
Abstract
Purpose
Tissue-resident memory T (TRM) cells are a newly described subset of memory T cells. The best characterized TRM cells are CD8+ and express CD103 and CD69. These cells are non-recirculating and persist long term in tissues, providing immediate protection against invading pathogens. However, their inappropriate activation might contribute to the pathogenesis of autoimmune and inflammatory disorders. In the skin, these cells have been described in psoriasis, vitiligo, and melanoma among other diseases.
Methods
Literature review was done to highlight what is currently known on the phenotype and function of TRM cells and summarizes the available data describing their role in various cutaneous conditions.
Results
Resolved psoriatic skin and disease-naïve non-lesional skin contain a population of IL-17-producing TRM cells with shared receptor sequences that recognize common antigens and likely contribute to disease recurrence after cessation of therapy. In vitiligo, TRM cells produce perforin, granzyme B, and interferon-γ following stimulation by interleukin-15 and collaborate with recirculating memory T cells to maintain disease. In melanoma, increased accumulation of TRM cells was recently shown to correlate with improved survival in patients undergoing therapy with immune checkpoint inhibitors.
Similar content being viewed by others

Introduction
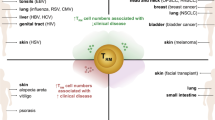
Healthy skin is populated by T cells. Following antigen exposure, naïve T cells differentiate into effector T cells capable of executing immune defense mechanisms. Most of these cells are short-lived and die following the immune response, but some remain and differentiate into memory T cells. Central memory T cells (TCM) traffic through lymphoid tissues while effector memory cells (TEM) circulate through peripheral tissues. Tissue-resident memory T (TRM) cells are a newly identified subset of memory T cells that persist long-term in tissues without recirculating in the blood thus providing a first line of adaptive cellular defense (Fig. 1) [1]. However, increasing evidence suggests that aberrant activation of these cells might contribute to the pathogenesis of autoimmune and inflammatory diseases making them a new therapeutic target [2]. It is estimated that healthy adult skin contains around 20 billion TRM cells [3]. This review summarizes the current data available on the protective and pathogenic roles of TRM cells in cutaneous disease.

Tissue-resident memory T (TRM) cell markers and the transcription factors involved in its development and survival. TRM cells are CD4+ or CD8+ T cells with a variable expression of different markers. CD103 binds E-cadherin on epithelial cells. CD69 blocks sphingosine 1-phosphate receptor 1 (S1PR1)-mediated egress from tissues. CD49 (α-subunit of the α1β1 integrin receptor) is another marker with important functional implications. The transcription factors Hobit, B lymphocyte-induced maturation protein-1 (Blimp-1), Runt Related Transcription Factor 3 (Runx3), Notch, aryl hydrocarbon receptor (AhR), eomesodermin (EOMES), transcription factor 1 (TCF1) and T-bet are all involved in the regulation of TRM cell differentiation and survival
Phenotype and functions
TRM cells have variable expression of different markers depending on the tissue of residence and the nature of the pathogen. Therefore, they might exist as different subsets with phenotypic heterogeneity (Table 1). The best characterized TRM cells are CD8+ cells that express CD103 and CD69 [4]. CD103 (αE integrin chain) binds E-cadherin potentially promoting retention within epithelial tissues [5]. CD69 is a T-cell activation marker and its expression mediates early T cell retention in the skin by blocking sphingosine 1-phosphate receptor 1 (S1PR1)-mediated egress from tissues [6]. The expression of CD69 precedes that of CD103. In fact, the former marker has a major influence on the early lodgment of TRM cells in tissue, whereas the latter is involved in their persistence long after reaching the skin. The absence of these surface molecules leads to a decrease, not a complete deletion, of the TRM cell population [7]. CD49 (α-subunit of the α1β1 integrin receptor) is another marker of TRM cells with important functional implications. In the skin, CD8+ CD49a+ TRM cells—which are abundant in vitiligo—produce perforin, granzyme B, and interferon (IFN)-γ following stimulation by interleukin (IL)-15 [8]. However, in the absence of external stimulation, these cells express only low levels of these cytolytic molecules and therefore differ from circulating CD8+ T cells [9]. CD8+ CD49a− TRM cells produce IL-17 and accumulate in psoriasis (Fig. 2) [8]. CD44 is a marker expressed on previously activated CD8+ effector and memory T cells. It is often retained in murine TRM cells and can bind to various proteins in the extracellular matrix [10]. CD4+ TRM cells are less well-studied but increasing evidence shows their potential role in cutaneous disease. These cells mediate protective immunity against skin infections such as leishmaniasis [11] and candidiasis [12]. In a study by Cheuk et al., IL-22-producing CD4+ TRM cells were shown to persist in psoriasis following the clinical resolution of lesions [13].

Role of TRM cells in a psoriasis and b vitiligo. a Resolved psoriatic skin and disease-naïve non-lesional skin contain a population of TRM cells which express CD103, CD69, and a psoriasis specific T-cell receptor (TCR). Upon antigen recognition, these cells secrete interleukin (IL)-17 and IL-22, leading to the appearance of cutaneous lesions. After stimulation by the proinflammatory cytokines IL-33, osteopontin (OPN), IL‐17, and tumor necrosis factor (TNF)‐α, TRM cells secrete pro‐osteoclastogenic factors, such as receptor activator of nuclear factor‐B ligand (RANKL), thus contributing to the bone damage in psoriatic arthritis (PsA). b In vitiligo, TRM cells express CD103, CD69, CD49a, as well as a melanocyte-specific TCR. In the presence of IL-15, these cells secrete interferon (IFN)-γ, perforin and granzyme B, possibly leading to melanocyte cytotoxicity. In addition, TRM cells secrete C–X–C Motif Chemokine Ligand (CXCL) 9 and CXCL10 which bind to C–X–C chemokine Receptor (CXCR) 3 on the surface of recirculating memory T (TRCM) cells to recruit them to the skin. TRM and TRCM cells then work together to maintain depigmentation
Several signaling pathways, including the mammalian target of rapamycin (mTOR) pathway, transforming growth factor-β (TGF-β) and IL-15 signaling, are believed to control the development and survival of memory T cells [14]. Specifically, the mTOR signaling pathway is activated early on during the immune response. Rapamycin (mTOR inhibitor) enhances the formation of memory CD8+ T cells in secondary lymphoid tissues but inhibits the formation of CD8+ TRM cells in the intestinal and vaginal mucosa. This inhibitory effect on the mucosal tissues markedly decreased the epithelial damage in a mouse model of cell-mediated intestinal autoimmunity [15]. Dysregulation of the mTOR pathway has been noted in several inflammatory and neoplastic conditions. Interestingly, several inflammatory skin diseases—such as psoriasis, allergic contact dermatitis, and atopic dermatitis—show an increased expression of the mTOR gene [16]. Given the potential role of TRM cells in these conditions (discussed below) and the influence of the mTOR pathway on these cells, targeting the mTOR pathway may alter the TRM cell population and provide a therapeutic benefit in these inflammatory dermatoses [17].
Several transcription factors regulate TRM cell differentiation and survival, including Hobit (Homolog of Blimp‐1 in T cells), B lymphocyte-induced maturation protein-1 (Blimp-1) [18], Runt Related Transcription Factor 3 (Runx3) [19], Notch [20], and the aryl hydrocarbon receptor (AhR) [21]. Eomesodermin (EOMES) and transcription factor 1 (TCF1) are upregulated in circulating long-lived memory cells but downregulated in TRM cells [14, 22]. Downregulation of T-bet is also essential but residual expression of this transcription factor is required for IL-15-mediated CD8+ CD103+ TRM cell survival [14]. Interestingly, Notch1 signalling was found to positively correlate with the severity of psoriasis [23] and AhR is believed to regulate immune balance in atopic dermatitis and psoriasis [24]. These effects may partly be mediated by TRM cells.
The ultimate role of TRM cells in protective immunity remains to be fully elucidated. These cells possess dendritic projections that extend into the basal cell layer of the epidermis, but not upward towards higher epidermal layers or downward towards the dermis. This dendritic morphology appears to be largely defined by the epidermal location of these cells and might not be a characteristic property of TRM cells. Regardless, these cells can navigate through the basal layer and detect antigens [21, 25]. However, their slow and random movement constrains them to a limited region of skin near the original site of formation [21]. TRM cells may also localize to distinct anatomical clusters containing antigen-presenting cells including the isthmus of the hair follicle [26]. Thus, they could act as a sensing and an alarming system capable of rapidly recruiting immune effectors through the secretion of cytokines and chemokines. Conversely, they might have direct cytotoxic effects allowing them to clear target pathogens [8, 27, 28] and are believed to provide superior protective immune responses against localized infections compared with circulating effector memory T cells [29]. In fact, they upregulate antiviral and antibacterial genes involved in broad-spectrum defense providing cross-protection against antigenically unrelated pathogens [30].
TRM cells have been reported to persist in tissues for variable periods of time. CD8+ T cells can be detected in the nasal tissues up to 3 months following total respiratory tract infection [31], and in the skin up to a year following herpes simplex virus (HSV) infection [21]. Recent studies have shown that following antigen re-encounter, pre-existing TRM cells divide giving rise to secondary TRM cells. Furthermore, the capacity of epidermal TRM niches are very large allowing for the accommodation of multiple TRM cells with different specificities without displacing previously established cells [1, 32, 33].
New TRM cells can also form in inflamed tissues even in the absence of specific antigenic stimulation. In a study by Mackay et al. conducted on mice, HSV-specific TRM cells were detected at sites treated with the contact-sensitizing agent 2,4-dinitrofluorobenzene without direct HSV antigen stimulation. These cells were capable of controlling local HSV infection once the mice were inoculated with the virus. In the absence of persisting antigenic stimulation, circulating CD8+ T cells lose the expression of homing molecules that drive their infiltration into tissues. Once the antigen has been re-encountered, these cells require some time to regain entry into tissues. The persistence of TRM cells in the skin following inflammation has the advantage of providing a fast means of achieving immune protection against peripheral pathogens, even those which have never been encountered before [29].
Fixed drug eruption
Fixed drug eruption (FDE) is characterized by relapse in the same location following the administration of the culprit drug. In resting (pigmented) FDE lesions, there is a predominance of CD8+ CD45RA+ CD69+ T cells in the lesional epidermis [34, 35]. In vitro, following expansion and stimulation, these cells display cytolytic functions against keratinocytes [36]. In vivo, most of them produce IFN-γ within 2–3 h of oral challenge with the causative drug. These results indicate that TRM cells play a major role in FDE. It has been noted that lesions can be induced by other unrelated drugs and trauma, a phenomenon called “polysensitivity”. This might be explained by the cross-reactive nature of these immune cells [34].
Psoriasis
Psoriatic lesions preferentially recur in previously affected areas indicating the possible role of immune memory. In a study in mice, grafts of non-lesional normal-appearing skin from psoriatic mice induced active lesions when transplanted into immunodeficient mice [37]. In fact, T cell-associated genes and inflammatory genes- such as IL-17 and IL-22-remain upregulated in resolved lesions three months following treatment with etanercept, a TNF-inhibitor [38]. CD8+ cells expressing CD69 are retained in resolved lesions several months after effective treatment with methotrexate [39]. CD4+ cells producing IL-22 and CD8+ cells producing IL-17 also remain in the epidermis of healed lesions [13]. Most of the IL-17-producing T cells are αβ T cells with unique psoriasis-specific T cell receptor (TCR) sequences that are not found in healthy skin [40]. These shared receptor sequences could be responsible for binding to specific antigens thus triggering the recurrence of inflammatory psoriatic lesions. In fact, several putative antigens have been identified in psoriasis, including the antimicrobial peptide LL37 [41], neolipid antigens [42], and a disintegrin and metalloproteinase with thrombospondin motifs-like protein 5 (ADAMTSL5) produced by melanocyte [43].
