
Abstract
Tumor mutations in the gene encoding estrogen receptor alpha (ESR1) have been identified in metastatic breast cancer patients with endocrine therapy resistance. However, relatively little is known about the occurrence of mutations in the progesterone receptor (PGR) gene in this population. The study objective was to determine the frequency and prognostic significance of tumor PGR mutations for patients with estrogen receptor (ER)-positive metastatic breast cancer. Thirty-five women with metastatic or locally recurrent ER+ breast cancer were included in this IRB-approved, retrospective study. Targeted next-generation sequencing of the PGR gene was performed on isolated tumor DNA. Associations between mutation status and clinicopathologic factors were analyzed as well as overall survival (OS) from time of metastatic diagnosis. The effect of the PGR variant Y890C (c.2669A>G) identified in this cohort on PR transactivation function was tested using ER−PR− (MDA-MB-231), ER+PR+ (T47D), and ER+PR− (T47D PR KO) breast cancer cell lines. There were 71 occurrences of protein-coding PGR variants in 67% (24/36; 95% CI 49–81%) of lesions. Of the 49 unique variants, 14 are single nucleotide polymorphisms (SNPs). Excluding SNPs, the median OS of patients with PGR variants was 32 months compared to 79 months with wild-type PGR (p = 0.42). The most frequently occurring (4/36 lesions) non-SNP variant was Y890C. Cells expressing Y890C had reduced progestin-stimulated PR transactivation compared to cells expressing wild-type PR. PGR variants occur frequently in ER+ metastatic breast cancer. Although some variants are SNPs, others are predicted to be functionally deleterious as demonstrated with Y890C PR.
Similar content being viewed by others


Introduction
Breast cancer remains a prevalent, potentially deadly disease despite advances in early detection and improved therapy. In 2019, breast cancer will comprise 268,600 of the 891,480 new cancers diagnosed and 41,760 of the 285,210 cancer deaths in women in the USA [1]. Approximately 70% of breast cancers express estrogen receptor alpha (ER) and the majority of deaths occur in patients with metastatic, therapy-resistant ER+ disease [2, 3]. For patients with ER+ breast cancer, endocrine therapy is preferred which targets the estrogen signaling pathway controlling tumor growth. Standard endocrine therapy agents include aromatase inhibitors (AIs), which decrease estrogen levels, or ER antagonist compounds, such as tamoxifen or fulvestrant, which directly bind to and inhibit ER function.
Endocrine therapy significantly reduces cancer recurrence, but many patients still relapse. Recently, several tumor mutations in the gene encoding ER (ESR1) have been identified in patients with endocrine-resistant metastatic ER+ breast cancer [4,5,6,7,8]. These gain-of-function mutations cluster in the ligand binding domain of ER, result in constitutively active receptor signaling, and are associated with reduced patient survival [4,5,6,7, 9, 10]. These mutated forms of ER cannot bind as well to fulvestrant or tamoxifen compared to the wild-type receptor and require higher doses to inhibit ER transcriptional function in cell culture models [6, 11,12,13].
An important downstream target of ER is progesterone receptor (PR), which has been shown to have increased expression in breast cancers that express these constitutively active variants of ER [14]. Like ER, PR is a ligand-activated transcription factor in the nuclear receptor superfamily that share a conserved modular structure [15]. Evidence is increasing that PR can modulate ER binding sites and transcriptional activity and that selective PR modulator drugs can synergize with tamoxifen to cause tumor regression in mice [16,17,18,19]. Thus, PR is important in breast cancer beyond simply its role as a gauge of ER functional activity. However, in contrast to ESR1, little is known regarding the occurrence of tumor mutations in the gene encoding PR (PGR) in patients with metastatic breast cancer.
The objective of this study was to determine the frequency of mutations in PGR in patients with metastatic ER+ breast cancer at our institution and to determine their association with patient survival. The study hypothesis was that similar to ESR1, PGR mutations occur in metastatic ER+ breast cancer and would be expected to be associated with poorer prognosis. We are particularly interested in mutations in the PR ligand binding domain that could impact binding of 18F-fluorofuranylnorprogesterone (18F-FFNP), which our studies suggest is an imaging biomarker of endocrine sensitivity [20,21,22].
