
Abstract
Translational genomics represents a broad field of study that combines genome and transcriptome-wide studies in humans and model systems to refine our understanding of human biology and ultimately identify new ways to treat and prevent disease. The approaches to translational genomics can be broadly grouped into two methodologies, forward and reverse genomic translation. Traditional (forward) genomic translation begins with model systems and aims at using unbiased genetic associations in these models to derive insight into biological mechanisms that may also be relevant in human disease. Reverse genomic translation begins with observations made through human genomic studies and refines these observations through follow-up studies using model systems. The ultimate goal of these approaches is to clarify intervenable processes as targets for therapeutic development. In this review, we describe some of the approaches being taken to apply translational genomics to the study of diseases commonly encountered in the neurocritical care setting, including hemorrhagic and ischemic stroke, traumatic brain injury, subarachnoid hemorrhage, and status epilepticus, utilizing both forward and reverse genomic translational techniques. Further, we highlight approaches in the field that could be applied in neurocritical care to improve our ability to identify new treatment modalities as well as to provide important information to patients about risk and prognosis.
Similar content being viewed by others


Introduction
Translational genomics represents a diverse collection of research approaches that leverage human genomics and model systems to identify new approaches to treat and prevent disease and improve healthcare (1, 2). Rooted by the central dogma of DNA to RNA to protein, genomic research examines the entire genome concurrently, and may include analyses of DNA variants in association with traits of interest as well as the impact of genomic variation on gene transcription and translation. Genomic research has been enabled by technological advances to accurately and cost-effectively study variation across the genome at scale, as well as computational techniques to store and analyze genomic data quickly and efficiently (3).
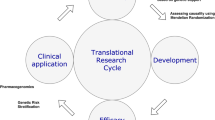
While translational research is often defined in terms of the traditional “bench to bedside” techniques that advance discoveries from model systems through biomarkers and mechanisms ultimately to clinical applications, genomic research offers a strong use-case for an alternative approach. Termed “reverse translation,” this approach starts with humans as the model system, utilizing genomic associations to derive new information about biological mechanisms that can be in turn studied further in vitro and in animal models for target refinement (Fig. 1). Both of these approaches possess advantages and drawbacks (4, 5).

A comparison of the approaches for forward and reverse genomic translational research. Forward translation begins with model systems, with later validation in humans. Reverse translation begins with human observations, with later exploration in model systems. Both serve the goal of identifying and refining therapeutic targets for human disease
Forward translation depends on the relevance of the model system to human disease, both in terms of the physiologic responses to disease or insult, as well as the approach taken to perturb the system. For instance, the human applicability of genomic studies of the response to traumatic brain injury (TBI) in a mouse model require that the mouse’s response to TBI is analogous to a human’s, and that the approach taken to create a TBI in the mouse provokes a similar pattern of injury seen in human TBI (6,7,8,9). As such, a great deal of careful work is required to demonstrate the validity of these model systems before the results arising from them can be judged relevant to human disease. The challenges of bridging this divide are illustrated by the universal failure of neuroprotection mechanisms that reached human trials in the last several decades, essentially all of which had promising model system data in preclinical development (10,11,12,13,14).
Reverse genomic translation, in contrast, begins with humans (Fig. 1). As such, there are few concerns as to the relevance of the system for discovery of biomarkers and mechanisms of disease. However, this approach carries a new series of challenges in study design and data acquisition (4). Compared to isogenic cell lines or carefully bred animals in a controlled setting, humans are highly variable in both their environmental and genetic exposures. This is advantageous in identifying genetic susceptibility to disease risk and outcomes, but teasing out these small genetic effects from highly variable non-genetic exposures requires both careful computational techniques as well as large sample sizes. Furthermore, because genomic data is both identifiable and can potentially lead to discrimination, human genomic studies require complex consent and data management procedures (15). In neurocritical care, the relative rarity of many of the diseases we encounter, coupled with the challenges of critical illness and surrogate consent make human genomic studies all the more difficult to execute effectively (16,17,18,19).
Neurointensivists routinely encounter diseases and complications for which there are a dearth of effective treatments, or even foundational knowledge of their underlying pathophysiologic mechanisms (20, 21). In this review, we will highlight some of the approaches being taken to apply translational genomics to the study of diseases commonly found in neurocritical care, utilizing both forward and reverse genomic translational techniques. Further, we will highlight some of the best practices in the field that could be applied in neurocritical care to improve our ability to identify new treatment modalities as well as risk and prognosis information to patients and their families.
Evolution of Genomic Research
Pre-GWAS Era
In advance of the Human Genome Project and the HapMap Consortium, genetic studies were confined to the study of candidate genes and lower-resolution genome-wide techniques such as categorization of restriction fragment length polymorphisms (RFLP), tandem repeats, and microsatellites (22). These genomic features enabled early efforts to perform linkage analyses in families with related traits and disorders, as well as selected populations of unrelated individuals. Careful work in this arena led to validated discoveries that have survived replication in the common era, such as Chromosome 19 in late-onset Alzheimer Disease (AD), ultimately mapped to the APOE locus that has become a target for a great deal of genetic research in AD, as well as many other diseases including TBI and intracerebral hemorrhage (ICH) (23,24,25,26).
Still, much of the preGWAS era was characterized by candidate gene studies that suffered from low statistical power and multiple sources of confounding that led to a failure to replicate many reported associations in the GWAS era that followed (27, 28). The most substantial source of confounding in candidate gene analyses is population stratification, in which differences in allele frequency due to ancestral imbalance between cases and controls introduces spurious associations (positive or negative) between genotype and trait based solely on these cryptic ancestral imbalances (29, 30). Even in studies of APOE in European ancestry populations, uncontrolled variation in the percentages of individuals of northern vs. southern European ancestry between cases and controls can mask true associations between APOE and ICH, for instance (31).
GWAS Era
The GWAS era, in which variants across the genome could be reliably genotyped and mapped to a common reference template by chromosomal location, ushered in a new system of best practices that could minimize the contribution of many of the sources of confounding in describing associations between genomic variation and traits or diseases. The International HapMap Consortium obtained genotypes on individuals across 11 ancestral populations around the globe, creating a resource that described the patterns of allele frequency variation across diverse populations (32). With these breakthroughs and a number of landmark evolutions that followed, case/control and population-based GWAS have led to the identification of over 11,000 associations with human diseases and other traits (https://www.ebi.ac.uk/gwas/). Obviously there is an enormous disconnect between the discovery of genetic loci and leveraging of this information for human benefit, which is where the translational genomic work that serves as the topic of the present review becomes relevant (32).
Post-GWAS Era
Post-GWAS, in addition to functional and translational efforts, the movement has been towards so-called “next-generation sequencing” methodologies consisting of whole exome sequencing (WES) and whole genome sequencing (WGS). Using these approaches, each nucleotide in the exome or genome is ascertained with high reliability, permitting the identification of rare and de novo variants that escape detection in traditional GWAS (33). WES captures within-gene coding variation only, offering detection of variants that may more directly impact protein structure and function than non-coding variation detected by WGS (34). Because the coding exome is only ~2% of the overall genome, it is more cost-effective than WGS, but debate continues as to which is the more appropriate tool for large-scale study of the human genome (35). Regardless, both WES and WGS remain orders of magnitude more expensive than traditional GWAS approaches at this time, and as such well-powered sequencing studies remain unreachable for many diseases in the current pricing models. Less common diseases and conditions that one may find in a neurocritical care unit are doubly disadvantaged, as even larger sample sizes are required for sequencing analyses than GWAS, due to the need for many observations to identify rare exonic or intronic variants associated with disease (17, 36). As pricing models improve and larger and larger community or hospital-based cohorts receive sequencing through clinical or biobanking efforts, it is hoped that even uncommon conditions such as subarachnoid hemorrhage or status epilepticus will benefit from the insights achievable through sequencing analysis, where case/control and smaller sequencing studies have shown promise (37, 38).
Genomics in Model Systems
Obviously genomic research need not be limited solely to human studies. A wealth of information about disease pathogenesis and response to injury can be gleaned from model systems of human conditions using genomic and transcriptomic approaches. Because animal models and isogenic tissue cultures are specifically designed to limit genetic differences between individual animals or plated cells, DNA-based association tests typically do not offer insight in the same way that they do in humans. As such, many model system studies start with RNA, examining how the genome responds to perturbation through the transcriptome. However, there are substantial genomic differences between model systems and humans, as coding sequences are not necessarily conserved, promoter and enhancer control of gene expression can vary, and in the case of immortalized tissue and cell-based assays, the chromosomal architecture itself can be quite different from the organism from which it was derived (39, 40). These differences can be highly relevant when determining whether observed transcriptomic and proteomic results from model systems are likely to be shared in humans. With those caveats, the dynamic nature of the transcriptome in model systems offers opportunities to assess the way in which the genome responds to noxious insults or drug exposures, and in animal models this can even be done across specific organs or tissues of interest (40). As one example, traumatic brain injury researchers have obtained insight into both the initial injury cascade as well as brain response to potential injury modulators such as valproate using animal models and transcriptional microarrays, in which RNA expression patterns in brain tissue can be rapidly and replicably assessed across the transcriptome (41). Using more recent technological advancements such as Drop-seq, RNA expression can be assessed in single cells, as has been done in individual hippocampal neurons in a mouse model of TBI (42). At a minimum, these elegant studies can help to identify relevant cell types important in the response to injury, highlighting testable hypotheses that may be important in human conditions, all with access to tissues and control over experimental conditions that would never be possible in human-based research. Given that diseases common to the neurocritical care population so rarely afford access to brain tissue for pathologic or genomic analysis antemortem, model system genomic studies offer an important adjunct for translational research.
Traditional (Forward) Genomic Translation
Forward genomic translation begins with model systems with the goal of using the measured associations in these models to derive insight into biological mechanisms that may also be relevant in human disease. Forward translation requires well-characterized models that are often designed to mimic the human exposures of interest as closely as possible. This is often challenging given the natural differences between humans and many of the animals chosen to serve as models. In this section, we will highlight several model systems in current use for translational genomics relevant to neurocritical care, but the field of translational modeling in neurologic disease is suitably large to prevent an exhaustive review herein.
Malignant Cerebral Edema
Malignant cerebral edema is a highly lethal complication of ischemic stroke, with mortality of 60–80% (43). Currently, hemicraniectomy is the only available option to prevent death and yet it does not address the underlying pathophysiology. Hyperosmolar therapy is potentially useful as a bridge to surgery. Preclinical data based on a forward translation approach has been useful in highlighting mechanisms underlying post-infarct edema as potential targets for therapeutic manipulation. The sulfonylurea receptor 1 (SUR1) is encoded by the ABCC8 gene that is upregulated after CNS injury, forming an ion channel in association with transient receptor potential melastatin (TRPM4). Continuous activation of this complex can lead to cytotoxic edema and neuronal cell death, which has been demonstrated in both animal and human models (44, 45). SUR1 is also found in pancreatic beta cells, constituting the target for the oral hypoglycemic agent, glyburide. Studies of rodent and porcine stroke models demonstrated that in the first few hours after an ischemic insult, both SUR1 and TRPM4 are upregulated (46, 47). Limited case series of human postmortem specimens also demonstrated upregulation of SUR1 in infarcted tissue (48). Therefore, intravenous glyburide has been proposed for treatment of malignant cerebral edema. Targeting Sur1 in rat models of ischemia have consistently resulted in reduced edema and better outcomes (49). In particular, glyburide infusion starting 6 h after complete middle cerebral artery occlusion resulted in decreased swelling by two thirds and 5% reduction in mortality (50).