In a study by Sérézal et al., stimulation of T cells within skin explants resulted in the upregulation of pathways induced by IFN-γ in both healthy and resolved psoriatic skin. However, IL-17A-driven pathways were only upregulated in resolved psoriatic skin. In addition, a dominant IL-17 tissue response correlated with early relapse following treatment with UVB [44]. In another study, the ratio of IL-17A-producing to IFN-γ-producing CD103+ CD8 TRM cells increased with disease duration in psoriatic disease-naïve non-lesional skin [45]. These findings suggest that earlier treatment might be associated with a lower risk of disease relapse. Similar to healed skin, never-lesional skin explants from patients with psoriasis also harbor resident CD8+ T cells poised to produce IL-22, and IL-17 and IFN-γ [45, 46]. The latter induces the expression of IFN-α in keratinocytes suggesting a new source of type I interferons capable of initiating new lesions of psoriasis. Keratinocytes from the same skin explants respond to fungal antigens with upregulation of the CCR6 ligand CCL20. Therefore, the accumulation of epidermal CCR6+ TRM cells might be attributed to an exaggerated and genetically determined keratinocyte CCL20 response to microbes [46].
Taken together, these studies suggest that resolved psoriatic skin and disease-naïve non-lesional skin contain a population of IL-17-producing TRM cells with shared receptor sequences that recognize common antigens and contribute to disease recurrence after cessation of therapy.
TRM cells might also be involved in the pathogenesis of psoriatic arthritis (PsA). Proinflammatory cytokines induce the release of pro-osteoclastogenic factors- such as Receptor Activator of Nuclear Factor Kappa-Β Ligand (RANKL)-from skin-resident T cells, potentially contributing to the bone damage in PsA [47].
Vitiligo
As in psoriasis, vitiligo lesions often recur in the same locations suggesting that autoimmune memory develops at these sites. Stable and active vitiligo perilesional skin contains a population of CD8+ CD69+ TRM cells. These cells persist in the skin for over a year and are particulary enriched within the epidermis [48] The TRM cell infiltrate is characterized by a combination of CD103+ and CD103− cells. These cells have moderate cytotoxic activity and secrete the proinflammatory cytokines IFN-γ and tumor necrosis factor-α (TNF-α). CD49 is also expressed in epidermal and dermal CD8+ TRM cells in vitiligo lesional skin. In the presence of IL-15, cells expressing this marker secrete IFN-γ, perforin and granzyme B [8]. These molecules are essential for inducing melanocyte apoptosis in vitiligo [49, 50]. Through their secretion of cytokines and chemokines, TRM cells might therefore have directs cytotoxic effects or play a role in the recruitment of other effector immune cells. Furthermore, these cells might also prevent the replenishment of melanocytes by inhibiting local regulatory T cells (Treg) which are critical for stem cell regeneration in the hair follicle [51].
However, TRM cells alone are not sufficient for the maintenance of vitiligo [48]. Autoreactive recirculating memory T cells (TRCM) have been detected in the blood of vitiligo patients. A recent study by Richmond et al. [52] found that TRCM and TRM cells work together to maintain depigmentation. Both cell types recognize auto-antigens in the skin and produce IFN-γ. Furthermore, TRM cells produce C–X–C Motif Chemokine Ligand (CXCL) 9 and CXCL10. These chemokines bind to C–X–C chemokine Receptor (CXCR) 3 on the surface of TRCM cells, potentially to recruit them to the skin. Interestingly, circulating CXCR3+ CD8+ TRCM cells in vitiligo patients have an enhanced proliferative capacity compared to cells in healthy controls [53]. In animals deficient in CXCL10, TRCM cells were more likely to engraft in the lymph node and less likely to be recruited to the epidermis. Furthermore, blocking TRCM access to the skin, or depleting these cells resulted in reversal of disease. These results highlight the importance of these chemokines and chemokine receptor in vitiligo [54].
Some studies have started looking into TRM cells as a potential therapeutic target in vitiligo. In a mouse model, long-term blockade of the Il-15 receptor with an anti-CD122 antibody was shown to deplete antigen-sepcific TRM cells from the skin, whereas short-term blockade reduced their secretion of IFN-γ. As a result, durable repigmentation occurred. Targeting Il-15 signaling might therefore be an effective and long-lasting treatment strategy for vitiligo [55].
Eczema
In a study of atopic dermatitis (AD), most top abundant T cell clones were shared between lesional and non-lesional skin. The infiltrate was maintained at least 4 months following successful treatment with a topical steroid [56]. Therefore, there is a potential population of pathogenic TRM cells that persists beyond treatment. In another study, CD4+ and CD8+ TRM cells expressing CD69 significantly infiltrated into AD skin compared to normal skin. These cells produced significant levels of the Th2 cytokines IL-4, IL-13, IL-17, and IL-22. Furthermore, thymic stromal lymphopoietin (TSLP), a known trigger for AD, increased the expression of CD69+ TRM cells. The authors concluded that TRM cells might be the main cause of AD recurrence [57, 58].
In a mouse model of allergic contact dermatitis, CD8+ CD69+ CD103+ T cells gradually accumulated in the epidermis during the acute contact hypersensitivity reaction. Their numbers stabilized for around 2 weeks following the resolution of skin inflammation and then gradually decreased up to 12 months after the challenge. 1 month following the initial acute contact hypersensitivity reaction, some of the mice were rechallenged with the same antigen and experienced disease flare-up with a more severe reaction compared to naive skin. The detected TRM cells were shown to express inhibitory checkpoint receptors, such as programmed cell death protein-1 (PD-1) and T cell immunoglobulin and mucin domain 3 (TIM-3). Blocking these inhibitory checkpoint receptors (ICRs) increased the magnitude and severity of the eczema flare-up [59]. Therefore, these receptors likely prevent excessive activation of TRM cells upon re-exposure to the antigen and are a potential therapeutic target in autoimmune and auto-inflammatory disorders. In another study in mice [60], the number of CD8+ TRM cells increased with the dose and the number of exposures to the allergen. This expansion was mediated by a combination of local proliferation and recruitment from the circulation. In addition, the magnitude of the contact hypersensitivity reaction directly correlated with the number of TRM cells.