Methods
Study Design and Patient Population
This study was approved by the Institutional Review Board (IRB Protocol #2014-1523) with waived informed consent and was Health Insurance Portability and Accountability Act compliant. The study design is an observational retrospective cohort analysis performed at a tertiary care institution. Search of the University of Wisconsin Carbone Cancer Center registry was performed from 1982 to 2015 to identify potentially eligible patients. Patients included were at least 18 years old with biopsy-proven ER+ metastatic breast cancer or locally recurrent disease, received at least 6 months of endocrine therapy in the adjuvant or metastatic setting, and had sufficient tissue for further processing (Fig. 1). Biopsy tissue obtained to confirm the diagnosis of metastatic disease was selected for the study. If insufficient tissue was present from the initial metastatic biopsy site, subsequent biopsy tissue re-demonstrating metastatic disease could be used. Dates of tissue sampling ranged from 2001 to 2015. For bone biopsy specimens, tissue blocks that were not decalcified were preferably selected for analysis. As part of routine processing of bone biopsies for metastatic cancer at the UW Pathology department, the soft tissue is separated from bony tissue at the grossing bench to ensure high-quality material for molecular analysis. Clinically approved primary monoclonal antibodies used during this study include 6F11 and SP1 for ER and 1A6 and 1E2 for PR. ER and PR positivity is defined as at least 1% positive cells according to the American Society of Clinical Oncology/College of American Pathologists guidelines for immunohistochemical testing of ER and PR in breast cancer [23]. Clinicopathologic data were obtained from the electronic medical record. Follow-up information was obtained from the electronic medical record (clinic notes, radiology, and pathology reports) and obituary records.

Flow diagram of study. MBC = metastatic breast cancer; UWCCC = University of Wisconsin Carbone Cancer Center; ER = estrogen receptor alpha
Genomic DNA Isolation and Sequencing
Archived formalin-fixed paraffin-embedded tissue specimens were requested and matched with the corresponding H&E slide. All slides and blocks were carefully screened and finally selected by our expert pathologist on the study to ensure adequacy for subsequent sequencing studies. A new H&E slide was made in cases where the archived slide was not representative of the remaining tissue on the block. The face of the block was wiped with RNase Away (#7002, Molecular BioProducts™) prior to taking the punches, and a sterile 2 mm disposable biopsy punch (#33-31, Integra®) was used to take two punches from each tumor tissue. Genomic DNA was extracted using the Maxwell® 16 MDx Instrument (AS3000, Promega). For MCF-7 and T47D breast cancer cell lines, genomic DNA was isolated using the DNeasy® Blood & Tissue Kit (Qiagen).
For targeted next-generation sequencing of PGR, libraries were prepared with the Illumina TruSeq Custom Amplicon v1.5 Library Preparation Kit using probes that tiled across the whole coding region of the PGR gene. For targeted next-generation sequencing of ESR1, libraries were prepared with the Illumina TruSeq Custom Amplicon v1.5 Library Preparation Kit using probes that tiled across the whole coding region and +3′ untranslated region of the ESR1 gene. According to the manufacturer’s instructions, sequencing libraries were prepared and probes containing oligonucleotide pairs specific to the genes were hybridized to each genomic DNA sample. Amplicons were generated by extension and ligation of the bound oligonucleotides, followed by PCR amplification. Products were amplified using primers that included individual per-sample indexes, as well as common adapters and stem sequences for sequencing cluster generation. After PCR cleanup, library quality was assessed using a Bioanalyzer High Sensitivity chip (Agilent Technologies, Santa Clara, CA). Libraries were normalized, and equal volumes of each sample were combined, diluted, and heat denatured prior to MiSeq sequencing, using a MiSeq 2 × 250 bp sequencing run and MiSeq 500 bp v2 kit. Images were analyzed using the standard Illumina Pipeline, version 1.8.2. Approximately 90k to 120k reads per sample were obtained.
For variant discovery analysis of the sequencing data, the method and best practices approach recommended by the Broad Institute were used. Sequencing reads were adapter and quality trimmed using the Skewer trimming program [24]. Flash was used to merge paired end reads into amplicon sequences [25]. Amplicons were then aligned to the reference genome, GRCh37.p13, using the Burrows-Wheeler Aligner, BWA-MEM [26], and local realignment performed using the Broad Institute’s Genome Analysis Toolkit (GATK) [27]. Variants were called using GATK HaplotypeCaller version 3.3 and annotated with SNPEff Version 4.1g, GRCh37.75 [28]. The NCBI Reference Sequence (RefSeq) mRNA used were NM_001202474 and NM_000926 for PGR and were NM_000125, NM_001291241, NM_001122741, and NM_001291230 for ESR1. The minimum confidence thresholds for calling and emitting were 30.0 and 10.0, respectively. The minimum base and mapping quality scores were 10 and 20, respectively. Variant allele fraction was calculated as the depth of the variant allele divided by the total depth.
Databases and Prediction Algorithms
The single nucleotide polymorphism database (dbSNP) [29] (http://www.ncbi.nlm.nih.gov/snp) was used to compare the identified missense mutations with reported single nucleotide polymorphisms from population databases. The National Cancer Institute’s Genome Data Commons (GDC) data portal (https://portal.gdc.cancer.gov/) was used to search The Cancer Genome Atlas (TCGA) [30, 31]. The cBioPortal for Cancer Genomics [32, 33] (http://www.cbioportal.org) was used to search the data released from the Metastatic Breast Cancer Project (Provisional, October 2018). Software used for computational prediction of the functional impact of the identified missense mutations included PROVEAN [34] (http://provean.jcvi.org/index.php) and PolyPhen-2 [35] (http://genetics.bwh.harvard.edu/pph2). PolyPhen-2 classifies mutations into three categories, benign, possibly damaging, and probably damaging, with the latter being the most deleterious.
Site-Directed Mutagenesis, Cell Culture, and Transfection
The Y890C mutation was introduced to wild-type PR-A and PR-B cDNA in a cloning vector (pBluescript; Stratagene) using QuikChange II XL site-directed mutagenesis kit (Agilent). Sense and antisense primers were 5′-CTTGTCAAACAGCTGCATCTGTGCTGCTTGAATACATTTATCCAG-3′ and 5′-CTGGATAAATGTATTCAAGCAGCACAGATGCAGCTGTTTGACAAG-3′. Y890C PR cDNA was then subcloned into an expression vector, LHLCA [36], to generate LHLCA Y890C PR-A and Y890C PR-B.
The ER-PR- breast cancer cell line MDA-MB-231 was cultured using Dulbecco’s modified Eagle’s medium (Corning), supplemented with 10% fetal bovine serum (VWR), and 1% penicillin/streptomycin (Gibco) at 37 °C and 10% CO2. MDA-MB-231 cells were transfected with expression plasmids containing either wild-type PR-A or PR-B [22], or Y890C PR-A or Y890C PR-B. Transfected cells were selected and maintained in 200 μg/mL hygromycin B (Life Technologies).
T47D PR knockout (KO) cells (clone #6) were a generous gift from J. Julie Kim, PhD (Northwestern University) [37]. T47D cells were obtained from the American Type Culture Collection (Manassas, VA). T47D cells were cultured using Roswell Park Memorial Institute medium (Corning), supplemented with 10% fetal bovine serum, and 1% penicillin/streptomycin at 37 °C and 5% CO2. Cells were authenticated using short tandem repeat analysis and tested negative for murine pathogens and Mycoplasma contamination (IDEXX BioResearch).
Transcriptional Reporter Gene Assay
Cells were placed in steroid hormone depleted media (phenol red free DMEM, 10% six-times charcoal/dextran stripped fetal bovine serum, 2% L-glutamine, and 1% penicillin/streptomycin) for 3 days. Cells were plated in 6-well plates, transfected with glucocorticoid/progesterone response element luciferase (0.75 μg) and CMV-β-galactosidase (0.25 μg) reporter plasmids [38, 39] using Lipofectamine 3000 (Life Technologies). For transient expression experiments, T47D PR KO cells were co-transfected with 100 ng LHLCA expression plasmid containing either wild-type PR-A or PR-B, or Y890C PR-A or PR-B along with the reporter plasmids. After 5 h, medium was changed. The next day, cells were treated with either ethanol (EtOH) or 10 nM R5020 (promegestone; Perkin Elmer) for 24 h. Luciferase (Promega) and β-galactosidase activity (Tropix) were assayed following manufacturer’s protocols.
Quantitative Real-Time PCR (qPCR)
Cells were deprived of steroid hormones for 3 days prior to experimentation. Cells were then seeded in 6-well plates and transiently transfected with 100 ng of LHLCA expression plasmids containing either wild-type PR-A or PR-B, or Y890C PR-A or PR-B or empty vector using Lipofectamine 3000. Media was changed after 5 h. The next day, cells were treated with either EtOH or 10 nM R5020 for 24 h. RNA was extracted using RNeasy kit (Qiagen) and cDNA was synthesized using iScript (Bio-Rad). Samples were run in duplicate and CT values normalized to the housekeeping gene RPLP0. Relative fold change was calculated using the formula 2−ΔΔCT. Primer sequences are as follows: RPLP0 forward 5′-GACAATGGCAGCATCTACAAC-3′ reverse 5′-GCAGACAGACACTGGCAAC-3′ and FKBP5 forward 5′-AGAACCAAACGGAAAGGAGAG-3′ reverse 5′-TCACCGCCTGCATGTATTT-3′.
Immunofluorescence
Cells were grown on chamber slides (Lab-Tek II) and then fixed with 3.7% formaldehyde, permeabilized with 0.5% Triton X-100 (Sigma), blocked with 10% goat serum for 1 h at 37 °C, and incubated with anti-PR antibody (1:100, NCL-L-PGR-312; Leica Biosystems) overnight at 4 °C. Next, the slides were incubated with Alexa Fluor 488 anti-mouse antibody (1:100, Life Technologies) for 1 h at room temperature, mounted with ProLong Gold Antifade with DAPI (Life Technologies), and imaged using confocal microscopy (Leica SP8 STED).
Western Blot Analysis
Whole cell lysates were prepared in RIPA buffer (Sigma) containing 2 mM sodium orthovanadate, protease inhibitor (1:500, Sigma), and phosphatase inhibitor (1:100, Sigma). Protein was quantified using Bradford reagent (Bio-Rad). Equal amounts of protein were run on 10% SDS-PAGE, transferred to PVDF membrane (Millipore), and probed with antibodies against PR (NCL-L-PGR-312, Leica Biosystems, 1:1000, 2 h at room temperature) and β-actin (clone AC-15, Sigma, 1:10,000, overnight at 4 °C). Blots were imaged using Clarity Western enhanced chemiluminescence substrate (Bio-Rad) and GeneSys GBox:XX6 (Syngene).
Statistical Methods
The frequencies of PGR variants were reported with percentages and exact 95% confidence intervals (CIs). The associations between mutation status and clinicopathologic factors were analyzed using Fisher’s exact test. The Kaplan-Meier method was used to analyze overall survival (OS), defined as date of metastatic diagnosis until death. Patients who were still living were censored at the date of last available follow-up. The log-rank test was used to compare OS by mutation status. Significance between WT and Y890C PR transcriptional activity was determined using unpaired t test, and results are presented as mean ± standard error. All statistical tests were two-sided, and 5% (p < 0.05) was set as the level of significance. Statistical analysis was done in R3.4.2, including the “Hmisc” and “survival” packages, Prism 6.04 (GraphPad), and IBM SPSS Statistics Version 25.
Results
Study Population
The study population consisted of 35 women (range 35–88 years old) with a total of 36 ER+ metastatic breast cancer lesions. One patient presented initially with a chest wall lesion and then subsequently developed a bone lesion; both lesions were used in the analyses. The remaining 34 women had one lesion each for analysis. Metastatic sites included bone (N = 13), brain (N = 6), chest wall (N = 3), liver (N = 4), lung/pleura (N = 4), and lymph nodes (N = 6) (Table 1).
Identification of PGR Variants
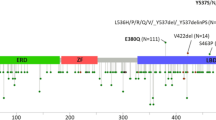
There were a total of 71 occurrences of protein-coding PGR variants (Fig. 2 and Supplemental Table 1). PGR variants were present in 67% of lesions (24/36; 95% CI 49–81%) and involved 5.3% (49/933; 95% CI 3.9–6.9%) of the amino acid residues of PR. Ten variants occurred in the first 164 amino acids that are only present in the PR-B isoform. Of the 49 distinct variants identified, six were present in more than one lesion and included L118P, S344T, F380S, A502del, V660L, and Y890C. Fourteen of the 49 PGR variants identified have been reported as SNPs (Supplemental Table 2). Ten of the lesions with PGR variants also contained ESR1 variants; however, none were the common hotspot mutations in the ligand binding domain of ESR1 [40].

Location and frequency of PGR variants. Schematic of PR protein. Number sign denotes SNPs previously reported; asterisk represents the number of occurrences of the variant across the study population. NTD = N-terminal domain; DBD = DNA-binding domain; H = hinge domain; LBD = ligand-binding domain; fs = frame shift; del = deletion; * = stop gained
Associations of PGR Variants with Key Clinicopathologic Factors
No significant association between PGR variants and sites of metastasis could be determined (Table 2). The type of endocrine therapy (9 treated with aromatase inhibitors, 14 with tamoxifen, and 12 with both) was also not associated with the presence of PGR variants (Table 2). Additionally, there were no significant associations between PGR variants and PR positivity by immunohistochemistry or HER2 status (Table 2).
Overall Survival
The median follow-up for the 17 survivors was 48.5 months with a range of 2 to 131 months. The median OS of patients with wild-type PGR lesions was 79 months and was 34 months for those with PGR variants (hazard ratio 0.71, 95% CI 0.27–1.9; p = 0.50) (Fig. 3a and Table 3). Of the 71 unique PGR gene variants identified, 14 have been reported as SNPs (Supplemental Table 2). Excluding the SNPs, the median OS of patients with wild-type PGR lesions was 79 months and was 32 months for those with PGR variants (hazard ratio 0.69, 95% CI 0.26–1.7; p = 0.42) (Fig. 3b).

Overall survival analysis. Kaplan-Meier survival plots comparing overall survival for subjects with a wild-type (WT) PGR (n = 12) or any variant (MT) PGR (n = 24) and for subjects with b wild-type (WT) PGR including SNPs (n = 21) or non-SNP variant (MT) PGR (n = 15)
Comparison with Public Databases
Search of The Cancer Genome Atlas (TCGA) for PGR yielded eight unique mutations. Of those mutations, E723K was present in our dataset and is also included in dbSNP (rs1175582391). The PGR variants identified in this study are distinct from the two (S42L and S166R) reported in the Metastatic Breast Cancer Project (237 samples from 180 patients as of the date of manuscript submission) contained within the cBioPortal for Cancer Genomics. Using cBioPortal, we were able to find other cancer types in which the PGR variants presented in Fig. 2 have been identified. These include uterine endometrioid carcinoma, pancreatic adenocarcinoma, colorectal adenocarcinoma, gliosarcoma, anaplastic oligoastrocytoma, head and neck squamous cell carcinoma, lung squamous cell carcinoma, and cutaneous melanoma (Supplemental Table 3).
Comparison with ER+ Metastatic Breast Cancer Cell Lines
Both MCF-7 and T47D cells were heterozygous for S344T and V660L, which are known SNPs. No other missense variants in PGR were identified in either cell line.
Prediction of Functional Impact
To investigate the potential impact of these missense PGR variants on PR protein function, two computational prediction algorithms were utilized. Provean classified mutations as either deleterious or neutral whereas PolyPhen-2 categorized mutations, in increasing severity, as benign, possibly damaging, or probably damaging. There were 11 PGR variants that had consensus between Provean and PolyPhen-2 (Supplemental Table 4). The ligand-binding domain contained six variants, the DNA-binding domain had two, and the N-terminal domain and hinge each had one deleterious variant. Additionally, two more variants were observed in the ligand-binding domain that were deleterious by Provean and possibly damaging by PolyPhen-2 analysis.
Reduced Progestin-Stimulated PR Transcriptional Activity of the PGR Variant Y890C
One variant, Y890C (c.2669A>G), was observed in 4 subjects, in multiple lesion locations (bone, brain, liver, lymph node), predicted to be functionally deleterious, and not previously reported as a SNP. The ER−PR− breast cancer cell line MDA-MB-231 was used to generate stable cells expressing either wild-type or Y890C PR. To determine how Y890C affects PR transcriptional activation, cells were transfected with a progesterone response element-driven luciferase reporter gene and were treated with the synthetic progestin, R5020. Cells expressing Y890C PR had reduced transcriptional activation in the presence of R5020 compared to cells expressing wild-type PR (relative fold change Y890C vs wild type: 0.2 ± 0.02 vs 1.0 ± 0.21, p = 0.004 for PR-A; and 0.08 ± 0.002 vs 1.0 ± 0.03, p = 0.001 for PR-B, Fig. 4a). Immunofluorescence confirmed appropriate nuclear localization of Y890C PR (Fig. 4b). Western blot analysis confirmed expression of PR protein in the stable cell lines (relative fold expression Y890C vs wild type: 1.04 ± 0.37, p = 0.70 for PR-A and 0.85 ± 0.07, p = 0.10 for PR-B, Fig. 4c). Expression of both PR-A and PR-B isoforms were observed in the cell line transfected with the PR-B expression plasmid.

Stable expression of Y890C mutation reduces PR transcriptional function. a MDA-MB-231 breast cancer cells (ER−PR−) stably transfected with either wild-type (WT) PR-A, WT PR-B, Y890C PR-A, or Y890C PR-B expression plasmids were deprived of steroid hormones for 3 days, transfected with reporter plasmids to measure PR transcriptional activation, then treated with 10 nM R5020 for 24 h. Values for luciferase activity normalized to β-galactosidase activity in the presence of R5020 are normalized to WT PR. * p < 0.05 compared with WT for each isoform. b Immunofluorescence of PR protein localization in the stable cell lines (green = Alexa Fluor 488 staining for PR, blue = DAPI nuclear staining). c Protein expression of WT and Y890C PR-A and PR-B in the stable cell lines
To independently confirm the inhibitory effect of Y890C mutation on PR transactivation function, we used a T47D ER+ cell line that has undergone CRISPR-Cas9 knockout of the PGR gene resulting in loss of endogenous PR protein expression [37]. T47D PR KO cells transiently expressing exogenous wild-type PR had increased luciferase reporter gene activity in the presence of R5020 compared to ethanol, whereas T47D PR KO cells expressing empty vector had no PR activation in the presence of agonist consistent with the lack of PR protein expression in this model system (Fig. 5a). Progestin-stimulated PR transactivation was reduced in cells expressing Y890C PR-A compared to wild-type PR-A (0.07 ± 0.04 vs 1.0 ± 0.17 relative fold change, p = 0.0064) and Y890C PR-B compared to wild-type PR-B (0.43 ± 0.12 vs 1.0 ± 0.13, p = 0.03) (Fig. 5b).

Transient expression of Y890C mutation reduces PR transcription in ER+ breast cancer cells. a T47D PR KO cells (ER+PR−) were deprived of steroid hormones for 3 days, transiently transfected with plasmids expressing either WT PR-A, WT PR-B, Y890C PR-A, Y890C PR-B, or empty vector along with reporter genes. Cells were treated with 10 nM R5020 or ethanol for 24 h. Values represent luciferase activity normalized to β–galactosidase activity. *p < 0.05 compared with EtOH for each receptor type. b Reporter gene assay results in T47D PR KO cells normalized to WT PR in the presence of R5020. c R5020 induction of FKBP5 expression obtained from qPCR in ER+PR+ T47D cells transiently transfected with Y890C PR-A, Y890C PR-B, or empty vector expression plasmids. Values were normalized to EtOH control and expressed as fold change relative to empty vector. Values represent the average ± SEM of 5 different experiments. *p < 0.05 compared with empty vector
Since the variant allele frequencies observed with Y890C in the patient-derived tumor samples (8.5 to 16.6%; Supplemental Table 4) suggests a heterozygous population of mutant and wild-type PR, the effect of Y890C on endogenous PR target gene regulation was also tested using ER+PR+ T47D cells. The progesterone regulated endogenous gene FKBP5 [41] was measured in response to 24-h treatment with ethanol or 10 nM R5020 after transient transfection with expression plasmids containing wild-type PR-A or PR-B, Y890C PR-A or PR-B, or empty vector. Agonist induction of FKBP5 normalized to ethanol control was reduced in cells expressing Y890C compared to wild-type PR. Cells expressing Y890C PR-A had 28.0 ± 7.8 fold induction of FKBP5 expression in response to R5020 compared to 145.1 ± 85.3 fold induction measured in cells expressing wild-type PR-A. Similarly, Y890C PR-B had a 20.8 ± 8.1 fold increase in FKBP5 expression compared to 77.4 ± 33.1 fold increase with wild-type PR-B. When compared to cells transfected with empty vector (1.0 ± 0.35), relative expression of FKBP5 was 0.3 ± 0.08 (p = 0.07) for Y890C PR-A and 0.2 ± 0.09 (p = 0.03) for Y890C PR-B (Fig. 5c). This finding implies a dominant negative effect of Y890C PR when expressed in cells with endogenous wild-type PR. Together, these results obtained using multiple cell lines support our overall conclusion that the Y890C mutation results in reduced PR transactivation function.
Overall Survival for Y890C PGR Variant Subset
The median OS for patients expressing the Y890C PGR variant was 19 months compared to 79 months for WT PGR (hazard ratio 0.50, 95% CI 0.06–2.8; p = 0.36, Fig. 6). None of the metastatic tumor samples containing the Y890C PGR variant contained common hotspot mutations in the ligand-binding domain of ESR1 (Supplemental Table 1).

Overall survival for Y890C PGR variant subset. Kaplan-Meier survival plots comparing overall survival for subjects with Y890C PGR (n = 4) or WT PGR which includes SNPs (n = 21)
Discussion
The primary purpose of this study was to identify the prevalent PGR gene mutations in metastatic ER+ breast cancer patients at our institution. Using a retrospective analysis of targeted next generation sequencing results from 36 lesions in 35 women, we identified 49 distinct PGR variants. One of these variants, Y890C, resulted in reduced progestin-stimulated PR transcriptional activity.
There is limited data regarding PGR mutations in breast cancer. Missense mutations reported in the Metastatic Breast Cancer Project include S42L and S166R and include C603R, E723K, R740Q, G762V, E833K, and A914V in the TCGA database. However, there have been several SNPs for PGR have been reported [42, 43]. The most common variant is referred to as “PROGINS,” which includes an Alu insertion in intron G, H770H, and V660L found in women with ovarian carcinoma and primary breast cancer [44,45,46]. Although some investigations have reported conflicting results regarding the association of V660L with either increased or decreased breast cancer risk, larger studies found no association [47,48,49,50,51]. Another SNP observed to be linked with V660L is S344T [52]. Our study confirms this linkage; 6 of the 9 patients with the V660L mutation also had S344T. The consequence of the PROGINS haplotype on PR functional activity remains uncertain with conflicting reports indicating increased, decreased, or similar transcriptional activation compared with wild-type PR [53,54,55].
Functional significance of the protein-coding PGR variants identified will be important to establish. Historical salient structural studies defining the functional domains in PR may provide insight into the functional role of the newly discovered PGR variants. For example, the N-terminal domain contains several sub-domains responsible for transcriptional activation and inhibition as well as multiple sites of posttranslational modification that can modulate PR transcriptional function [56,57,58,59]. We identified two patients with L118P variants, which maps to a leucine-rich LXXLL nuclear receptor box important for activation function-3 in PR-B [60]. Furthermore, the hinge region has been shown to be multi-functional and mutation of key residues in this domain could positively or negatively regulate gene expression, suggesting that this domain could regulate hormone responsiveness in either direction [61]. Further structure-function analysis studies of select PGR variants are planned.
The most frequently observed variant in the PR ligand binding domain in our study was Y890C. Y890 is located within alpha helix 11 and is a highly conserved amino acid residue across many species (human, mouse, rat, rabbit, and sheep) [62, 63]. The Y890 residue is important for ligand binding as indicated by one in vitro mutagenesis study in which mutation of the tyrosine to phenylalanine resulted in a marked decrease in relative binding to progesterone compared to wild-type PR [62]. Our computational prediction algorithms suggest that the Y890C mutation will have a similar negative effect on ligand binding as the published Y890F mutation. Consistent with this prediction, we showed that Y890C PR has reduced transcriptional activity compared to wild-type PR in the presence of R5020. Further studies are needed to fully understand the ramifications Y890C PR has on cross-talk with ER function and endocrine therapy resistance.
An interesting observation was that three of the four samples with Y890C PGR displayed a PR-negative phenotype by immunohistochemistry. Given the small sample size, we are hesitant to speculate regarding the biologic significance of this finding—e.g., possibility that Y890C PGR mutation leads to loss of PR protein expression through clonal selection. Overall, there was no significant association between PR positivity by immunohistochemistry and the presence of PGR variants (Table 2). Further research with larger sample sizes is needed to directly test a potential association of Y890C mutation with a PR-negative phenotype. The biology of PR loss/silencing in breast cancer is complex with many potential molecular mechanisms, reviewed by Cui et al. [64], including nonfunctional ER, low levels of estrogen, PGR promoter hypermethylation, loss of PGR gene locus heterozygosity, growth factor downregulation of PR, non-genomic ER activity, and altered ER coregulator protein level or activity. It is possible that one or more of these mechanisms could be contributing to PR protein expression loss in samples with Y890C PGR.
Due to the retrospective nature of this study, a paired germline sample (e.g., blood) was not available for definitive separation of germline findings from somatic acquired variants. Although desirable to clarify interpretation, paired analysis “is not always practical and should not be required” according to current guidelines for reporting sequence variants in cancer [65]. Germline variants can be suspected based on allele fractions between 40 and 60% for a heterozygous variant and by comparison with public databases such as dbSNP. Furthermore, only one subject had primary breast tumor tissue available for analysis. Thus, our study was not able to compare how PGR variants may differ between the primary and metastatic disease settings.
This study expands the existing literature by identification of PGR variants in patients with ER+ metastatic breast cancer. In contrast to ESR1, the clinical significance of PGR mutations is not currently known. However, the accumulating evidence that PR can modulate ER function and increasing interest in combining selective PR modulator drugs with standard of care ER-targeted agents [16,17,18,19] points to the need for future studies to elucidate the functional consequences of the variants we have identified. Determining the mechanistic impact of PGR variants on ER and PR cross-talk in breast cancer may potentially improve our understanding of tumor evolution and endocrine resistance. Furthermore, testing for PGR mutations may help better select patients for new and ongoing clinical trials evaluating progestin-based endocrine therapies.
Conclusion
PGR variants occur frequently in patients with metastatic ER+ breast cancer in our study population. Although some variants are SNPs, others are predicted to be functionally deleterious as we have demonstrated with Y890C PR which has reduced transcriptional activity in the presence of R5020 compared to wild-type PR. The functional impact of these PGR variants will be important to investigate given the cross-talk between ER and PR in breast cancer and the ongoing clinical trials evaluating progestin-based endocrine therapies.
References
Siegel RL, Miller KD, Jemal A (2019) Cancer statistics, 2019. CA Cancer J Clin 69:7–34
Osborne CK, Schiff R (2011) Mechanisms of endocrine resistance in breast cancer. Annu Rev Med 62:233–247
Jatoi I, Chen BE, Anderson WF, Rosenberg PS (2007) Breast cancer mortality trends in the United States according to estrogen receptor status and age at diagnosis. J Clin Oncol 25:1683–1690
Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, He X, Liu S, Hoog J, Lu C, Ding L, Griffith OL, Miller C, Larson D, Fulton RS, Harrison M, Mooney T, McMichael JF, Luo J, Tao Y, Goncalves R, