in this study are shown in Table 2.

Construction of recombinant plasmid pOJ260-Δ0101. The upstream and downstream of the gene SACE_0101 are inserted into HindIII and EcoRI site of the pOJ260 vector, respectively

a Genotype verification primers. PCR product would be 650 bp by the primers 0101C1 and 5DC3 when genomic DNA of WT-Δ0101 was used as the template while WT was 0 bp. PCR product would be 470 bp by the primers 5DCF and 0101C2 when genomic DNA of WT-Δ0101 was used as the template while WT was 0 bp. PCR product would be 1250 bp by the primers 0101C1 and 0101C2 when genomic DNA of WT-Δ0101 was used as the template while WT was 1120 bp. b Genotype verification for SACE_0101 gene inactivation in mutant strain WT-Δ0101. Lane 1: PCR product (650 bp) by the primers 0101C1 and 5DC3 when genomic DNA of WT-Δ0101 was used as the template. Lane 2: PCR product (0 bp) by the primers 0101C1 and 5DC3 when genomic DNA of wild type (WT) was used as the template. Lane 3: PCR product (470 bp) by the primers 5DCF and 0101C2 when genomic DNA of WT-Δ0101 was used as the template. Lane 4: PCR product (0 bp) by the primers 5DCF and 0101C2 when genomic DNA of WT was used as the template. Lane 5: PCR product (1250 bp) by the primers 0101C1 and 0101C2 when genomic DNA of WT-Δ0101 was used as the template. Lane 6: PCR product (1120 bp) by the primers 0101C1 and 0101C2 when genomic DNA of WT was used as the template. Lane 7: DL 2000 marker. Lane 8: PCR product (760 bp) by the primers neoF and neoR when genomic DNA of WT-Δ0101 was used as the template. Lane 9: PCR product (0 bp) by the primers neoF and neoR when genomic DNA of WT was used as the template
Determination of growth parameter
All fermentation cultures were cultivated at 34 °C while shaking (220 rpm). All experiments were performed in 3 replicates to normalize for sample variation. Growth curve was determined by measuring the absorbance at OD600. A total of 1 mL culture was collected at different time-points, washed three times with ultrapure water, and resuspended to a final volume of 1 mL, except in the case of high biomass where an appropriate dilution was adapted accordingly. Diethyl pyrocarbonate (DEPC)-treated water was used as a control, and the absorbance at OD600 was measured after shaking for 30 s. Erythromycin production was determined using a phosphoric acid colorimetry measurement (Zou et al. 2009).
Gene function prediction
The Basic Local Alignment Search Tool (BLAST) was used to explore similarities of the DNA sequence of interest with the available data from the National Center for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov/) (Hoshino et al. 1999; Altschul et al. 1990). We performed a tblastn (protein-translated nucloetide) analysis of the TCS SACE_0101/0102 using the genome of S. coelicolor as a reference.
Real-time quantitative polymerase chain reaction (RT-qPCR)
RT-qPCR was used to analyze the relative transcript levels of target genes. The 16srRNA gene in S. erythraea was used as the positive internal control. RT-qPCR was performed in 25 μL mixtures using a CFX96™ real-time PCR detection system (Bio-Rad, Hercules, CA, USA) (Lu et al. 2007; Rozas et al. 2012; Yu et al. 2012). After optimizing the Tm value and Ct value, each sample was analyzed in three replicates. The transcript levels of target genes were expressed as fold change relative to 16srRNA transcript level (set to 1.0). The relative transcript levels were calculated using the 2−ΔΔCT method (Schmittgen and Livak 2007).
Results
Functional prediction of TCS SACE_0101/0102
We conducted a preliminary study on the function of TCS gene SACE_0101, a point mutant gene found in E3. After homologous alignment, the DNA sequence of SACE_0101 showed 64.22% similarity to a response regulator TCS gene involved in the regulation of intracellular heavy metal ions in S. coelicolor (Additional file 1: Table S1). Simultaneously, we analyzed another TCS gene (SACE_0102) that was mutated in another high-yield S. erythraea strain called HL1075 E4. We observed that SACE_0102 showed high homology (48.19%) with histidine kinase (HK), a gene encoding for an enzyme involved in S. coelicolor copper metabolism (Additional file 1: Table S2). Based on these findings, we hypothesized that SACE_0101 and SACE_0102 function as a TCS involving copper homeostasis in S. erythraea.