Melanoma
TRM cells are increasingly recognized for their role in infectious, autoimmune and auto-inflammatory diseases but their involvement in cancer immunity remains unclear. CD103+ TRM cells accumulate in several human solid tumors—such as lung, liver, and breast cancers—and are often indicative of a good prognosis [61,62,63]. Similarly, increased accumulation of these cells was recently shown to correlate with improved survival in melanoma patients undergoing therapy with immune checkpoint inhibitors. Local IL-15 expression levels strongly correlates with TRM cell numbers and therefore seems to be essential for their retention in the tumor microenvironment [64]. Another study showed that expression of CD49a by vaccine‐induced TRM cells also predicts a prolonged overall and disease‐free survival. In addition, in vivo blockade of this marker as well as CD103 significantly impairs control of subcutaneous tumor [65].
Melanoma antigen-specific CD8+ TRM cells generated in vitiligo produce IFN-γ and are critical for protection against melanoma rechallenge. These cells reside mostly around depigmented hair follicles [66]. Furthermore, following the administration of ovalbumin-expressing vaccines, generated CD8+ TRM cells provide effective anti-tumor immunity against OVA-expressing melanoma independently of circulating CD8+ T cells [67]. In cases where some cancer cells resist initial eradication, TRM cells were shown to promote long-term melanoma-immune equilibrium by controlling the outgrowth and spread of these residual tumor cells [68]. In a study by Rosato et al. [69], antiviral memory CD8+ T cells were reactivated by viral peptides injected into the tumors of mouse models of melanoma. Within 12 h, these cells expressed IFN-γ, CD25, and granzyme B. Innate and adaptive immune response mechanisms were activated leading to an arrest in tumor growth. In addition, peptide therapy extended the range of tumors responding to checkpoint blockers. In a study of metastatic melanoma by Boddupalli et al. [70], tumor associated TRM cells were shown to express the highest levels of immune checkpoints among tumor-infiltrating lymphocytes. Another study reported that these immune checkpoints were mainly enriched in TRM cells expressing CD103 and that the use of checkpoint inhibitors expanded the number of this TRM cell population. Taken together, these studies suggest that immune profiling of TRM cells prior to treatment could help design more effective strategies for immune checkpoint blockade in melanoma [64].
The exact mechanism of action for the anti-tumor effects of TRM cells is still unknown. Activated tumor-specific TRM cells produce IFN-γ and TNF-α and promote the maturation of dermal dendritic cells (DCs) and their trafficking to draining lymph nodes. These DCs are required for the expansion of CD8+ T cells which can respond not only to the TRM cell-targeted antigen but also to other tumor-derived neo- and self-antigens [71]. In addition, CD103 was found to concentrate in the synapse formed between immune and tumor cells and initiate E-cadherin-dependent signaling pathways which enhance the effector functions of cytotoxic T lymphocytes [72]. Effective tumor eradication likely involves an interplay between multiple immune cells and a higher density of peri-tumoral TRM cells.
Others
TRM cells possibly play a role in many other skin diseases. These cells persist in the skin following the resolution of cutaneous viral infections such as HSV and vaccinia virus and provide rapid viral clearance upon reinfection [73, 74]. In murine models of graft-versus-host disease, TRM cells were detected in the epidermis, dermis, intestinal tract, and spleen [75]. In cutaneous T-cell lymphoma (CTCL), low-dose alemtuzumab depleted all circulating and recirculating T cells including central and effector memory T cells but spared TRM cells. It effectively treated leukemic-CTCL but not mycosis fungoides (MF) indicating that TRM cells might contribute to the pathogenesis of the latter [76]. In fact, T cells isolated from MF lesions strongly express C–C chemokine receptor (CCR) 4 but not l-selectin (CD62L) or CCR7. This phenotype is suggestive of TRM cells [77]. In a study of actinic keratoses, a 4-day course of calcipotriol plus 5-fluorouracil treatment induced TRM formation and lowered the risk of development of squamous cell carcinoma development after a follow-up period of 3 years [78].
Discussion and conclusion
In the coming years, it will be critical to develop better techniques for detecting TRM cells in tissues and determine with better accuracy the factors involved in their generation, metabolism and survival. The identification of diseases with TRM cell involvement will allow for the development of new and improved therapies. However, many important points should be kept in mind when designing new treatment strategies. TRM cells are heterogeneous, and different subsets with distinct expressions of surface markers and effector functions are likely involved in different diseases and/or anatomical locations. Therefore, a TRM cell-targeted treatment that works for one disease may not work for another. In addition, special attention should be given to the potential cross-talk between the different TRM cell subtypes and between TRM cells and other cells of the immune system [79]. The interplay between environmental factors and TRM cells should also be kept in mind [80]. For instance, following ultraviolet radiation (UVR), skin-resident T cells are activated and protect keratinocytes from UVR-induced DNA damage [81]. TRM cells may have been one of the unrecognized targets of phototherapy in several skin diseases. Similarly, it is likely that the local microbiome has an influence on the generation, compartmentalization, diversity, and function of these cells [80].
In disorders where TRM cells play a protective role, vaccination can be used as a tool to amplify the immune response. TRM cells have been generated in various sites by the local delivery of vaccine vectors [82]. Conversely, other approaches have focused on inducing inflammation in target tissues to recruit T cells generated at remote sites. This vaccine protocol termed ‘prime and pull’ relies on two successive steps: the first is the administration of the vaccine parenterally, and the second is the application of a chemokine to the target tissue thus attracting immune cells [83, 84]. Another therapeutic strategy is to release TRM cells from immune checkpoints. The number of TRM cells correlates with improved survival in melanoma patients undergoing therapy with immune checkpoint inhibitors [64]. In fact, among tumor-infiltrating lymphocytes, TRM cells seem to be particularly rich in immune checkpoints [70] and the use of checkpoint inhibitors leads to a significant expansion of these cells [64]. Therefore, TRM cells may be the major target of immune checkpoint inhibitors [85].
In diseases where TRM cells play a pathogenic role, the long-lived nature of these cells becomes a problem. Early treatment might be critical to prevent their accumulation during disease flare-up. Maintenance therapy is also necessary for the same reason. However, the side effects that could appear following the blockade of these cells should be carefully assessed. Although not fully elucidated yet, TRM cells rely on multiple cytokines and chemokines for their development, survival, and activation. These include TGF-β, TNF-α, IL-33, type I interferons, IL-15, and others. Possible future treatments could target these factors [51, 55] or the markers expressed on these cells, such as ICRs [59, 86]. A unique characteristic of TRM cells is their dependence on exogenous lipid uptake for survival. In fact, cells lacking the fatty-acid-binding proteins 4 and 5 (FABP4 and FABP5) died prematurely. Furthermore, the administration of pharmacologic agents that block lipid metabolism (etomoxir or trimetazidine) was shown to decrease the survival of these cells [87, 88]. Therefore, the oxidative metabolism of exogenous fatty acid is essential for the survival of TRM cells. Furthermore, targeting the metabolic pathways of these cells appears to be a promising therapeutic strategy.
References
Takamura S. Niches for the long-term maintenance of tissue-resident memory T cells. Front Immunol. 2018;9:1214. https://doi.org/10.3389/fimmu.2018.01214.
Wu H, Liao W, Li Q, Long H, Yin H, Zhao M, et al. Pathogenic role of tissue-resident memory T cells in autoimmune diseases. Autoimmun Rev. 2018;17(9):906–11. https://doi.org/10.1016/j.autrev.2018.03.014.
Clark RA, Chong B, Mirchandani N, Brinster NK, Yamanaka K, Dowgiert RK, et al. The vast majority of CLA+ T cells are resident in normal skin. J Immunol. 2006;176(7):4431–9.
Mueller SN, Zaid A, Carbone FR. Tissue-resident T cells: dynamic players in skin immunity. Front Immunol. 2014;5:332. https://doi.org/10.3389/fimmu.2014.00332.
Corgnac S, Boutet M, Kfoury M, Naltet C, Mami-Chouaib F. The emerging role of CD8(+) tissue resident memory T (TRM) cells in antitumor immunity: a unique functional contribution of the CD103 integrin. Front Immunol. 2018;9:1904. https://doi.org/10.3389/fimmu.2018.01904.
Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, Bedoui S, et al. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J Immunol. 2015;194(5):2059–63. https://doi.org/10.4049/jimmunol.1402256.
Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol. 2013;14(12):1294–301. https://doi.org/10.1038/ni.2744.
Cheuk S, Schlums H, Gallais Sérézal I, Martini E, Chiang SC, Marquardt N, et al. CD49a expression defines tissue-resident CD8(+) T cells poised for cytotoxic function in human skin. Immunity. 2017;46(2):287–300. https://doi.org/10.1016/j.immuni.2017.01.009.
Seidel JA, Vukmanovic-Stejic M, Muller-Durovic B, Patel N, Fuentes-Duculan J, Henson SM, et al. Skin resident memory CD8(+) T cells are phenotypically and functionally distinct from circulating populations and lack immediate cytotoxic function. Clin Exp Immunol. 2018;194(1):79–92. https://doi.org/10.1111/cei.13189.
Topham DJ, Reilly EC. Tissue-resident memory CD8(+) T cells: from phenotype to function. Front Immunol. 2018;9:515. https://doi.org/10.3389/fimmu.2018.00515.
Glennie ND, Volk SW, Scott P. Skin-resident CD4+ T cells protect against Leishmania major by recruiting and activating inflammatory monocytes. PLoS Pathog. 2017;13(4):e1006349. https://doi.org/10.1371/journal.ppat.1006349.
Park CO, Fu X, Jiang X, Pan Y, Teague JE, Collins N, et al. Staged development of long-lived T-cell receptor alphabeta TH17 resident memory T-cell population to Candida albicans after skin infection. J Allergy Clin Immunol. 2018;142(2):647–62. https://doi.org/10.1016/j.jaci.2017.09.042.
Cheuk S, Wiken M, Blomqvist L, Nylen S, Talme T, Stahle M, et al. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol. 2014;192(7):3111–200. https://doi.org/10.4049/jimmunol.1302313.
Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, et al. T-box transcription factors combine with the cytokines TGF-beta and IL-15 to control tissue-resident memory T cell fate. Immunity. 2015;43(6):1101–11. https://doi.org/10.1016/j.immuni.2015.11.008.
Sowell RT, Rogozinska M, Nelson CE, Vezys V, Marzo AL. Cutting edge: generation of effector cells that localize to mucosal tissues and form resident memory CD8 T cells is controlled by mTOR. J Immunol. 2014;193(5):2067–71. https://doi.org/10.4049/jimmunol.1400074.
Balato ADCR, Lembo S, Mattii M, Megna M, Schiattarella M, et al. Mammalian target of rapamycin in inflammatory skin conditions. Eur J Inflam. 2014;12(2):341–50.
Buerger C. Epidermal mTORC1 signaling contributes to the pathogenesis of psoriasis and could serve as a therapeutic target. Front Immunol. 2018;9:2786. https://doi.org/10.3389/fimmu.2018.02786.
Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science. 2016;352(6284):459–63. https://doi.org/10.1126/science.aad2035.
Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, et al. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature. 2017;552(7684):253–7. https://doi.org/10.1038/nature24993.
Hombrink P, Helbig C, Backer RA, Piet B, Oja AE, Stark R, et al. Programs for the persistence, vigilance and control of human CD8(+) lung-resident memory T cells. Nat Immunol. 2016;17(12):1467–78. https://doi.org/10.1038/ni.3589.
Zaid A, Mackay LK, Rahimpour A, Braun A, Veldhoen M, Carbone FR, et al. Persistence of skin-resident memory T cells within an epidermal niche. Proc Natl Acad Sci USA. 2014;111(14):5307–12. https://doi.org/10.1073/pnas.1322292111.
Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity. 2010;33(2):229–40. https://doi.org/10.1016/j.immuni.2010.08.002.
Ma L, Xue H, Gao T, Gao M, Zhang Y. Notch1 signaling regulates the Th17/Treg immune imbalance in patients with psoriasis vulgaris. Mediators Inflamm. 2018;2018:3069521. https://doi.org/10.1155/2018/3069521.
Furue M, Hashimoto-Hachiya A, Tsuji G. Aryl hydrocarbon receptor in atopic dermatitis and psoriasis. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20215424.
Ariotti S, Beltman JB, Chodaczek G, Hoekstra ME, van Beek AE, Gomez-Eerland R, et al. Tissue-resident memory CD8+ T cells continuously patrol skin epithelia to quickly recognize local antigen. Proc Natl Acad Sci USA. 2012;109(48):19739–44. https://doi.org/10.1073/pnas.1208927109.
Collins N, Jiang X, Zaid A, Macleod BL, Li J, Park CO, et al. Skin CD4(+) memory T cells exhibit combined cluster-mediated retention and equilibration with the circulation. Nat Commun. 2016;7:11514. https://doi.org/10.1038/ncomms11514.
Hartana CA, Bergman EA, Broome A, Berglund S, Johansson M, Alamdari F, et al. Tissue-resident memory T cells are epigenetically cytotoxic with signs of exhaustion in human urinary bladder cancer. Clin Exp Immunol. 2018;194(1):39–533. https://doi.org/10.1111/cei.13183.
Steinbach K, Vincenti I, Kreutzfeldt M, Page N, Muschaweckh A, Wagner I, et al. Brain-resident memory T cells represent an autonomous cytotoxic barrier to viral infection. J Exp Med. 2016;213(8):1571–87. https://doi.org/10.1084/jem.20151916.
Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, et al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci USA. 2012;109(18):7037–42. https://doi.org/10.1073/pnas.1202288109.
Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song JY, et al. T cell memory. Skin-resident memory CD8(+) T cells trigger a state of tissue-wide pathogen alert. Science. 2014;346(6205):101–5. https://doi.org/10.1126/science.1254803.
Pizzolla A, Nguyen THO, Smith JM, Brooks AG, Kedzieska K, Heath WR, et al. Resident memory CD8(+) T cells in the upper respiratory tract prevent pulmonary influenza virus infection. Sci Immunol. 2017. https://doi.org/10.1126/sciimmunol.aam6970.
Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B, et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat Immunol. 2018;19(2):183–91. https://doi.org/10.1038/s41590-017-0027-5.
Beura LK, Mitchell JS, Thompson EA, Schenkel JM, Mohammed J, Wijeyesinghe S, et al. Intravital mucosal imaging of CD8(+) resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat Immunol. 2018;19(2):173–82. https://doi.org/10.1038/s41590-017-0029-3.
Shiohara T, Mizukawa Y, Teraki Y. Pathophysiology of fixed drug eruption: the role of skin-resident T cells. Curr Opin Allergy Clin Immunol. 2002;2(4):317–23.
Teraki Y, Shiohara T. IFN-gamma-producing effector CD8+ T cells and IL-10-producing regulatory CD4+ T cells in fixed drug eruption. J Allergy Clin Immunol. 2003;112(3):609–15.
Komatsu T, Moriya N, Shiohara T. T cell receptor (TCR) repertoire and function of human epidermal T cells: restricted TCR V alpha-V beta genes are utilized by T cells residing in the lesional epidermis in fixed drug eruption. Clin Exp Immunol. 1996;104(2):343–50.
Boyman O, Hefti HP, Conrad C, Nickoloff BJ, Suter M, Nestle FO. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-alpha. J Exp Med. 2004;199(5):731–6. https://doi.org/10.1084/jem.20031482.
Suarez-Farinas M, Fuentes-Duculan J, Lowes MA, Krueger JG. Resolved psoriasis lesions retain expression of a subset of disease-related genes. J Invest Dermatol. 2011;131(2):391–400. https://doi.org/10.1038/jid.2010.280.
Torres-Alvarez B, Castanedo-Cazares JP, Fuentes-Ahumada C, Moncada B. The effect of methotrexate on the expression of cell adhesion molecules and activation molecule CD69 in psoriasis. J Eur Acad Dermatol Venereol. 2007;21(3):334–9. https://doi.org/10.1111/j.1468-3083.2006.01916.x.
Matos TR, O'Malley JT, Lowry EL, Hamm D, Kirsch IR, Robins HS, et al. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing alphabeta T cell clones. J Clin Invest. 2017;127(11):4031–41. https://doi.org/10.1172/JCI93396.
Lande R, Botti E, Jandus C, Dojcinovic D, Fanelli G, Conrad C, et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat Commun. 2014;5:5621. https://doi.org/10.1038/ncomms6621.
Cheung KL, Jarrett R, Subramaniam S, Salimi M, Gutowska-Owsiak D, Chen YL, et al. Psoriatic T cells recognize neolipid antigens generated by mast cell phospholipase delivered by exosomes and presented by CD1a. J Exp Med. 2016;213(11):2399–412. https://doi.org/10.1084/jem.20160258.
Arakawa A, Siewert K, Stohr J, Besgen P, Kim SM, Ruhl G, et al. Melanocyte antigen triggers autoimmunity in human psoriasis. J Exp Med. 2015;212(13):2203–12. https://doi.org/10.1084/jem.20151093.
Gallais Sérézal I, Classon C, Cheuk S, Barrientos-Somarribas M, Wadman E, Martini E, et al. Resident T cells in resolved psoriasis steer tissue responses that stratify clinical outcome. J Invest Dermatol. 2018;138(8):1754–63. https://doi.org/10.1016/j.jid.2018.02.030.
Vo S, Watanabe R, Koguchi-Yoshioka H, Matsumura Y, Ishitsuka Y, Nakamura Y, et al. CD8 resident memory T cells with IL-17A-producing potential are accumulated in disease-naive non-lesional sites of psoriasis possibly in correlation with disease duration. Br J Dermatol. 2019. https://doi.org/10.1111/bjd.17748.
Gallais Sérézal I, Hoffer E, Ignatov B, Martini E, Zitti B, Ehrstrom M, et al. A skewed pool of resident T cells triggers psoriasis-associated tissue responses in never-lesional skin from patients with psoriasis. J Allergy Clin Immunol. 2019;143(4):1444–54. https://doi.org/10.1016/j.jaci.2018.08.048.
Raimondo A, Lembo S, Di Caprio R, Donnarumma G, Monfrecola G, Balato N, et al. Psoriatic cutaneous inflammation promotes human monocyte differentiation into active osteoclasts, facilitating bone damage. Eur J Immunol. 2017;47(6):1062–74. https://doi.org/10.1002/eji.201646774.
Riding RL, Harris JE. The role of memory CD8(+) T cells in vitiligo. J Immunol. 2019;203(1):11–9. https://doi.org/10.4049/jimmunol.1900027.
Harris JE, Harris TH, Weninger W, Wherry EJ, Hunter CA, Turka LA. A mouse model of vitiligo with focused epidermal depigmentation requires IFN-gamma for autoreactive CD8(+) T-cell accumulation in the skin. J Invest Dermatol. 2012;132(7):1869–76. https://doi.org/10.1038/jid.2011.463.
Yang L, Wei Y, Sun Y, Shi W, Yang J, Zhu L, et al. Interferon-gamma inhibits melanogenesis and induces apoptosis in melanocytes: a pivotal role of CD8+ cytotoxic T lymphocytes in vitiligo. Acta Derm Venereol. 2015;95(6):664–70. https://doi.org/10.2340/00015555-2080.
Boniface K, Seneschal J. Vitiligo as a skin memory disease: the need for early intervention with immunomodulating agents and a maintenance therapy to target resident memory T cells. Exp Dermatol. 2019. https://doi.org/10.1111/exd.13879.
Richmond JM, Strassner JP, Rashighi M, Agarwal P, Garg M, Essien KI, et al. Resident memory and recirculating memory T cells cooperate to maintain disease in a mouse model of vitiligo. J Invest Dermatol. 2019;139(4):769–78. https://doi.org/10.1016/j.jid.2018.10.032.
Boniface K, Jacquemin C, Darrigade AS, Dessarthe B, Martins C, Boukhedouni N, et al. Vitiligo skin is imprinted with resident memory CD8 T cells expressing CXCR3. J Invest Dermatol. 2018;138(2):355–64. https://doi.org/10.1016/j.jid.2017.08.038.
Rashighi M, Agarwal P, Richmond JM, Harris TH, Dresser K, Su MW, et al. CXCL10 is critical for the progression and maintenance of depigmentation in a mouse model of vitiligo. Sci Transl Med. 2014;6(223):223ra23. https://doi.org/10.1126/scitranslmed.3007811.
Richmond JM, Strassner JP, Zapata L Jr, Garg M, Riding RL, Refat MA, et al. Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Sci Transl Med. 2018. https://doi.org/10.1126/scitranslmed.aam7710.
Brunner PM, Emerson RO, Tipton C, Garcet S, Khattri S, Coats I, et al. Nonlesional atopic dermatitis skin shares similar T-cell clones with lesional tissues. Allergy. 2017;72(12):2017–25. https://doi.org/10.1111/all.13223.
Kim SPC, Shin J, Noh J, Kim H, Kim J, et al. 049 Multicytokine-producing tissue resident memory (TRM) cells in atopic dermatitis patient. J Investig Dermatol. 2016;136(5):S9.
Kim SKJ, Park C, Kupper T, Lee K. 22 Distinct transcriptome signature of skin-resident memory T cells and migratory memory T cells in atopic dermatitis. J Investig Dermatol. 2018;138(5):S4.
Gamradt P, Laoubi L, Nosbaum A, Mutez V, Lenief V, Grande S, et al. Inhibitory checkpoint receptors control CD8(+) resident memory T cells to prevent skin allergy. J Allergy Clin Immunol. 2019. https://doi.org/10.1016/j.jaci.2018.11.048.
As OG, Jee MH, Funch AB, Alhede M, Mraz V, Weber JF, et al. Pathogenic CD8(+) epidermis-resident memory T cells displace dendritic epidermal T cells in allergic dermatitis. J Invest Dermatol. 2019. https://doi.org/10.1016/j.jid.2019.07.722.
Ganesan AP, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol. 2017;18(8):940–50. https://doi.org/10.1038/ni.3775.
Lim CJ, Lee YH, Pan L, Lai L, Chua C, Wasser M, et al. Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus-related hepatocellular carcinoma. Gut. 2019;68(5):916–27. https://doi.org/10.1136/gutjnl-2018-316510.
Wang ZQ, Milne K, Derocher H, Webb JR, Nelson BH, Watson PH. CD103 and intratumoral immune response in breast cancer. Clin Cancer Res. 2016;22(24):6290–7. https://doi.org/10.1158/1078-0432.CCR-16-0732.
Edwards J, Wilmott JS, Madore J, Gide TN, Quek C, Tasker A, et al. CD103(+) tumor-resident CD8(+) t cells are associated with improved survival in immunotherapy-naive melanoma patients and expand significantly during anti-PD-1 treatment. Clin Cancer Res. 2018;24(13):3036–45. https://doi.org/10.1158/1078-0432.CCR-17-2257.
Murray T, Fuertes Marraco SA, Baumgaertner P, Bordry N, Cagnon L, Donda A, et al. Very late antigen-1 marks functional tumor-resident CD8 T cells and correlates with survival of melanoma patients. Front Immunol. 2016;7:573. https://doi.org/10.3389/fimmu.2016.00573.
Malik BT, Byrne KT, Vella JL, Zhang P, Shabaneh TB, Steinberg SM, et al. Resident memory T cells in the skin mediate durable immunity to melanoma. Sci Immunol. 2017. https://doi.org/10.1126/sciimmunol.aam6346.
Enamorado M, Iborra S, Priego E, Cueto FJ, Quintana JA, Martinez-Cano S, et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8(+) T cells. Nat Commun. 2017;8:16073. https://doi.org/10.1038/ncomms16073.
Park SL, Buzzai A, Rautela J, Hor JL, Hochheiser K, Effern M, et al. Tissue-resident memory CD8(+) T cells promote melanoma-immune equilibrium in skin. Nature. 2019;565(7739):366–71. https://doi.org/10.1038/s41586-018-0812-9.
Rosato PC, Wijeyesinghe S, Stolley JM, Nelson CE, Davis RL, Manlove LS, et al. Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nat Commun. 2019;10(1):567. https://doi.org/10.1038/s41467-019-08534-1.
Boddupalli CS, Bar N, Kadaveru K, Krauthammer M, Pornputtapong N, Mai Z, et al. Interlesional diversity of T cell receptors in melanoma with immune checkpoints enriched in tissue-resident memory T cells. JCI Insight. 2016;1(21):e88955. https://doi.org/10.1172/jci.insight.88955.
Menares E, Galvez-Cancino F, Caceres-Morgado P, Ghorani E, Lopez E, Diaz X, et al. Tissue-resident memory CD8(+) T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells. Nat Commun. 2019;10(1):4401. https://doi.org/10.1038/s41467-019-12319-x.
Gauthier L, Corgnac S, Boutet M, Gros G, Validire P, Bismuth G, et al. Paxillin binding to the cytoplasmic domain of CD103 promotes cell adhesion and effector functions for CD8(+) resident memory T cells in tumors. Cancer Res. 2017;77(24):7072–82. https://doi.org/10.1158/0008-5472.CAN-17-1487.
Zhu J, Peng T, Johnston C, Phasouk K, Kask AS, Klock A, et al. Immune surveillance by CD8alphaalpha+ skin-resident T cells in human herpes virus infection. Nature. 2013;497(7450):494–7. https://doi.org/10.1038/nature12110.
Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature. 2012;483(7388):227–31. https://doi.org/10.1038/nature10851.
Sacirbegovic F, Zhu J, Liu J, Rosenberger S, Shlomchik MJ, Shlomchik WD. Identifying tissue-resident memory T cells in graft-versus-host disease. Blood. 2016;128(22):4544. https://doi.org/10.1182/blood.V128.22.4544.4544.
Clark RA, Watanabe R, Teague JE, Schlapbach C, Tawa MC, Adams N, et al. Skin effector memory T cells do not recirculate and provide immune protection in alemtuzumab-treated CTCL patients. Sci Transl Med. 2012;4(117):117ra7. https://doi.org/10.1126/scitranslmed.3003008.
Campbell JJ, Clark RA, Watanabe R, Kupper TS. Sezary syndrome and mycosis fungoides arise from distinct T-cell subsets: a biologic rationale for their distinct clinical behaviors. Blood. 2010;116(5):767–71. https://doi.org/10.1182/blood-2009-11-251926.
Rosenberg AR, Tabacchi M, Ngo KH, Wallendorf M, Rosman IS, Cornelius LA, et al. Skin cancer precursor immunotherapy for squamous cell carcinoma prevention. JCI Insight. 2019. https://doi.org/10.1172/jci.insight.125476.
Chen L, Shen Z. Tissue-resident memory T cells and their biological characteristics in the recurrence of inflammatory skin disorders. Cell Mol Immunol. 2019. https://doi.org/10.1038/s41423-019-0291-4.
Patra V, Laoubi L, Nicolas JF, Vocanson M, Wolf P. A perspective on the interplay of ultraviolet-radiation, skin microbiome and skin resident memory TCRalphabeta+ cells. Front Med (Lausanne). 2018;5:166. https://doi.org/10.3389/fmed.2018.00166.
MacLeod AS, Rudolph R, Corriden R, Ye I, Garijo O, Havran WL. Skin-resident T cells sense ultraviolet radiation-induced injury and contribute to DNA repair. J Immunol. 2014;192(12):5695–702. https://doi.org/10.4049/jimmunol.1303297.
Gebhardt T, Palendira U, Tscharke DC, Bedoui S. Tissue-resident memory T cells in tissue homeostasis, persistent infection, and cancer surveillance. Immunol Rev. 2018;283(1):54–76. https://doi.org/10.1111/imr.12650.
Shin H, Iwasaki A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature. 2012;491(7424):463–7. https://doi.org/10.1038/nature11522.
Fernandez-Ruiz D, Ng WY, Holz LE, Ma JZ, Zaid A, Wong YC, et al. Liver-resident memory CD8(+) T cells form a front-line defense against malaria liver-stage infection. Immunity. 2016;45(4):889–902. https://doi.org/10.1016/j.immuni.2016.08.011.
Willemsen M, Linkute R, Luiten RM, Matos TR. Skin-resident memory T cells as a potential new therapeutic target in vitiligo and melanoma. Pigment Cell Melanoma Res. 2019;32(5):612–22. https://doi.org/10.1111/pcmr.12803.
Sun H, Sun C, Xiao W, Sun R. Tissue-resident lymphocytes: from adaptive to innate immunity. Cell Mol Immunol. 2019;16(3):205–15. https://doi.org/10.1038/s41423-018-0192-y.
Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature. 2017;543(7644):252–6. https://doi.org/10.1038/nature21379.
Pan Y, Kupper TS. Metabolic reprogramming and longevity of tissue-resident memory T cells. Front Immunol. 2018;9:1347. https://doi.org/10.3389/fimmu.2018.01347.
Steinbach K, Vincenti I, Merkler D. Resident-memory T cells in tissue-restricted immune responses: for better or worse? Front Immunol. 2018;9:2827. https://doi.org/10.3389/fimmu.2018.02827.
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Additional information
Responsible Editor: John Di Battista.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Khalil, S., Bardawil, T., Kurban, M. et al. Tissue-resident memory T cells in the skin. Inflamm. Res. 69, 245–254 (2020). https://doi.org/10.1007/s00011-020-01320-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-020-01320-6