One desirable characteristic of glyburide is that it cannot penetrate intact blood-brain barrier, but that is facilitated following brain injury (51). The effect of glyburide for treatment of cerebral edema has also been studied in TBI with promising data obtained from animal studies (52). Limited randomized trials in human using oral glyburide have shown promising results; however, use of oral formulation and study design limitations prohibit generalizability of results (52, 53).
Building on this preclinical data, the phase 2 randomized clinical trial (GAMES-RP) showed that the IV preparation of glyburide, glibenclamide, is associated with reduction in edema-related deaths, less midline shift, and reduced rate of NIH stroke scale deterioration. However, it did not significantly affect the proportion of patients developing malignant edema (54). The Phase 3 CHARM trial, sponsored by Biogen, is currently enrolling patients with large hemispheric infarction to determine whether IV glibenclamide improves 90-day modified Rankin scale scores. If this trial proves successful, this vignette will represent a dramatic success story for the forward translation paradigm in genomic research.
In the light of recent advances in revascularization therapy, the National Institute of Neurological Disorders and Stroke has supported an initiative aiming to develop neuroprotective agents to be used as adjunctive therapy to extend the time window for reperfusion and to improve long-term functional outcome. This Stroke Preclinical Assessment Network (SPAN) supports late-stage preclinical studies of putative neuroprotectants to be administered prior to or at the time of reperfusion, with long-term outcomes and comorbidities constituting the endpoint. The goal is to determine if an intervention can improve outcome as compared to reperfusion alone and/or extend the therapeutic window for reperfusion. SPAN directly applies to forward translation efforts in preclinical models of neuroprotection after stroke and is an outstanding opportunity to stimulate research efforts in a field more remembered for its past failures than the promise it holds for the future of therapeutic development in the area.
Other societies have also begun to endorse more comprehensive modeling approaches in areas with few therapeutic options with the hope of implementing a paradigm shift. For example, the Neurocritical Care Society has initiated “Curing Coma” campaign with the 10- to 20-year mission to improve the understanding of the mechanisms and to ultimately develop preventative and therapeutic measures.
Traumatic Brain Injury
Traumatic brain injury (TBI) is among the leading causes of disability and death worldwide, particularly in the young. The type of TBI is in part determined by the attributes of mechanical forces, including objects or blasts striking the head, rapid acceleration-deceleration forces, or rotational impacts. Following the primary injury, an intricate cascade of neurometabolic and physiological processes initiates that can cause secondary or additional injury (55, 56). Intensive care management has improved the prognosis of TBI patients; however, specific targeted treatments informed by pathophysiology could have a tremendous impact on recovery. The period of secondary tissue injury is the window of opportunity when patients would potentially benefit from targeted interventions, given that in TBI, the primary injury cannot be intervened upon by the neurologist or intensivist. The goal of therapy is therefore to reduce secondary damage and enhance neuroplasticity.
The utility of animal models of TBI primarily depends on the research question, as each model emulates specific aspects of injury and has selective advantages and disadvantages. These include biomechanics of initial injury, molecular mechanisms of tissue response, and suitability for high-throughput testing of therapeutic agents, to name a few. Although phylogenetically higher species are likely more representative models for human TBI, rodent models are more commonly used given the feasibility to generate and measure outcomes, as well as ethical and financial limitations of higher-order models. Table 1 summarizes some common and representative TBI models [Table 1].
In contrast with the rodent models described in Table 1, other model systems in TBI have been selected specifically to study other aspects of the physiologic response to TBI. For example, a swine model of controlled cortical impact offers the opportunity to readily monitor systemic physiologic parameters such as tissue oxygen and acid-base status while investigating therapeutic interventions, which is argued to provide greater insight into human response to injury (66).
Translation of preclinical studies using these animal TBI models to humans is inherently challenging. Differences in brain structure, including geometry, craniospinal angle, gyral complexity, and white-gray matter ratio, particularly in the rodent models, can result in different responses to trauma (65). The limitation of extrapolating animal studies to human is also manifested at the genetic level, as differences in gene structure, function, and expression levels may suggest genetic mechanisms that are incompletely correlated with humans. As an example, female sex may be associated with better outcome through the neuroprotective effect of progesterone in animal models, but these observations did not carry over to humans in the ProTECT-III trial (67, 68). Variable outcome measures, including neurobehavioral functional tests, Glasgow outcome scale correlates, and high-resolution MRI have been used in attempts to correlate animal responses to injury with those of humans. The lack of a large cache of standardized tools further limits comparison or pooling the results of different studies that use variable models of TBI or outcome measurement.
Transcriptomics, a genomic technique in which global RNA expression is quantified through either expression microarrays or RNA sequencing, has been employed to characterize specific inflammatory states following TBI. Many studies have assessed the transcriptome in the acute post-TBI interval within 1–2 days after injury, with some showing upregulation of inflammation and apoptosis genes. Gene ontology analysis at 3 months post-TBI have shown similar changes, with upregulation of inflammatory and immune-related genes (69). Importantly, late downregulation of ion channel expression in the peri-lesional cortex and thalamus suggests that this delayed examination of the transcriptome could be valuable for revealing mechanisms relevant to chronic TBI morbidities, including epileptogenesis and prolonged cognitive impairment (70). In addition, tissue-specific analysis of gene expression across cell types in brain could provide useful insight into cell-specific pathways. For example, temporal trending of microglial expression profile indicates a biphasic inflammatory pattern that transitions from downregulation of homeostasis genes in the early stages to a mixed proinflammatory and anti-inflammatory states at subacute and chronic phases (71).
Status Epilepticus
The list of antiepileptic drugs has expanded significantly in the past decade, reflecting substantial investment in the search for new therapeutics with better efficacy and tolerability. However, the list of options with demonstrated efficacy in status epilepticus (SE) has remained limited. The utility of benzodiazepines, often deployed in the field as a first-line agent, decreases with increasing duration of SE. In addition, 9–31% of patients with SE develop refractory SE when they fail to respond to first- and second-line therapy, posing a significant management and prognostic challenge (72).
The development of AEDs has relied substantially on preclinical animal models to establish efficacy and safety prior to proceeding to human trials. Different epilepsy models exist that are each useful for different aspects of drug development and no model is suitable for all purposes. The majority of animal models induce epilepsy using electroshock or chemical seizure induction. Nearly all recent AEDs have been discovered by the same conventional models, and the reliance on these common screening models has been implicated as one of the reasons for the low yield of drugs with efficacy in refractory epilepsy (73).
The pros and cons for each epilepsy model are discussed in detail in several excellent reviews (74, 75). Some of the chemicals used include kainic acid, pilocarpine, lithium, organophosphates, and flurothyl (76). Sustained electrical stimulation to specific sites, including the perforant path, the ventral hippocampus, the anterior piriform cortex can induce SE (77).
The latency, length, and mortality of convulsive SE are more variable in chemoconvulsant as compared to electrical models, which are in turn determined by the drug and route of administration, species, sex, age, strain, and genetic background among other factors (78). It should also be noted that the presence of behavioral convulsion does not correlate fully with the electrographic data and vice versa. This can have critical implications when studying drugs for pharmacoresistant SE. Therefore, it has been suggested that electroencephalographic quantification be used to measure the severity of SE (78). Furthermore, the genetic background and expressivity of animals can have a significant effect on seizure susceptibility, even between batches of inbred mice (79).
Proteomic and transcriptomic approaches have been utilized for assessment of alterations in expression profile following SE, demonstrating that certain subsets of genes are upregulated at each timepoint following the onset of SE. Specifically, upregulation of genes regulating synaptic physiology and transcription, homeostasis and metabolism and, cell excitability and morphogenesis occur at immediate, early, and delayed timepoints. In addition, related studies have demonstrated changes in expression of microRNAs related to epileptogenesis, including miRNA-124 and mi-RNA-128 following SE (80, 81). Selective RNA editing post-transcription is yet another potential source of proteomic diversity in preclinical models of SE, and merits further investigation as a modulator of protein levels that may be less closely tethered to gene expression (82).
Subarachnoid Hemorrhage
Aneurysmal subarachnoid hemorrhage (SAH) has an earlier age of onset and is associated with higher morbidity compared with other stroke subtypes. The pathophysiology of insult has traditionally been studied under two time-intervals, early brain injury (EBI) and, cerebral vasospasm (CV) and delayed cerebral ischemia (DCI). The prime goal of translational research in this arena is to identify the mechanisms and targets related to the risk, severity, evolution and outcome.
About 15% of patients die immediately following SAH (83). Thereafter, early brain injury within the first 3 days, followed by DCI are the most feared complications. CV is the phenomenon with strongest association with the development of DCI, which 30–70% of patients experience between day 4 to 14 (84). The underlying mechanisms leading to CV remain poorly understood and have therefore been a prime focus of preclinical studies. The majority have used rodent models, but primate, swine, and dog models have also been employed (85).
Cerebral aneurysms are difficult to model and hence two common approaches to modeling SAH use alternative strategies. The first is direct injection of blood into the subarachnoid space, specifically into either the prechiasmatic cistern or cisterna magna, to generate SAH predominantly in the anterior or posterior circulation territories, respectively (86). The second model, endovascular suture, passes a suture or filament through the internal carotid artery, creating a hole in one of the major branches resulting in egress of variable amount of blood into the subarachnoid space (87).
Variations in some parameters of the first method, including injected blood volume, CSF removal prior to injection to prevent egress of blood into the spinal canal, and replenishing intravascular volume to keep cerebral perfusion pressure constant through maintenance of mean arterial pressure, as well as the rapidity of injection have raised questions about comparability and biofidelity of the results (88,89,90,91). The latter model appears to remove some of the mentioned confounding factors, as the hemorrhage occurs at physiologic mean arterial pressure (MAP) and intracranial pressure (ICP), but is limited by variable puncture site and ultimate hemorrhage volume. Another potential drawback is the period of ischemia caused by the intraluminal suture, although the occlusion period is typically not judged to be long enough to cause significant ischemia.
The missing element in these models is the absence of aneurysm formation and rupture, and consequently the vascular processes intrinsic to the aneurysm itself that influence DCI. As such, some studies have used combinations of interventions to generate aneurysms, including induced hypertension via unilateral nephrectomy and administration of angiotension II or deoxycorticosterone acetate, as well as elastase injection. The downside of these models is that the timing of aneurysm rupture cannot be reliably predicted, which limits close monitoring and physiologic assessments in the early phase following SAH, blurring the timing of DCI (92,93,94).
The immediate hemodynamic changes following the hemorrhage are monitored via a variety of methods. Regardless of the method chosen, reports on the direction and range of values of CPP, CBF, and MAP can be quite variable, both within the same model and between different models. A common technique to measure blood flow is laser doppler flowmetry that provides a continuous measure of cortical perfusion. Although it does not measure global cerebral blood flow and has spatial limitation, it appears to be relatively reliable and technically reproducible. Other methods of flow measurement include radiolabeling methods and MRI with the latter has the advantage of capturing the dynamic nature of the condition, as well as global and region-specific blood flows.
As noted, CV and DCI are responsible for delayed morbidity and mortality. Given that these manifestations typically occur while patients are inpatient for care of their SAH, therapeutic interventions are more feasible compared to the hyperacute phase when the processes leading to initial damage may have already occurred. However, monitoring for CV in animal models is not straightforward. One method of identifying CV is measuring the intraluminal diameter of vessels on histological samples. In addition to being an end-measure and therefore precluding measurements at different time points in the same animal, varying degrees of tissue desiccation among samples may yield numbers different from actual in vivo values. Digital subtraction angiography and magnetic resonance angiography can provide a real-time evaluation, but the severity of CV and its timing, as well as neuronal cell death varies depending on the model and the affected vessels (86).
The foundational molecular pathways that orchestrate CV are complex and remain incompletely elucidated. However, translational research using many of the above models has demonstrated that endothelin-1, nitric oxide, and an inflammatory cascade ignited by breakdown of blood products play predominant roles. Endothelin-1 is a potent vasoconstrictor produced by infiltrated leukocytes, and based on this notion, clazosentan was developed as an endothelin-1 receptor antagonist to combat CV. In human trials, clazosentan was found to significantly reduce the incidence of the DCI without improving the functional outcome, and this or a related approach could ultimately prove beneficial if off-target drug effects, including pulmonary complications, hypotension, and anemia can be mitigated (95, 96). Hemoglobin and its degradation products are also a strong stimulus for CV through direct oxidative stress on arterial smooth muscle, decreased nitric oxide production and, increasing endothelin and free radical production (97). This suggests that facilitating clearance of hemoglobin degradation products from the CSF may be a potential therapeutic target. Modulating the intense inflammatory response is also intuitive and while preclinical results support this notion in general, the evidence has thus far not been judged adequate to justify clinical trials. For example, IL-1 receptor antagonist (IL-1Ra) reduces blood-brain barrier (BBB) breakdown, a biomarker that is itself correlated with the severity of brain injury, and work continues to determine whether this or related pathways mediating BBB permeability might have therapeutic promise (98). Given these numerous and likely interconnected mechanisms of delayed brain injury, further research is needed to understand their relative applicability to humans, and whether targeting a single pathway or a number of pathways simultaneously is likely to be the most adaptive strategy to reduce CV and DCI in humans.
The results of genome-wide RNA sequencing analysis have supported the primary role of neuroinflammation in the pathogenesis of early brain injury. Some studies have specifically found a key role for long non-coding RNA (lncRNA), a type of RNA without protein-coding potential that are particularly abundant in the brain, in modulating the inflammatory behaviors of microglial cells (82). High-throughput mass spectrometry has also been utilized in demonstration of differential expression of proteins in the cerebral vessels after SAH, as well as for monitoring the effect of experimental therapeutics (99). We will not cover these proteomic studies in detail here, as they typically fall outside the rubric of what is classically considered “genomics”, but their approach, which leverages global protein signatures rather than restricting observations to specific compounds, shares many similarities with genomics.
Reverse Genomic Translation
As mentioned above, reverse genomic translation refers to an approach to the study of a disease by starting with humans using either cohort-based or case/control genomic studies. The observations made through the course of these studies then inform on the best approach for target validation and refinement to prioritize candidate mechanisms and related endophenotypes for therapeutic development. It has been shown that candidate compounds with independent confirmation of their therapeutic target via human genomics are more than twice as likely to prove effective in clinical trials (100). Therefore, the reverse translation approach would seem an adaptive strategy to identify disease-associated mechanisms and therapeutic targets with the best chance of impacting clinical care in the near term. However, the approach to reverse translation requires large sample sizes with well-characterized patient data in order to achieve a statistically confident result. These large sample sizes raise the issue of variability in risk and treatment exposures between participants, which could impact patient outcomes independently of genomic effects and therefore erode power to detect genetic risk.
The utility of reverse translation in target refinement and mechanism exploration in model systems can be highlighted using an example from the stroke community. Recent GWAS and subsequent meta-analyses of ischemic stroke and stroke subtypes in very large case/control datasets have validated the histone deacetylase 9 (HDAC9) region in chromosome 7p21.1 as a major risk locus for stroke due to large artery atheroembolism (LAA). This locus was also previously discovered in association with coronary artery disease (CAD) (101, 102). Based on these findings, Azghandi et al. sought to investigate the role of the leading single nucleotide polymorphism (SNP) in this genomic region (rs2107595) in increasing LAA stroke risk (103). They found that rs2107595, both in heterozygotes as well as in homozygote human carriers, is associated with increased expression of HDAC9 in peripheral blood mononuclear cells on a dose-dependent manner, suggesting that the effect of this locus in stroke risk may be mediated by increased HDAC9 expression. Additionally, they demonstrated that HDAC9 deficiency in mice is associated with smaller and less advanced atherosclerotic lesions in the aortic valves, curvature, and branching arteries, suggesting that HDAC9 may increase atherogenesis and therefore represents a novel target for atherosclerosis and LAA stroke prevention. Notably, recent studies have suggested that both non-specific (e.g. sodium valproate) as well as specific HDAC9 inhibitors can have a positive impact on both stroke recurrence risk, as well as other phenotypes, including cancer. This highlights the central role that reverse translation can have in therapeutic target investigation and refinement, with potential beneficial off-target properties (104, 105).
While acute stroke care is a vital component of neurocritical care at many institutions, reverse genomic translation successes in other relevant traits also merit mention. Acute respiratory distress syndrome (ARDS) is a frequent complication of severe neurologic injury due to SAH or neurotrauma. In a recent GWAS by Bime et al., variation in the Selectin P Ligand Gene (SELPG), encoding P-selectin Glycoprotein Ligand 1 (PSGL-1) was found to be associated with increased susceptibility to ARDS (106). The most significant SNP in this locus, rs2228315, which results in a missense mutation, has been successfully replicated in independent cohorts. Further functional analyses have demonstrated that SELPG expression was significantly increased in mice with ventilator (VILI)- and lipoprotein (LPS)-induced lung injury, and that PSGL-1 inhibition with a neutralizing polyclonal antibody led to an attenuation of inflammatory response and lung injury. In SELPG knockout mice, inflammatory response as well as lung injury scores were significantly reduced compared to wild-type mice (106). These results highlight the value of reverse genomic translation in first identifying human-relevant genetic risk factors for disease, and using model systems to understand the pathways impacted by their introduction to select rationally-informed modalities for potential treatment.
Intracranial aneurysms (IA) are commonly encountered in the neurocritical care setting, albeit most commonly after rupture. Even so, inroads leading to a better understanding of aneurysm formation may ultimately reveal opportunities for treatments to prevent acute re-rupture or prevent future aneurysm formation after SAH. The strongest associations with IA have been reported in the region near CDKN2A/CDKN2B in 9p21.3 as well as in a nearby intragenic region known as CDKN2BAS or ANRIL (107, 108). ANRIL is a long non-coding region responsible for the regulation of CDKN2A and CDKN2B and has also been implicated in the pathogenesis of CAD and atherosclerosis, among other traits (109). Overexpression of ANRIL in mouse models of CAD has been associated with negative atherosclerosis outcomes including increased atherosclerosis index, unfavorable lipid profiles, thrombus formation, endothelial cell injury, overexpression of inflammatory factors in vascular endothelial cells, increased apoptosis of endothelial cells, and upregulation of apoptosis-related genes. Notably, reduced ANRIL expression has been associated with reduced inflammatory, biochemical and molecular markers of atherosclerosis, indicating a potential target for atherosclerosis and IA prevention (110).
When utilizing the reverse translation approach in genomic studies, the aforementioned examples highlight two distinct but equally important considerations for a successful implementation of such approaches. The first major consideration is that large populations of well-characterized individuals must be selected to ensure adequate statistical power to detect meaningful associations. Thorough and standardized phenotyping of study subjects is one of the main predictors of the success of a GWAS (111, 112). Careful assignment of cases based on strict phenotypic criteria permits well-executed GWAS even in diseases with heterogeneous presentations and multiple pathogenic features, such as multiple sclerosis (MS) and stroke (113). In neurocritical care populations where subtle characteristics of disease presentation and intermediate outcomes may represent important phenotypes for genomic investigation, such as SAH, these traits should be closely defined and recorded to the greatest degree possible in all participants. This initial step is critically important in the greater scheme of reverse translational genomics, as these associations with subclasses and endophenotypes of disease often provide the biological insights needed to continue translational efforts using model systems tailored to refine observations. The second major consideration is that the execution of genomic studies needs to be comprehensive and thorough so as to permit association testing in a hypothesis-free environment. At the moment, GWAS array-based studies seem to remain a favorable option of the genome, considering the lower cost associated with their utilization and proven track record in discovery, but over time, WES and WGS studies will become reachable even on more modest research budgets. For the transcriptome, RNA sequencing and RNA microarrays both offer unbiased surveys of global transcriptional variation, but because gene expression varies substantially by tissue it is critical that rational choices are made regarding the suitability of specific tissues for specific conditions. In uncommon conditions with necessarily small sample sizes, including neurocritical care-relevant diseases like SE, SAH, and ARDS, external validation studies can strengthen associations from an initial small discovery dataset, and in many cases these follow-up studies can make use of freely available resources. For example, in a recent expression-based GWAS (eGWAS), microarray data for ARDS from the Gene Expression Omnibus (GEO) were collected and combined in an effort to identify novel genetic targets (114). The study not only validated previously known lung injury- and ARDS-related genes, but also discovered 14 new candidate genes that may prove to be useful in future translational work.
Modeling of Human Genomic Discoveries
Identifying loci, variants, expression patterns, and gene-networks with the use of human genomic studies is only the initial step in the reverse translation process. These discoveries must inform and guide the research to further understand and refine the phenotypic effects of these variants in model systems, including some of those described above. There are several techniques with which we can utilize the discoveries made from case/control genomic studies to build or modify model systems.
One approach is transgenesis, in which a larger DNA sequence including a human gene containing a mutation of interest, called a transgene, is injected into the pronucleus of a mouse fertilized egg. The fertilized egg is then inserted into the oviduct of a pseudopregnant female mouse, which is a female who has been mated with vasectomized male in order to achieve the hormonal profile of a pregnancy state. The offspring produced from this female can create an animal line that contains the human gene and allele of interest (115). However, because the transgene is inserted randomly at one or more genetic locations as either one or more copies, the level of expression and regulatory influences of the gene of interest may not initially be well-controlled across animals. As such, there are several intermediate steps that can allow more specific genetic alteration using transgenesis, involving embryonic stem cells (ESCs). The first step is the introduction of regulatory sequences (such as expression cassettes) into ESCs. Then, by injecting the transgene first into these modified ESCs, gene expression can be more closely controlled. The ESCs with the transgene can then be inserted into blastocysts and give rise to new strains, using the same methods previously described (116). There are multiple variations on the transgenic approach which are uniquely suited to the model system being employed and can give rise to models that express transgenes in response to a particular stimulus, or in particular tissues of interest.
A newer method utilizing programmable endonucleases has allowed researchers to bypass more traditional ESC-based methods for direct and precise gene editing. Endonucleases are enzymes that cause double stranded DNA (dsDNA) breaks that can further be repaired either with non-homologous end-joining (NHEJ), an imprecise method for rejoining the DNA breaks that involves various enzymes and may result in inactivating mutations, or with homology-directed repair (HDR), in which the DNA breaks are repaired based on a co-injected template. Four categories of programmable endonucleases have been used for direct and precise gene editing: homing endonucleases (HE), zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and the clustered regularly interspaced short palindromic repeats/CRISPR-associated 9 (CRISPR/Cas9) system. The common characteristic of these enzymes is that they possess sequence-specific nuclease activity, allowing researchers to cleave dsDNA at desired, pre-specified sites. The CRISPR/Cas9 system has proven to be the most successful so far, in terms of efficiency, cost, and simplicity of use. Perhaps the most important advantage of this approach is that programmable endonucleases do not require the use of ESCs and can directly be inserted into one- or two-cell stage embryos, thus allowing more specific and direct gene-editing in a single step (116). Drawbacks include enzymatic limitations as to where DNA breaks can be reliably introduced, as well as off-target endonuclease activity at other sites across the genome which can disrupt gene activity in unintended ways. Work is ongoing to refine these tools, improving the number of sites where gene editing can occur while also improving the specificity of the system (117).
One illustrative example of human genomic studies being used to refine models to understand disease processes is the case of human ICH-associated mutations in COL4A1 and COL4A2. COL4A1 and COL4A2 are the most abundant proteins in basement membranes. They form heterotrimers consisting of one COL4A1 and two COL4A2 peptides and are produced and modified in the endoplasmic reticulum (ER). After their production, they are packaged into vesicles in the Golgi apparatus and transferred to vascular endothelial basement membranes (118,119,120). The initial identification of mutations in this region in familial forms of cerebral small vessel disease, coupled with the subsequent detection of common COL4A1/COL4A2 variants associated with sporadic deep ICH led to the development of animal model systems to explore their effects (121,122,123,124). Through mouse models, representative COL4A1/COL4A2 mutations were found to recapitulate human disease phenotypes, with multifocal ICH in subcortical regions of the forebrain and the cerebellum, as well as porencephaly, small vessel disease, recurrent intraparenchymal and intraventricular hemorrhages, age-related ICH, and macro-angiopathy (125, 126). Using cellular assays and tissue derived from mouse models, mutations in COL4A1/COL4A2 have been associated with decreased ratio of intracellular to extracellular Col4a2, retention of abnormal collagen proteins in the ER, ER stress, and activation of the unfolded protein response (126,127,128), suggesting that the intracellular accumulation and ER-stress could be an important molecular mechanism underlying ICH related to COL4A1 and COL4A2 mutations. Notably, treatment with the molecular chaperone sodium 4-phenylbutyrate resulted in decreased intracellular accumulation and significant decrease of ICH severity in vivo, which could point the way towards eventual forms of treatment for both familial and sporadic COL4A1- and COL4A2-associated ICH (125).
Another recent example of model system refinement for neurocritical care-relevant disorders is status epilepticus (SE). Pyridoxal phosphate binding protein (PLPBP) variants have been associated with a rare form of B6-dependent epilepsy, which, if left untreated can lead to SE. In a recent study, Johnstone et al. utilized CRISPR/Cas9 to create a zebrafish model lacking its encoded protein (129). They observed that PLPBP-deficient zebrafish experienced significantly increased epileptic activity compared to their wild type counterparts, in terms of physical activity (high-speed movements), biochemistry (c-fos expression) and electrophysiologically-recorded neuronal activity. Additionally, treatment of plpbp−/− larvae with PLP and pyridoxine led to an increase in their lifespan, and a decrease in their epileptic movements and neuronal activity. Lastly, in these PLPBP-deficient zebrafish, systemic concentrations of PLP and pyridoxine were significantly reduced, as well as concentrations of PLP-dependent neurotransmitters. Collectively, these results provide insights for biomarker development and preclinical target refinement in B6-dependent epilepsy. Understanding how novel treatments might impact rare disease presentations could ultimately lead to new insights for common forms of disease as well, just as the discovery of rare PCSK9 variants in patients with very low cholesterol ultimately led to PCSK9-inhibitors to treat more common forms of familial hypercholesterolemia.
However, the use of animal models is not always the ideal approach to describing the effects of genetic variation, as the phenotypic alterations may be too subtle to observe or require impractical prolonged observation in late-life animals to ultimately exhibit relevant phenotypes. In these cases, tissue-based systems can provide a useful tool to study these effects. For example, IA formation, as previously described, has been associated with variants in ANRIL. Although the direct impact of these variants in human tissue or animal models is difficult to discern, work with mutations of ANRIL in endothelial models have provided valuable insight. Specifically, upregulation of ANRIL has been associated with increased expression of inflammatory and oxidative markers in the vascular tissue such as IL-6, IL-8, NF-κB, TNF-a, iNOS, ICAM-1, VCAM-1, and COX-2 (130, 131). These observations provide vital information about cellular mechanisms impacted by human disease-associated genetic risk factors without requiring the expense and time investment of creating, validating, and studying animal models. Ultimately such models may still be required, but prior knowledge about cellular phenotypes associated with genetic variation may be highly valuable in choosing the right model system and selecting efficient approaches to validate these systems.
The aforementioned examples highlight significant contributions of the field of translational genomics in identifying novel therapeutic targets, developing biomarkers of disease severity and elucidating disease-relevant pathophysiology. Undoubtedly, these contributions are valuable in application to existing model systems of disease, or through refinement of models informed by the reverse translation process. Given that many of our current models have proven to be ineffective in many cases, the reverse translation approach offers a significant advantage in that the translational discoveries arising in established or refined model systems have already been proven to be relevant to human disease. This advantage provides us with reasonable expectation that observed effects in model systems will also remain relevant to human disease, providing a substrate for therapeutic development.
Improving Human Health
Certainly, the ultimate goal of translational genomics is to be able to transfer the discoveries found from experimental models into clinically useful information in order to improve human health. This aim, with regard to the translational genomic approach, can be satisfied with two distinct approaches. One is concerned with improving our understanding of the mechanisms of disease, providing novel targets for therapeutic development. The other is concerned with leveraging the conclusions of translational genomics through more direct applications to clinical care. We will discuss these in order.
Once genomic discovery and translational exploration have confirmed the mechanism and relevance of a particular genomic association, translational genomics offers the opportunity to use these same translational approaches to derive high-throughput assays for screening of compound libraries, which are collections of small molecules useful for early-stage drug-discovery (132). The same in vitro assays used to identify cellular phenotypes associated with genetic risk factors can be tested for amelioration or “rescue” of wild-type features after exposure to library compounds. This is particularly advantageous for the reverse genomic translation approach, as these assays are often critical components of the overall discovery cycle, and with optimization to provide ideal readouts, screening can proceed quickly. An example of success here is the identification of molecular chaperones that can ameliorate the unfolded protein response detrimental to cell survival in COL4A1-mediated cerebrovascular disease (125). Identifying hits in these assays has the potential to accelerate drug-discovery, provided that the mechanism can be targeted by a small molecule and not a designed biologic entity such as a monoclonal antibody. While screening can be performed using novel compound libraries, it can also be accomplished using libraries containing already-approved drugs, providing an innovative way for compound repurposing based on genetic interactions. Numerous tools already exist for in silico evaluation of existing compounds based on known mechanisms, so this step can begin even in the genomic discovery phase prior to translational validation (133).
The second approach in which translational genomics has proven to be of great potential is the rapidly evolving and highly anticipated field of precision medicine. The observations arising from translational genomics, even when not informing us about the specific mechanisms associated with the phenotype in question, may be of predictive value. This finds application in two relevant translational genomics tools: polygenic risk scores (PRS) and biomarker development based on RNA expression profiles.
Polygenic Risk Scores
While common genetic variation can provide valuable information about disease-relevant mechanisms and help refine disease models, they are relatively weak in explaining a significant proportion of the genetic basis of complex polygenic disorders, such as CAD, diabetes, stroke, or SAH (111). By summarizing the impact of many variants of small effect across the genome simultaneously, a polygenic risk score (PRS) can be developed which explains far more of the genetic risk of a disease than any common variant can individually (Fig. 2). Application of these PRS in independent clinical populations as a predictive tool represents a novel translational approach. In a recent study examining stroke, a PRS combining SNPs associated with atrial fibrillation (AF) was found to be significantly associated with cardioembolic (CE) stroke risk and no other stroke subtypes, paving the way for a potentially useful tool to discriminate CE stroke from other etiologies without reliance on expert adjudication or longitudinal monitoring (134). Another recent study compiled a PRS of CAD, demonstrating that individuals in the highest quantiles of the PRS exhibited CAD risk on par with known Mendelian cardiac diseases (135). These studies highlight the potential uses of PRS as a genetic biomarker of disease, capturing orthogonal risk information compared to clinical risk factors alone. Much work is still needed in this arena, ranging from derivation of readily accessible clinical genomic testing, dissemination of PRS results in an interpretable format, disclosure of off-target results that may be clinically meaningful in their own right, and, critically, the validation of PRS in ancestrally-diverse populations (136). Despite these challenges, utilization of polygenic risk data to directly inform patient risk independent of our understanding of the underlying mechanisms is an exciting and rapidly evolving use-case for translational genomics.

Polygenic risk scores (PRS). A PRS for each individual in a population is calculated based on the summing the content of a large group of single nucleotide polymorphisms (SNPs) weighted by the effect size of each SNP on the disease of interest. Each individual is assigned a PRS percentile across the population distribution. Plotting percentiles by disease risk, patients in higher PRS percentiles (red dots) are at correspondingly highest risk for the disease
Biomarkers
Development of biomarkers is another approach in translational genomics that focuses more on predictive utilization than on elucidating mechanisms, and critical care has seen some early potential applications of this approach. In sepsis, where clinical outcomes are highly heterogenous, tools that might identify patients who are more likely to respond to certain treatments or identify individuals at highest risk for morbidity and mortality would be highly useful. In a recent study by Scicluna et al., the authors categorized sepsis patients based on peripheral blood-derived genome-wide expression profiles and identified four distinct molecular endotypes (Mars1–4) (137). The Mars1 expression profile was the only category that was significantly associated with 28-day and 1-year mortality. In addition, combination of the APACHE IV clinical score with this genetic scoring system resulted in significant improvement in 28-day mortality risk prediction, compared to APACHE IV alone. To further aid translation to clinical application, the authors used expression ratios of combinations of genes to stratify patients to the four molecular endotypes. Bisphosphoglycerate mutase (BPGM): Transporter 2, ATP binding cassette subfamily B member (TAP2) ratio reliably stratified patients to Mars1 endotype; while other protein ratios were able to assign individuals to the other three MARS categories. Using this approach, not only could BPGM and TAP2 transcripts potentially be used to identify patients with increased risk of mortality, but if these categorizations can be demonstrated to be causal, these molecular pathways could also be explored for therapeutic target identification and validation. Further work is required to extend these findings across clinical populations, but this approach could ultimately yield new tools for prognostication in sepsis.
In ischemic stroke, tissue plasminogen activator (t-PA) response and risk for hemorrhagic transformation (HT) are highly correlated with functional outcomes, and biomarkers to predict each of these would have obvious clinical utility. In a recent study, del Rio-Espinola et al. found that two genetic variations (rs669 and rs1801020) were associated with increased risk for HT and mortality after t-PA administration in stroke patients (138). Specifically, rs669 in A2M was associated with HT and rs1801020 was associated with in-hospital mortality. In a subsequent validation study, researchers created a genetic-clinical regression score that was successfully used to stratify stroke patients treated with t-PA based on risk for HT and parenchymal hemorrhage (PH) (139). While in the current clinical landscape the vast majority of patients do not have readily accessible genome-wide genotypes prior to events like acute stroke, increasing uptake of clinical genomics and genomically-enabled electronic health record systems could soon enable real-time risk prediction calculations incorporating both clinical and genetic information, providing more accurate tools for clinicians to incorporate into medical decision-making.
Whole Blood Transcriptomics
A separate set of tools that could potentially become diagnostically useful in the clinical setting is the transcriptomic approaches to identify biomarkers, using array-based screening or RNA sequencing. In a recent systematic review, a total of 22 miRNAs were reported to be differentially expressed in the blood cells of patients with acute ischemic stroke within 24 h after stroke (140). Some studies reported the area under the curve (AUC) ranging from 0.76 to 0.987, indicating a potential for clinical utility as early diagnostic markers when neuroimaging is not immediately available or is limited by feasibility. Subsequent studies were able to partially replicate these findings, showing three miRNAs (miR-125a-5p, miR-125b-5p, and miR-143-3p) that were upregulated in the acute post-stroke period, an effect independent of stroke pathophysiology and infarct volume (141). These transcripts were associated with an AUC of 0.90 in differentiating ischemic stroke and healthy controls, a metric that significantly outperformed computed tomography, as well as previously reported blood-based biomarkers.
In ICH, a recent report identified up to 489 and 256 transcripts from whole blood that are differentially expressed between ICH and controls and, ICH and ischemic stroke, respectively (142). When comparing ICH and ischemic stroke transcriptomes in the first 24 h, 2667 and 311 transcripts were differentially expressed compared to controls, respectively. ICH transcriptome was over-represented by T cell receptor genes compared to none for ischemic stroke and under-represented by non-coding and antisense transcripts. T cell receptor expression successfully differentiated between ICH, ischemic stroke, and controls. Similarly, RNA-seq of whole blood RNA successfully differentiated between not only ICH, ischemic stroke and controls, but also between different stroke subtypes (143).
Next-Generation Sequencing for Status Epilepticus
The list of genetic mutations that can cause SE is extensive with most genes are associated with infantile-onset or childhood epilepsy syndromes. Only a minority are seen in adult-onset status epilepticus (144). The patients in the former group usually have accompanying intellectual disability related to their epilepsy syndromes. However, the evidence supporting a genetic etiology in the latter group may be absent, posing a diagnostic challenge. The available options include gene panel sequencing, whole exome sequencing, or whole genome sequencing. Sequencing a pre-selected panel of genes is more common, but with decreasing cost, exome and genome sequencing are being used with increasing frequency. Bioinformatic filtering and genotype-phenotype correlation are the main challenges, particularly with the large number of genetic variants identified during whole exome or genome sequencing. The yield of sequencing studies depends on pretest probability that is determined by early age of onset, consanguinity, or affected siblings. As such, to date, only a few genes associated with adult-onset SE have been identified, posing a practical limitation that predominantly limits next-generation sequencing to pediatric patients at present (144). As clinical tools for determination of putative functional significance and deleteriousness of variants identified through sequencing are refined, it is hoped that sequencing approaches find a home in the armamentarium of the clinicians treating refractory or recurrent SE in the neurocritical care unit.
Next Steps
Translational genomics undoubtedly represents an important component to overall efforts to improve our understanding of the diseases we treat, and in principle should improve our ability to identify therapeutic approaches to improve outcomes and, in some cases, prevent disease altogether. Given the inherent complexity and inaccessibility of the human brain and its tissues, combined with the relative infrequency of the conditions we treat at the overall population level, progress has been modest when compared to conditions such as hyperlipidemia (145), coronary artery disease (146), or atrial fibrillation (147). Nevertheless, the observation-based, hypothesis-free experimental process inherent to translational genomics lends itself well to conditions such as stroke and TBI in which the search for the “master regulator” that governs response to injury has remained elusive despite carefully designed and executed hypothesis-driven studies.
An important component to future translational genomic studies in neurocritical care is the pressing need for collaboration across centers with access to large, well-characterized patient populations. The success of the International Stroke Genetics Consortium, and the TRACK-TBI and CENTER-TBI consortia in amassing large human populations with stroke and traumatic brain injury, respectively, is a proven model to accelerate the human genomic studies that serve as the basis for reverse genomic translational research (112, 148,149,150). Similar efforts through the Critical Care EEG Monitoring Research Consortium and other partners could lead to biorepositories of specific conditions relevant to neurocritical care that could provide sample sizes sufficient to drive unbiased genomic discoveries (151).
Close alliances with model systems researchers are another critical component to accelerating translational genomics in neurocritical care. As characterization of human disease through multimodal and continuous physiological monitoring, electrophysiology, medical imaging, and biomarker sampling continues to evolve, it is imperative that this information is shared and explored with allied model systems researchers to ensure that models are re-evaluated for their correlation with these endophenotypes, and potentially for dedicated exploration of how these human-derived phenotypes inform on the utility of specific model systems to investigate disease.
Finally, building relationships with biotechnology and pharmaceutical industry partners will be essential to efforts to extend therapeutic targets arising from translational genomic discoveries towards drug development (152). While repurposing existing drug compounds for new indications is an important consideration, small molecule and biologic targets are likely to require extensive research and development in the preclinical and clinical space, and industry partners are often optimized for these phases of the therapeutic development process (153, 154). Relatedly, development of polygenic risk scores for assessment of risk, prognosis, or treatment response will also require commercial investment and infrastructure, as few academic environments exist that can manage CLIA-certified genotyping, quality control, and result reporting and interpretation for on-target and clinically relevant secondary results (155, 156). Particularly in rarer or particularly challenging disease indications like those commonly encountered in neurocritical care populations, academic-industry partnerships are important to raise awareness of and interest in important clinical indications where investment could yield a large impact on a relatively small population of patients.
Conclusion
Translational genomics, in which genomic associations with risk, outcome, or treatment response are systematically identified and explored for functional relevance in humans or model systems of disease, is a valuable tool for identification of mechanisms, risk factors, therapeutic targets, and risk estimates in multiple diseases that are highly relevant to clinicians and scientists operating in the neurocritical care space. While there are undoubtedly challenges to studying some of the most complex diseases that affect the most complex organ in the body, translational genomic approaches may be uniquely suited to this task. Coordinated investments in the collaborations, consortia, and infrastructures that enable these studies are likely to contribute to the novel treatments and biomarkers that are so sorely needed in the highly morbid and often poorly understood conditions in the patient populations we serve.
References
Kussmann M, Kaput J. Translational genomics. Appl Transl Genom. 2014;3(3):43–7.
Collins FS. Reengineering translational science: the time is right. Sci Transl Med. 2011;3(90):90cm17.
Green ED, Watson JD, Collins FS. Human Genome Project: Twenty-five years of big biology. Nature. 2015;526(7571):29–31.
Shakhnovich V. It's Time to Reverse our Thinking: The Reverse Translation Research Paradigm. Clin Transl Sci. 2018;11(2):98–9.
Rubio DM, Schoenbaum EE, Lee LS, Schteingart DE, Marantz PR, Anderson KE, et al. Defining translational research: implications for training. Acad Med. 2010;85(3):470–5.
Mayer AR, Dodd AB, Vermillion MS, Stephenson DD, Chaudry IH, Bragin DE, et al. A systematic review of large animal models of combined traumatic brain injury and hemorrhagic shock. Neurosci Biobehav Rev. 2019;104:160–77.
Rubin TG, Lipton ML. Sex Differences in Animal Models of Traumatic Brain Injury. J Exp Neurosci. 2019;13:1179069519844020.
Ma X, Aravind A, Pfister BJ, Chandra N, Haorah J. Animal Models of Traumatic Brain Injury and Assessment of Injury Severity. Molecular neurobiology. 2019;56(8):5332–45.
Song YM, Qian Y, Su WQ, Liu XH, Huang JH, Gong ZT, et al. Differences in pathological changes between two rat models of severe traumatic brain injury. Neural Regen Res. 2019;14(10):1796–804.
Neuhaus AA, Couch Y, Hadley G, Buchan AM. Neuroprotection in stroke: the importance of collaboration and reproducibility. Brain. 2017;140(8):2079–92.
Cheng YD, Al-Khoury L, Zivin JA. Neuroprotection for ischemic stroke: two decades of success and failure. NeuroRx : the journal of the American Society for Experimental NeuroTherapeutics. 2004;1(1):36–45.
Gladstone DJ, Black SE, Hakim AM, Heart, Stroke Foundation of Ontario Centre of Excellence in Stroke R. Toward wisdom from failure: lessons from neuroprotective stroke trials and new therapeutic directions. Stroke. 2002;33(8):2123–36.
Wright DW, Yeatts SD, Silbergleit R, Palesch YY, Hertzberg VS, Frankel M, et al. Very early administration of progesterone for acute traumatic brain injury. N Engl J Med. 2014;371(26):2457–66.
Stein DG. Embracing failure: What the Phase III progesterone studies can teach about TBI clinical trials. Brain Inj. 2015;29(11):1259–72.
Guo Y, Yang TL, Liu YZ, Shen H, Lei SF, Yu N, et al. Mitochondria-wide association study of common variants in osteoporosis. Annals of human genetics. 2011;75(5):569–74.
Auer PL, Stitziel NO. Genetic association studies in cardiovascular diseases: Do we have enough power? Trends Cardiovasc Med. 2017;27(6):397–404.
Kosmicki JA, Churchhouse CL, Rivas MA, Neale BM. Discovery of rare variants for complex phenotypes. Hum Genet. 2016;135(6):625–34.
Dichgans M, Pulit SL, Rosand J. Stroke genetics: discovery, biology, and clinical applications. Lancet Neurol. 2019;18(6):587–99.
Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet. 2013;14(10):681–91.
Becker AJ. Review: Animal models of acquired epilepsy: insights into mechanisms of human epileptogenesis. Neuropathol Appl Neurobiol. 2018;44(1):112–29.
Penn DL, Witte SR, Komotar RJ, Sander Connolly E, Jr. Pathological mechanisms underlying aneurysmal subarachnoid haemorrhage and vasospasm. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia. 2015;22(1):1–5.
Southern E. Tools for genomics. Nat Med. 2005;11(10):1029–34.
Tanzi RE. A brief history of Alzheimer's disease gene discovery. J Alzheimers Dis. 2013;33 Suppl 1:S5–13.
Marini S, Crawford K, Morotti A, Lee MJ, Pezzini A, Moomaw CJ, et al. Association of Apolipoprotein E With Intracerebral Hemorrhage Risk by Race/Ethnicity: A Meta-analysis. JAMA Neurol. 2019;76(4):480–91.
McFadyen CA, Zeiler FA, Newcombe V, Synnot A, Steyerberg E, Gruen RL, et al. Apolipoprotein E4 Polymorphism and Outcomes from Traumatic Brain Injury: A Living Systematic Review and Meta-Analysis. Journal of neurotrauma. 2019.
Muza P, Bachmeier C, Mouzon B, Algamal M, Rafi NG, Lungmus C, et al. APOE Genotype Specific Effects on the Early Neurodegenerative Sequelae Following Chronic Repeated Mild Traumatic Brain Injury. Neuroscience. 2019;404:297–313.
Farrell MS, Werge T, Sklar P, Owen MJ, Ophoff RA, O'Donovan MC, et al. Evaluating historical candidate genes for schizophrenia. Mol Psychiatry. 2015;20(5):555–62.
Falcone GJ, Malik R, Dichgans M, Rosand J. Current concepts and clinical applications of stroke genetics. Lancet Neurol. 2014;13(4):405–18.
Devlin B, Roeder K, Bacanu SA. Unbiased methods for population-based association studies. Genet Epidemiol. 2001;21(4):273–84.
Conomos MP, Miller MB, Thornton TA. Robust inference of population structure for ancestry prediction and correction of stratification in the presence of relatedness. Genet Epidemiol. 2015;39(4):276–93.
Biffi A, Sonni A, Anderson CD, Kissela B, Jagiella JM, Schmidt H, et al. Variants at APOE influence risk of deep and lobar intracerebral hemorrhage. Ann Neurol. 2010;68(6):934–43.
International HapMap C. The International HapMap Project. Nature. 2003;426(6968):789–96.
Levy SE, Myers RM. Advancements in Next-Generation Sequencing. Annu Rev Genomics Hum Genet. 2016;17:95–115.
Petersen BS, Fredrich B, Hoeppner MP, Ellinghaus D, Franke A. Opportunities and challenges of whole-genome and -exome sequencing. BMC Genet. 2017;18(1):14.
Belkadi A, Bolze A, Itan Y, Cobat A, Vincent QB, Antipenko A, et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci U S A. 2015;112(17):5473–8.
Sham PC, Purcell SM. Statistical power and significance testing in large-scale genetic studies. Nat Rev Genet. 2014;15(5):335–46.
Bourcier R, Le Scouarnec S, Bonnaud S, Karakachoff M, Bourcereau E, Heurtebise-Chretien S, et al. Rare Coding Variants in ANGPTL6 Are Associated with Familial Forms of Intracranial Aneurysm. Am J Hum Genet. 2018;102(1):133–41.
Belal H, Nakashima M, Matsumoto H, Yokochi K, Taniguchi-Ikeda M, Aoto K, et al. De novo variants in RHOBTB2, an atypical Rho GTPase gene, cause epileptic encephalopathy. Hum Mutat. 2018;39(8):1070–5.
Liu Y, Mi Y, Mueller T, Kreibich S, Williams EG, Van Drogen A, et al. Multi-omic measurements of heterogeneity in HeLa cells across laboratories. Nat Biotechnol. 2019;37(3):314–22.
Vierstra J, Rynes E, Sandstrom R, Zhang M, Canfield T, Hansen RS, et al. Mouse regulatory DNA landscapes reveal global principles of cis-regulatory evolution. Science. 2014;346(6212):1007–12.
Bambakidis T, Dekker SE, Sillesen M, Liu B, Johnson CN, Jin G, et al. Resuscitation with Valproic Acid Alters Inflammatory Genes in a Porcine Model of Combined Traumatic Brain Injury and Hemorrhagic Shock. Journal of neurotrauma. 2016;33(16):1514–21.
Arneson D, Zhang G, Ying Z, Zhuang Y, Byun HR, Ahn IS, et al. Single cell molecular alterations reveal target cells and pathways of concussive brain injury. Nat Commun. 2018;9(1):3894.
Hacke W, Schwab S, Horn M, Spranger M, De Georgia M, von Kummer R. 'Malignant' middle cerebral artery territory infarction: clinical course and prognostic signs. Archives of neurology. 1996;53(4):309–15.
Loh KP, Ng G, Yu CY, Fhu CK, Yu D, Vennekens R, Nilius B, Soong TW, Liao P. TRPM4 inhibition promotes angiogenesis after ischemic stroke. Pflugers Arch. 2014;466(3):563–76. https://doi.org/10.1007/s00424-013-1347-4.
Mehta RI, Tosun C, Ivanova S, Tsymbalyuk N, Famakin BM, Kwon MS, Castellani RJ, Gerzanich V, Simard JM. Sur1-Trpm4 Cation Channel Expression in Human Cerebral Infarcts. Journal of neuropathology and experimental neurology. 2015;74(8):835–49. https://doi.org/10.1097/NEN.0000000000000223.
Arikan F, Martinez-Valverde T, Sanchez-Guerrero A, Campos M, Esteves M, Gandara D, Torne R, Castro L, Dalmau A, Tibau J, Sahuquillo J. Malignant infarction of the middle cerebral artery in a porcine model. A pilot study. PloS one. 2017;12(2):e0172637. https://doi.org/10.1371/journal.pone.0172637.
Simard JM, Chen M, Tarasov KV, Bhatta S, Ivanova S, Melnitchenko L, Tsymbalyuk N, West GA, Gerzanich V. Newly expressed SUR1-regulated NC(Ca-ATP) channel mediates cerebral edema after ischemic stroke. Nat Med. 2006;12(4):433–40. https://doi.org/10.1038/nm1390.
Mehta RI, Ivanova S, Tosun C, Castellani RJ, Gerzanich V, Simard JM. Sulfonylurea receptor 1 expression in human cerebral infarcts. Journal of neuropathology and experimental neurology. 2013;72(9):871–83. https://doi.org/10.1097/NEN.0b013e3182a32e40.
Simard JM, Geng Z, Silver FL, Sheth KN, Kimberly WT, Stern BJ, Colucci M, Gerzanich V. Does inhibiting Sur1 complement rt-PA in cerebral ischemia? Annals of the New York Academy of Sciences. 2012;1268:95–107. https://doi.org/10.1111/j.1749-6632.2012.06705.x.
Simard JM, Tsymbalyuk N, Tsymbalyuk O, Ivanova S, Yurovsky V, Gerzanich V. Glibenclamide is superior to decompressive craniectomy in a rat model of malignant stroke. Stroke; a journal of cerebral circulation. 2010;41(3):531–7. https://doi.org/10.1161/STROKEAHA.109.572644.
Ortega FJ, Gimeno-Bayon J, Espinosa-Parrilla JF, Carrasco JL, Batlle M, Pugliese M, Mahy N, Rodriguez MJ. ATP-dependent potassium channel blockade strengthens microglial neuroprotection after hypoxia-ischemia in rats. Exp Neurol. 2012;235(1):282–96. https://doi.org/10.1016/j.expneurol.2012.02.010.
Stokum JA, Keledjian K, Hayman E, Karimy JK, Pampori A, Imran Z, Woo SK, Gerzanich V, Simard JM. Glibenclamide pretreatment protects against chronic memory dysfunction and glial activation in rat cranial blast traumatic brain injury. Behav Brain Res. 2017;333:43–53. https://doi.org/10.1016/j.bbr.2017.06.038.
Khalili H, Derakhshan N, Niakan A, Ghaffarpasand F, Salehi M, Eshraghian H, Shakibafard A, Zahabi B. Effects of Oral Glibenclamide on Brain Contusion Volume and Functional Outcome of Patients with Moderate and Severe Traumatic Brain Injuries: A Randomized Double-Blind Placebo-Controlled Clinical Trial. World Neurosurg. 2017;101:130–6. https://doi.org/10.1016/j.wneu.2017.01.103.
Kimberly WT, Bevers MB, von Kummer R, Demchuk AM, Romero JM, Elm JJ, Hinson HE, Molyneaux BJ, Simard JM, Sheth KN. Effect of IV glyburide on adjudicated edema endpoints in the GAMES-RP Trial. Neurology. 2018;91(23):e2163-e9. https://doi.org/10.1212/WNL.0000000000006618.
Reilly PL. Brain injury: the pathophysiology of the first hours.'Talk and Die revisited'. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia. 2001;8(5):398–403.
Stoica BA, Faden AI. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2010;7(1):3–12.
Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA, et al. Lateral fluid percussion brain injury: a 15-year review and evaluation. Journal of neurotrauma. 2005;22(1):42–75.
Hicks R, Soares H, Smith D, McIntosh T. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta neuropathologica. 1996;91(3):236–46.
Albert-Weissenberger C, Siren AL. Experimental traumatic brain injury. Experimental & translational stroke medicine. 2010;2(1):16.
Cernak I. Animal models of head trauma. NeuroRx : the journal of the American Society for Experimental NeuroTherapeutics. 2005;2(3):410–22.
Marmarou A, Foda MA, van den Brink W, Campbell J, Kita H, Demetriadou K. A new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. Journal of neurosurgery. 1994;80(2):291–300.
Feeney DM, Boyeson MG, Linn RT, Murray HM, Dail WG. Responses to cortical injury: I. Methodology and local effects of contusions in the rat. Brain research. 1981;211(1):67–77.
Williams AJ, Hartings JA, Lu XC, Rolli ML, Dave JR, Tortella FC. Characterization of a new rat model of penetrating ballistic brain injury. Journal of neurotrauma. 2005;22(2):313–31.
Leung LY, VandeVord PJ, Dal Cengio AL, Bir C, Yang KH, King AI. Blast related neurotrauma: a review of cellular injury. Molecular & cellular biomechanics : MCB. 2008;5(3):155–68.
Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nature reviews Neuroscience. 2013;14(2):128–42.
Alessandri B, Heimann A, Filippi R, Kopacz L, Kempski O. Moderate controlled cortical contusion in pigs: effects on multi-parametric neuromonitoring and clinical relevance. Journal of neurotrauma. 2003;20(12):1293–305.
Stein DG. Progesterone in the treatment of acute traumatic brain injury: a clinical perspective and update. Neuroscience. 2011;191:101–6.
Espinoza TR, Wright DW. The role of progesterone in traumatic brain injury. The Journal of head trauma rehabilitation. 2011;26(6):497–9.
Samal BB, Waites CK, Almeida-Suhett C, Li Z, Marini AM, Samal NR, et al. Acute Response of the Hippocampal Transcriptome Following Mild Traumatic Brain Injury After Controlled Cortical Impact in the Rat. Journal of molecular neuroscience : MN. 2015;57(2):282–303.
Lipponen A, Paananen J, Puhakka N, Pitkanen A. Analysis of Post-Traumatic Brain Injury Gene Expression Signature Reveals Tubulins, Nfe2l2, Nfkb, Cd44, and S100a4 as Treatment Targets. Sci Rep. 2016;6:31570.
Izzy S, Liu Q, Fang Z, Lule S, Wu L, Chung JY, et al. Time-Dependent Changes in Microglia Transcriptional Networks Following Traumatic Brain Injury. Frontiers in cellular neuroscience. 2019;13:307.
Claassen J, Hirsch LJ, Emerson RG, Mayer SA. Treatment of refractory status epilepticus with pentobarbital, propofol, or midazolam: a systematic review. Epilepsia. 2002;43(2):146–53.
Loscher W. Animal Models of Seizures and Epilepsy: Past, Present, and Future Role for the Discovery of Antiseizure Drugs. Neurochemical research. 2017;42(7):1873–88.
Reddy DS, Kuruba R. Experimental models of status epilepticus and neuronal injury for evaluation of therapeutic interventions. International journal of molecular sciences. 2013;14(9):18284–318.
Loscher W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure. 2011;20(5):359–68.
Nirwan N, Vyas P, Vohora D. Animal models of status epilepticus and temporal lobe epilepsy: a narrative review. Reviews in the neurosciences. 2018;29(7):757–70.
Morimoto K, Fahnestock M, Racine RJ. Kindling and status epilepticus models of epilepsy: rewiring the brain. Progress in neurobiology. 2004;73(1):1–60.
Sharma S, Puttachary S, Thippeswamy A, Kanthasamy AG, Thippeswamy T. Status Epilepticus: Behavioral and Electroencephalography Seizure Correlates in Kainate Experimental Models. Frontiers in neurology. 2018;9:7.
Muller CJ, Groticke I, Hoffmann K, Schughart K, Loscher W. Differences in sensitivity to the convulsant pilocarpine in substrains and sublines of C57BL/6 mice. Genes, brain, and behavior. 2009;8(5):481–92.
Araujo MA, Marques TE, Octacilio-Silva S, Arroxelas-Silva CL, Pereira MG, Peixoto-Santos JE, et al. Identification of microRNAs with Dysregulated Expression in Status Epilepticus Induced Epileptogenesis. PloS one. 2016;11(10):e0163855.
Brennan GP, Dey D, Chen Y, Patterson KP, Magnetta EJ, Hall AM, et al. Dual and Opposing Roles of MicroRNA-124 in Epilepsy Are Mediated through Inflammatory and NRSF-Dependent Gene Networks. Cell reports. 2016;14(10):2402–12.
Srivastava PK, Bagnati M, Delahaye-Duriez A, Ko JH, Rotival M, Langley SR, et al. Genome-wide analysis of differential RNA editing in epilepsy. Genome research. 2017;27(3):440–50.
Leclerc JL, Garcia JM, Diller MA, Carpenter AM, Kamat PK, Hoh BL, et al. A Comparison of Pathophysiology in Humans and Rodent Models of Subarachnoid Hemorrhage. Frontiers in molecular neuroscience. 2018;11:71.
Izzy S, Muehlschlegel S. Cerebral vasospasm after aneurysmal subarachnoid hemorrhage and traumatic brain injury. Current treatment options in neurology. 2014;16(1):278.
Zibly Z, Fein L, Sharma M, Assaf Y, Wohl A, Harnof S. A novel swine model of subarachnoid hemorrhage-induced cerebral vasospasm. Neurology India. 2017;65(5):1035–42.
Prunell GF, Mathiesen T, Diemer NH, Svendgaard NA. Experimental subarachnoid hemorrhage: subarachnoid blood volume, mortality rate, neuronal death, cerebral blood flow, and perfusion pressure in three different rat models. Neurosurgery. 2003;52(1):165–75; discussion 75-6.
Schüller K, Bühler D, Plesnila N. A murine model of subarachnoid hemorrhage. J Vis Exp. 2013(81):e50845-e.
Jeon H, Ai J, Sabri M, Tariq A, Macdonald RL. Learning deficits after experimental subarachnoid hemorrhage in rats. Neuroscience. 2010;169(4):1805–14.
Lin CL, Calisaneller T, Ukita N, Dumont AS, Kassell NF, Lee KS. A murine model of subarachnoid hemorrhage-induced cerebral vasospasm. Journal of neuroscience methods. 2003;123(1):89–97.
Raslan F, Albert-Weissenberger C, Westermaier T, Saker S, Kleinschnitz C, Lee JY. A modified double injection model of cisterna magna for the study of delayed cerebral vasospasm following subarachnoid hemorrhage in rats. Experimental & translational stroke medicine. 2012;4(1):23.
Munoz-Sanchez MA, Egea-Guerrero JJ, Revuelto-Rey J, Moreno-Valladares M, Murillo-Cabezas F. A new percutaneous model of Subarachnoid Haemorrhage in rats. Journal of neuroscience methods. 2012;211(1):88–93.
Makino H, Tada Y, Wada K, Liang EI, Chang M, Mobashery S, et al. Pharmacological stabilization of intracranial aneurysms in mice: a feasibility study. Stroke. 2012;43(9):2450–6.
Nuki Y, Tsou TL, Kurihara C, Kanematsu M, Kanematsu Y, Hashimoto T. Elastase-induced intracranial aneurysms in hypertensive mice. Hypertension (Dallas, Tex : 1979). 2009;54(6):1337–44.
Tada Y, Kanematsu Y, Kanematsu M, Nuki Y, Liang EI, Wada K, et al. A mouse model of intracranial aneurysm: technical considerations. Acta neurochirurgica Supplement. 2011;111:31–5.
Macdonald RL, Higashida RT, Keller E, Mayer SA, Molyneux A, Raabe A, Vajkoczy P, Wanke I, Bach D, Frey A, Marr A, Roux S, Kassell N. Clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: a randomised, doubleblind, placebo-controlled phase 3 trial (CONSCIOUS–2). Lancet neurology. 2011;10(7):618–25. https://doi.org/10.1016/S1474-4422(11)70108-9.
Macdonald RL, Higashida RT, Keller E, Mayer SA, Molyneux A, Raabe A, Vajkoczy P, Wanke I, Bach D, Frey A, Nowbakht P, Roux S, Kassell N. Randomized trial of clazosentan in patients with aneurysmal subarachnoid hemorrhage undergoing endovascular coiling. Stroke; a journal of cerebral circulation. 2012;43(6):1463–9. https://doi.org/10.1161/STROKEAHA.111.648980.
Pluta RM, Hansen-Schwartz J, Dreier J, Vajkoczy P, Macdonald RL, Nishizawa S, et al. Cerebral vasospasm following subarachnoid hemorrhage: time for a new world of thought. Neurological research. 2009;31(2):151–8.
Greenhalgh AD, Brough D, Robinson EM, Girard S, Rothwell NJ, Allan SM. Interleukin-1 receptor antagonist is beneficial after subarachnoid haemorrhage in rat by blocking haem-driven inflammatory pathology. Disease Models & Mechanisms. 2012;5(6):823–33.
Muller AH, Edwards AVG, Larsen MR, Nielsen J, Warfvinge K, Povlsen GK, et al. Proteomic Expression Changes in Large Cerebral Arteries After Experimental Subarachnoid Hemorrhage in Rat Are Regulated by the MEK-ERK1/2 Pathway. Journal of molecular neuroscience : MN. 2017;62(3–4):380–94.
Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, et al. The support of human genetic evidence for approved drug indications. Nature genetics. 2015;47:856.
Traylor M, Farrall M, Holliday EG, Sudlow C, Hopewell JC, Cheng YC, et al. Genetic risk factors for ischaemic stroke and its subtypes (the METASTROKE collaboration): a meta-analysis of genome-wide association studies. Lancet Neurol. 2012;11(11):951–62.
Bellenguez C, Bevan S, Gschwendtner A, Spencer CC, Burgess AI, Pirinen M, et al. Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nature genetics. 2012;44(3):328–33.
Azghandi S, Prell C, van der Laan SW, Schneider M, Malik R, Berer K, et al. Deficiency of the stroke relevant HDAC9 gene attenuates atherosclerosis in accord with allele-specific effects at 7p21.1. Stroke. 2015;46(1):197–202.
Brookes RL, Crichton S, Wolfe CDA, Yi Q, Li L, Hankey GJ, et al. Sodium Valproate, a Histone Deacetylase Inhibitor, Is Associated With Reduced Stroke Risk After Previous Ischemic Stroke or Transient Ischemic Attack. Stroke; a journal of cerebral circulation. 2018;49(1):54–61.
Xiong K, Zhang H, Du Y, Tian J, Ding S. Identification of HDAC9 as a viable therapeutic target for the treatment of gastric cancer. Exp Mol Med. 2019;51(8):100.
Bime C, Pouladi N, Sammani S, Batai K, Casanova N, Zhou T, et al. Genome-Wide Association Study in African Americans with Acute Respiratory Distress Syndrome Identifies the Selectin P Ligand Gene as a Risk Factor. American journal of respiratory and critical care medicine. 2018;197(11):1421–32.
Foroud T, Koller DL, Lai D, Sauerbeck L, Anderson C, Ko N, et al. Genome-wide association study of intracranial aneurysms confirms role of Anril and SOX17 in disease risk. Stroke. 2012;43(11):2846–52.
Yasuno K, Bilguvar K, Bijlenga P, Low SK, Krischek B, Auburger G, et al. Genome-wide association study of intracranial aneurysm identifies three new risk loci. Nature genetics. 2010;42(5):420–5.
Kong Y, Hsieh CH, Alonso LC. ANRIL: A lncRNA at the CDKN2A/B Locus With Roles in Cancer and Metabolic Disease. Frontiers in endocrinology. 2018;9:405.
Song CL, Wang JP, Xue X, Liu N, Zhang XH, Zhao Z, et al. Effect of Circular ANRIL on the Inflammatory Response of Vascular Endothelial Cells in a Rat Model of Coronary Atherosclerosis. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2017;42(3):1202–12.
Bush WS, Moore JH. Chapter 11: Genome-wide association studies. PLoS computational biology. 2012;8(12):e1002822.
Majersik JJ, Cole JW, Golledge J, Rost NS, Chan YF, Gurol ME, et al. Recommendations from the international stroke genetics consortium, part 1: standardized phenotypic data collection. Stroke. 2015;46(1):279–84.
Habek M, Brinar VV, Borovecki F. Genes associated with multiple sclerosis: 15 and counting. Expert review of molecular diagnostics. 2010;10(7):857–61.
Grigoryev DN, Cheranova DI, Chaudhary S, Heruth DP, Zhang LQ, Ye SQ. Identification of new biomarkers for Acute Respiratory Distress Syndrome by expression-based genome-wide association study. BMC pulmonary medicine. 2015;15:95.
Justice MJ, Siracusa LD, Stewart AF. Technical approaches for mouse models of human disease. Disease models & mechanisms. 2011;4(3):305–10.
Gurumurthy CB, Lloyd KCK. Generating mouse models for biomedical research: technological advances. Disease models & mechanisms. 2019;12(1). https://doi.org/10.1242/dmm.029462.
Peng R, Lin G, Li J. Potential pitfalls of CRISPR/Cas9-mediated genome editing. FEBS J. 2016;283(7):1218–31. https://doi.org/10.1111/febs.13586.
Fatemi SH. The role of secretory granules in the transport of basement membrane components: radioautographic studies of rat parietal yolk sac employing 3H-proline as a precursor of type IV collagen. Connective tissue research. 1987;16(1):1–14.
Mayne R, Wiedemann H, Irwin MH, Sanderson RD, Fitch JM, Linsenmayer TF, et al. Monoclonal antibodies against chicken type IV and V collagens: electron microscopic mapping of the epitopes after rotary shadowing. The Journal of cell biology. 1984;98(5):1637–44.
Trueb B, Grobli B, Spiess M, Odermatt BF, Winterhalter KH. Basement membrane (type IV) collagen is a heteropolymer. The Journal of biological chemistry. 1982;257(9):5239–45.
Lanfranconi S, Markus HS. COL4A1 mutations as a monogenic cause of cerebral small vessel disease: a systematic review. Stroke. 2010;41(8):e513–8.
Rannikmae K, Davies G, Thomson PA, Bevan S, Devan WJ, Falcone GJ, et al. Common variation in COL4A1/COL4A2 is associated with sporadic cerebral small vessel disease. Neurology. 2015;84(9):918–26.
Yamamoto Y, Craggs L, Baumann M, Kalimo H, Kalaria RN. Review: molecular genetics and pathology of hereditary small vessel diseases of the brain. Neuropathol Appl Neurobiol. 2011;37(1):94–113.
Chung J, Marini S, Pera J, Norrving B, Jimenez-Conde J, Roquer J, et al. Genome-wide association study of cerebral small vessel disease reveals established and novel loci. Brain. 2019.
Jeanne M, Jorgensen J, Gould DB. Molecular and Genetic Analyses of Collagen Type IV Mutant Mouse Models of Spontaneous Intracerebral Hemorrhage Identify Mechanisms for Stroke Prevention. Circulation. 2015;131(18):1555–65.
Jeanne M, Labelle-Dumais C, Jorgensen J, Kauffman WB, Mancini GM, Favor J, et al. COL4A2 mutations impair COL4A1 and COL4A2 secretion and cause hemorrhagic stroke. Am J Hum Genet. 2012;90(1):91–101.
Firtina Z, Danysh BP, Bai X, Gould DB, Kobayashi T, Duncan MK. Abnormal expression of collagen IV in lens activates unfolded protein response resulting in cataract. The Journal of biological chemistry. 2009;284(51):35872–84.
Gould DB, Marchant JK, Savinova OV, Smith RS, John SW. Col4a1 mutation causes endoplasmic reticulum stress and genetically modifiable ocular dysgenesis. Human molecular genetics. 2007;16(7):798–807.
Johnstone DL, Al-Shekaili HH, Tarailo-Graovac M, Wolf NI, Ivy AS, Demarest S, et al. PLPHP deficiency: clinical, genetic, biochemical, and mechanistic insights. Brain. 2019;142(3):542–59.
Dai W, Lee D. Interfering with long chain noncoding RNA ANRIL expression reduces heart failure in rats with diabetes by inhibiting myocardial oxidative stress. Journal of cellular biochemistry. 2019;120(10):18446–56.
Guo F, Tang C, Li Y, Liu Y, Lv P, Wang W, et al. The interplay of LncRNA ANRIL and miR-181b on the inflammation-relevant coronary artery disease through mediating NF-kappaB signalling pathway. Journal of cellular and molecular medicine. 2018;22(10):5062–75.
Gong Z, Hu G, Li Q, Liu Z, Wang F, Zhang X, et al. Compound Libraries: Recent Advances and Their Applications in Drug Discovery. Current drug discovery technologies. 2017;14(4):216–28.
Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nature genetics. 2018;50(4):524–37.
Pulit SL, Weng LC, McArdle PF, Trinquart L, Choi SH, Mitchell BD, et al. Atrial fibrillation genetic risk differentiates cardioembolic stroke from other stroke subtypes. Neurology Genetics. 2018;4(6):e293.
Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, Natarajan P, Lander ES, Lubitz SA, Ellinor PT, Kathiresan S. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nature genetics. 2018;50(9):1219–24. https://doi.org/10.1038/s41588-018-0183-z.
Martin AR, Daly MJ, Robinson EB, Hyman SE, Neale BM. Predicting Polygenic Risk of Psychiatric Disorders. Biological psychiatry. 2019;86(2):97–109.
Scicluna BP, van Vught LA, Zwinderman AH, Wiewel MA, Davenport EE, Burnham KL, et al. Classification of patients with sepsis according to blood genomic endotype: a prospective cohort study. The Lancet Respiratory medicine. 2017;5(10):816–26.
del Rio-Espinola A, Fernandez-Cadenas I, Giralt D, Quiroga A, Gutierrez-Agullo M, Quintana M, et al. A predictive clinical-genetic model of tissue plasminogen activator response in acute ischemic stroke. Ann Neurol. 2012;72(5):716–29.
Carrera C, Cullell N, Torres-Aguila N, Muino E, Bustamante A, Davalos A, et al. Validation of a clinical-genetics score to predict hemorrhagic transformations after rtPA. Neurology. 2019;93(9):e851-e63.
Dewdney B, Trollope A, Moxon J, Thomas Manapurathe D, Biros E, Golledge J. Circulating MicroRNAs as Biomarkers for Acute Ischemic Stroke: A Systematic Review. Journal of stroke and cerebrovascular diseases : the official journal of National Stroke Association. 2018;27(3):522–30.
Tiedt S, Prestel M, Malik R, Schieferdecker N, Duering M, Kautzky V, et al. RNA-Seq Identifies Circulating miR-125a-5p, miR-125b-5p, and miR-143-3p as Potential Biomarkers for Acute Ischemic Stroke. Circulation research. 2017;121(8):970–80.
Stamova B, Ander BP, Jickling G, Hamade F, Durocher M, Zhan X, et al. The intracerebral hemorrhage blood transcriptome in humans differs from the ischemic stroke and vascular risk factor control blood transcriptomes. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2019;39(9):1818–35.
Dykstra-Aiello C, Jickling GC, Ander BP, Zhan X, Liu D, Hull H, et al. Intracerebral Hemorrhage and Ischemic Stroke of Different Etiologies Have Distinct Alternatively Spliced mRNA Profiles in the Blood: a Pilot RNA-seq Study. Transl Stroke Res. 2015;6(4):284–9.
Bhatnagar M, Shorvon S. Genetic mutations associated with status epilepticus. Epilepsy Behav. 2015;49:104–10. https://doi.org/10.1016/j.yebeh.2015.04.013.
Ference BA, Kastelein JJP, Ginsberg HN, Chapman MJ, Nicholls SJ, Ray KK, et al. Association of Genetic Variants Related to CETP Inhibitors and Statins With Lipoprotein Levels and Cardiovascular Risk. JAMA. 2017;318(10):947–56.
Shapiro MD, Tavori H, Fazio S. PCSK9: From Basic Science Discoveries to Clinical Trials. Circ Res. 2018;122(10):1420–38.
Ahlberg G, Refsgaard L, Lundegaard PR, Andreasen L, Ranthe MF, Linscheid N, et al. Rare truncating variants in the sarcomeric protein titin associate with familial and early-onset atrial fibrillation. Nat Commun. 2018;9(1):4316.
Battey TW, Valant V, Kassis SB, Kourkoulis C, Lee C, Anderson CD, et al. Recommendations from the international stroke genetics consortium, part 2: biological sample collection and storage. Stroke. 2015;46(1):285–90.
Lingsma HF, Yue JK, Maas AI, Steyerberg EW, Manley GT, Investigators T-T. Outcome prediction after mild and complicated mild traumatic brain injury: external validation of existing models and identification of new predictors using the TRACK-TBI pilot study. Journal of neurotrauma. 2015;32(2):83–94.
Burton A. The CENTER-TBI core study: The making-of. Lancet Neurol. 2017;16(12):958–9.
Struck AF, Rodriguez-Ruiz AA, Osman G, Gilmore EJ, Haider HA, Dhakar MB, et al. Comparison of machine learning models for seizure prediction in hospitalized patients. Ann Clin Transl Neurol. 2019;6(7):1239–47.
Berry SA, Coughlin CR, 2nd, McCandless S, McCarter R, Seminara J, Yudkoff M, LeMons C. Developing interactions with industry in rare diseases: lessons learned and continuing challenges. Genetics in medicine : official journal of the American College of Medical Genetics. 2020;22(1):219–26. https://doi.org/10.1038/s41436-019-0616-9.
Cha Y, Erez T, Reynolds IJ, Kumar D, Ross J, Koytiger G, et al. Drug repurposing from the perspective of pharmaceutical companies. Br J Pharmacol. 2018;175(2):168–80.
Paranjpe MD, Taubes A, Sirota M. Insights into Computational Drug Repurposing for Neurodegenerative Disease. Trends Pharmacol Sci. 2019;40(8):565–76.
Wynn J, Lewis K, Amendola LM, Bernhardt BA, Biswas S, Joshi M, et al. Clinical providers' experiences with returning results from genomic sequencing: an interview study. BMC Med Genomics. 2018;11(1):45.
Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19(2):249–55.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Funding
Drs. Anderson and Myserlis were supported by NINDS R01NS103924, and Dr. Anderson was further supported by the American Heart Association Strategically Focused Research Network for Atrial Fibrillation, and grants from the Massachusetts General Hospital Center for Genomic Medicine and Center for Neuroscience for this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosures
Dr. Anderson receives sponsored research support from the National Institutes of Health of the United States, the American Heart Association, Bayer AG, and Massachusetts General Hospital, and has consulted for ApoPharma, Inc.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 1225 kb)
Rights and permissions
About this article

Cite this article
Myserlis, P., Radmanesh, F. & Anderson, C.D. Translational Genomics in Neurocritical Care: a Review. Neurotherapeutics 17, 563–580 (2020). https://doi.org/10.1007/s13311-020-00838-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-020-00838-1