Phenotypes and antibiotic production of WT-Δ0101 and the wild type (WT)
Diagnostic PCR showed that we successfully constructed a SACE_0101-disrupted mutant of S. erythraea (hereafter indicated as WT-Δ0101) (Fig. 2b). In comparison to the wild-type (WT) strain, the WT-Δ0101 exhibited no visible morphological alteration (as the top side of the plate showed) and displayed pigment production (as the bottom side of the plate showed) when cultured for 5 days on YS medium (Additional file 1: Figure S3).
The erythromycin production of the WT-Δ0101 was higher than the WT throughout the whole fermentation process, although the growth of the two strains showed no significant difference (Fig. 3a). The yield of erythromycin in WT-Δ0101 (61.45 μg/mL) increased by 2.1-fold compared with that in WT strain (29.30 μg/mL), indicating that the deletion of this gene was beneficial to erythromycin synthesis (Fig. 3b).

Time course of OD600 (a) and erythromycin production (b) in wild-type strain WT (white square) and mutant strain WT-Δ0101(square with upper right to lower left fill)
Influence of Cu2+ on erythromycin production of WT, E3, and WT-Δ0101
In the previous comparison between WT and E3 genome, we found that the mutant gene SACE_0101 was associated with copper homeostasis. To identify whether Cu2+ could affect the yield of erythromycin in WT, E3, and WT-Δ0101, we added different Cu2+ concentrations (0–2.0 mmol/L, final concentration) and monitored the growth of these strains. The effect of Cu2+ on the cell growth of WT is shown in Fig. 4. The addition of Cu2+ showed no significant effect on the cell growth at lag stage (10 h before); however, 0.4, 0.6, and 1.0 mmol/L could slightly promote the growth in the logarithmic phase. Of note, the growth of WT was completely inhibited when the concentration reached 2.0 mmol/L. Based on the results, we selected 0.4 mmol/L concentration to examine the effect of Cu2+ on erythromycin synthesis.

Influence of different Cu2+ concentrations on the growth of WT
The addition of 0.4 mmol/L Cu2+ to the medium when cells were grown to 36 h increased the yield of erythromycin by 16.73% in the E3 strain (from 623.856 to 728.236 μg/mL), but not in WT (from 29.1 to 27.6 μg/mL). When 0.4 mmol/L Cu2+ was added to the medium for 36 h, the erythromycin production in WT-Δ0101 increased by 33.49% (from 61.95 to 82.70 μg/mL) compared to the control culture (no additional Cu2+) (Fig. 5).

The relative increase in erythromycin production in strains WT, E3, and WT-Δ0101 after adding Cu2+. WT showed no visible change, while E3 and WT-Δ0101, respectively, increased 16.73% and 33.49%
Determination of the relative transcript levels of copper-related genes in WT and WT-Δ0101
To maintain the copper homeostasis, cytosolic copper is precisely regulated by resistance proteins. The two-component system was previously confirmed as the key regulatory system for the extra-cytoplasmic sensing of elevated copper ion and can regulate the expression of related genes to elicit adaptive responses to changes in the concentration of copper (Chaplin et al. 2015; Singh et al. 2014). Here, we found a total of 8 genes in the genome of S. erythraea NRRL23338 that are potentially involved in copper metabolism. The Gene IDs and gene functions are listed in Table 3.
RT-qPCR was performed to determine the relative transcript levels of these genes in WT and WT-Δ0101 with or without adding Cu2+. Results showed that the relative transcript levels of 5 genes were extremely low; these genes were SACE_0073, SACE_0677, SACE_3010, SACE_3011, and SACE_6621. The other 3 genes, i.e., SACE_0528, SACE_1321, and SACE_2475, showed a significant level of mRNA transcripts (Fig. 6).

Evaluation of the transcript levels of three genes SACE_0528, SACE_1321, and SACE_2475 by RT-qPCR of the control strain WT (white square), WT cultured with 0.4 mmol/L Cu2+ (square with upper right to lower left fill), WT-Δ0101 (square with diagonal crosshatch fill), and WT-Δ0101 cultured with 0.4 mmol/L Cu2+ (square with horizontal fill). Genes expression in each sample were calculated as the fold expression ratio after normalization to the 16srRNA transcript as a reference control. The values are averages of three independent RT-qPCR experiments; error bars indicate standard deviation
The gene SACE_0528 putatively encodes a copper homeostasis protein CutC. In WT, the relative transcript level of this gene increased slightly after adding Cu2+. On the other hand, the inactivation of SACE_0101 doubled the transcript level of SACE_0528, which was much higher after the addition of Cu2+. In bacteria, there are two separate systems responsible for maintaining intracellular copper balance, i.e., Cop and Cut family proteins (Mauricio et al. 2011). Indeed, our results indicate that CutC (encoded by SACE_0528) may be involved in regulating intracellular copper ions as CopR (encoded by SACE_0101) in S. erythraea, and that SACE_0101 may act as a response regulator affecting the transcript level of SACE_0528. The gene SACE_1321 is presumed to encode a copper-resistant protein. The transcription of SACE_1321 increased by 1.5-fold after the addition of Cu2+ to the medium of WT. As for WT-Δ0101, the transcription level of SACE_1321 hardly changed regardless of whether or not Cu2+ was added to the medium. Our results further showed that SACE_0101 partially responded to extracellular copper concentration by directly regulating the expression of gene SACE_1321.
The gene SACE_2475, which encodes a multicopper oxidase that oxidizes Cu+ to Cu2+, showed an increase level of mRNA transcripts in WT and WT-Δ0101 after the addition of Cu2+ to the medium. The required amount of copper oxidase decreased with increasing Cu2+. As shown in Fig. 6, when SACE_0101 was disrupted in WT-Δ0101, the transcriptional level of SACE_2475 decreased significantly in the presence of Cu2+ due to its noninvolvement in the regulation to Cu2+.
The CopS in the CopRS two-component system could detect copper ions in vitro and bind to it (Joaquin et al. 2012). CopS is activated to induce CopR and regulates the expression of a series of genes on the genome. Based on RT-qPCR results, we proposed a simple model of copper homeostasis mechanism regulated by SACE_0101/SACE_0102 in S. erythraea (Fig. 7). The protein encoded by SACE_0102 (similar to CopS) recognizes the extracellular Cu2+, then autophosphorylates and phosphorylates the response regulator (encoded by SACE_0101, similar to CopR), thus affecting or regulating the transcriptional level of three downstream genes SACE_0528, SACE_1321, and SACE_2475.

Simple model of copper homeostasis mechanism in S. erythraea. SACE_0528 and SACE_1321 were up-regulated with the deletion of SACE_0101, while SACE_2475 down-regulated. The protein encoded by SACE_0102 recognizes extracellular Cu2+, followed by autophosphorylation and phosphotransfer to the response regulator (encoded by SACE_0101). Phosphorylated regulator affects or regulates the transcriptional level of three downstream genes. This resulted in a transcriptional activation of SACE_0528 and SACE_1321 and inactivation of SACE_2475
Because of copper’s ability to cycle between Cu2+ and Cu+ at biologically relevant redox potentials, it has become a cofactor for over 30 known enzymes in organisms. Prominent examples are cytochrome c oxidase as a terminal electron acceptor of the respiratory chain, and superoxide dismutase, which is required for defense against oxidative damage (Worrall and Vijgenboom 2010). In this study, the transcriptional regulation of several genes by Cu2+ has been identified; however, its effect on the relevant enzyme activity remains unknown.
Discussion
Saccharopolyspora erythraea is a significant industrial producer of broad-spectrum macrolide antibiotic erythromycin. In recent years, many efforts have been made to explore the mechanism of high-yield erythromycin producer (Carata et al. 2009; Marcellin et al. 2013; Li et al. 2013). One of these mechanisms may include the two-component system (TCS), which is one of the most important signal transduction systems in Streptomyces. However, TCS is hardly investigated in S. erythraea. There are 84 putative histidine kinase genes and 80 response regulator genes in the genome of S. coelicolor, and 67 typical TCSs are known (Bentley et al. 2002). Several of them have been identified to affect the regulation of antibiotic biosynthesis, such as RapA1A2, PhoRP, and AfsQ1Q2. Despite knowing that copper plays a critical role in the developmental cycle of Streptomyces, the detailed information remains lacking on the molecular and genetic level, especially regarding the function and regulation of copper (Solioz et al. 2010).
The availability of the whole genome sequence of S. erythraea provided the opportunity to deeply investigate the molecular mechanism that controls the production of erythromycin. By comparing the mutant strain E3 with the parental strain NRRL23338, we found that their genomes are highly conserved, including the GC content, gene classification, and core gene types. Genes that code for transcriptional regulatory factors accounted for a large proportion of the mutant genes.
Since copper ions would increase the erythromycin production in E3 and that SACE_0101 was identified as one of the positive mutant TCS genes in this strain, we hypothesized that the regulation of copper homeostasis could be associated with erythromycin synthesis. Our study on the function of SACE_0101 indicated that this two-component system gene played a crucial role in regulating copper homeostasis in S. erythraea. Therefore, deleting SACE_0101 would result in the dysregulation of copper homeostasis, which then leads to the failure of completely regulating Cu2+. This phenomenon might lead to Cu2+ overflow in other pathways that are related to erythromycin synthesis. Cytochrome bc1–aa3 supercomplex, one of the terminal oxidases during aerobic respiration that transfers electrons from menaquinol to oxygen, requires Cu2+ to enhance its activity (Fujimoto et al. 2016). In the case of WT-Δ0101 strain, its system most likely allows higher intracellular copper ion concentration, making oxidative phosphorylation more active thus generating more energy. Also, we observed a positive correlation between intracellular energy level and erythromycin synthesis (unpublished work), explaining the high erythromycin yield in WT-Δ0101 and E3 after the addition of Cu2+. Therefore, future undertakings should look into the details of the two-component regulation system in S. erythraea.
Conclusion
In the E3 strain, the mutation in the TCS gene SACE_0101 is involved in regulating copper homeostasis. The mutation in SACE_0101 correlates with high erythromycin yield, especially upon the addition of copper ions. Therefore, the two-component system gene SACE_0101 plays a crucial role in regulating copper homeostasis and most likely affecting erythromycin synthesis in S. erythraea.
Availability of data and materials
The data presented in the manuscript were reviewed and concluded from the earlier reported studies and mostly presented in the form of text. All the figures in the manuscript have been drawn by the authors themselves.
Abbreviations
- E3:
-
S. erythraea HL3168 E3
- TCS:
-
Two-component system
- WT:
-
Wild-type strain
- HK:
-
Histidine kinase
- RR:
-
Response regulator
- CDS:
-
Coding sequence
- RT-qPCR:
-
Real-Time Quantitative Polymerase Chain Reaction
- BLAST:
-
Basic Local Alignment Search Tool
References
Altschul SF, Gish W, Miller W, Eugene W, Myers Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Bentley SD, Chater KF, Cerdeno-Tarraga A et al (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141–147
Bijlsma JJE, Groisman EA (2003) Making informed decisions: regulatory interactions between two-component systems. Trends Microbiol 11:359–366
Carata E, Peano C, Tredici SM et al (2009) Phenotypes and gene expression profiles of Saccharopolyspora erythraea rifampicin-resistant (rif) mutants affected in erythromycin production. Microb Cell Fact 8:18
Chaplin AK, Tan BG, Vijgenboom E, Chaplin AK, Tan BG, Vijgenboom E, Worrall JAR (2015) Copper trafficking in the CsoR regulon of Streptomyces lividans. Metallomics 7(1):145–155
Couronne O, Poliakov A, Bray N, Ishkhanov T, Ryaboy D, Rubin E, Pachter L, Dubchak I (2003) Strategies and tools for whole genome alignments. Genome Res 13(1):73–80
Fujimoto M, Chijiwa M, Nishiyama T, Takano H, Ueda K (2016) Developmental defect of cytochrome oxidase mutants of Streptomyces coelicolor A3(2). Microbiology 162:1446–1455
González-Quiñónez N, Corte-Rodríguez M et al (2019) Cytosolic copper is a major modulator of germination, development and secondary metabolism in Streptomyces coelicolor. Sci Rep 9(1):1–8
Hodgson DA (1982) Glucose repression of carbon source uptake in Streptomyces coelicolor A3(2) and its perturbation in mutants resistant to 2-deoxyglucose. J Gen Appl Microbiol 128:2417–2430
Hopwood DA (1985) Genetic manipulation of Streptomyces: a laboratory manual. Biochem Educ 14(4):196
Hoshino N, Kimura T, Yamaji A, Ando T (1999) Damage to the cytoplasmic membrane of Escherichia coli by catechin–copper (II) complexes. Free Radic Biol Med 27:1245–1250
Hou YH, Li FC, Wang SJ, Qin S, Wang QF (2008) Intergeneric conjugation in holomycin-producing marine Streptomyces sp. strain M09. Microbiol Res 163:96–104
Joaquin GL, Luis LM, Jose CR, Francisco JF (2012) The CopRS two-component system is responsible for resistance to copper in the cyanobacterium Synechocystis sp. PCC 6803. Plant Physiol 159:1806–1818
Li YY, Chang X, Yu WB et al (2013) Systems perspectives on erythromycin biosynthesis by comparative genomic and transcriptomic analyses of S. erythraea E3 and NRRL23338 strains. BMC Genom 14:523
Liu Y, Ye RF, Zheng L et al (2005) Effect of n-propanol, copper ions and nicotinamide on the synthesis of erythromycin. J East China Univ Sci Technol 06:808–811
Lu YH, Wang WH, Shu D, Zhang W, Chen L, Qin Z, Yang S, Jiang W (2007) Characterization of a novel two-component regulatory system involved in the regulation of both actinorhodin and a type I polyketide in Streptomyces coelicolor. Appl Microbiol Biotechnol 77:625–635
Marcellin E, Mercer TR, Licona CC, Palfreyman RW, Dinger ME, Steen JA, Mattick JS, Nielsen LK (2013) Saccharopolyspora erythraea’s genome is organised in high-order transcriptional regions mediated by targeted degradation at the metabolic switch. BMC Genomics 14:15
Mascher T, Helmann JD, Unden G (2016) Stimulus perception in bacterial signal-transducing histidine kinases. Microbiol Mol Biol Rev 70:910–938
Mauricio L, Felipe O, Angelica RJ, Guadalupe L, Mauricio G (2011) CutC is induced late during copper exposure and can modify intracellular copper content in Enterococcus faecalis. Biochem Biophys Res Commun 06:633–637
Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S, Haydock SF, Leadlay PF (2007) Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat Biotechnol 25(4):447–453
Peano C, Talà A, Corti G, Pasanisi D, Durante M, Mita G, Bicciato S, Bellis GD, Alifano P (2012) Comparative genomics and transcriptional profiles of Saccharopolyspora erythraea NRRL2338 and a classically improved erythromycin over-producing strain. Microb Cell Fact 11:32
Rozas D, Gullon S, Mellado RP (2012) A novel two-component system involved in the transition to secondary metabolism in Streptomyces coelicolor. PLoS ONE 7:1–10
Schmittgen TD, Livak KJ (2007) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108
Singh K, Senadheera DB, Cvitkovitch DG (2014) An intimate link: two-component signal transduction systems and metal transport systems in bacteria. Fut Microbiol 9(11):1283–1293
Solioz M, Abicht HK, Mermod M (2010) Response of gram-positive bacteria to copper stress. J Biol Inorg Chem 15(1):3–14
Worrall JAR, Vijgenboom E (2010) Copper mining in Streptomyces: enzymes, natural products and development. Nat Prod Rep 27(5):742–756
Yu L, Yan X, Wang L, Chu J, Zhuang Y, Zhang S, Guo M (2012) Molecular cloning and functional characterization of an ATP-binding cassette transporter OtrC from Streptomyces rimosus. BMC Biotechnol 12:52
Zou X, Hang HF, Chu J, Zhuang YP, Zhang SL (2009) Enhancement of erythromycin A production with feeding available nitrogen sources in erythromycin biosynthesis phase. Biores Technol 100:3358–3365
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21276081) and the National Scientific and Technological Major Special Project (Significant Creation of New drugs), No. 2011ZX09203-001-03. We also acknowledge our colleague Dr. Liming Ouyang at ECUST China for her constructive discussion on this work.
Funding
This work was financially supported by the National Natural Science Foundation of China (No. 21276081) and the National Scientific and Technological Major Special Project (Significant Creation of New drugs), No. 2011ZX09203-001-03.
Author information
Authors and Affiliations
Contributions
LJQ, XBL, and XK were in charge of the experiments and manuscript writing. JC directed the study as the tutors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent for publication
All of the authors have read and approved to submit it to bioresources and bioprocessing.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Sequence alignment of gene SACE_0101 in WT and E3.There was a point mutant in E3 compared with WT. Figure S2. Electrophoresis pattern. A, PCR product 0101UH and DH. Lane 1: DL5000 Marker. Lane 2: 0101UH (4200 bp). Lane 3: 0101DH (4200 bp). B, The colony PCR identification map of the recombinant plasmid pOJ260-Δ0101. Lane 1: 0101UH (0 bp) when plasmid pOJ260 was used as the template. Lane 2: 0101DH (0 bp) when plasmid pOJ260 was used as the template. Lane 3: DL5000 Marker. Lane 4: 0101UH (4200 bp). Lane 5: 0101DH (4200 bp). Figure S3. Phenotypes of WT and WT-Δ0101 cultivated 5 days on YS agar media. It was the same piece of plate showing the top and bottom sides and there were no significant differences in morphology and culture pigmentation. Table S1. Sequences producing significant alignments with SACE_0101. Table S2. Sequences producing significant alignments with SACE_0102.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qiao, L., Li, X., Ke, X. et al. A two-component system gene SACE_0101 regulates copper homeostasis in Saccharopolyspora erythraea. Bioresour. Bioprocess. 7, 12 (2020). https://doi.org/10.1186/s40643-020-0299-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40643-020-0299-8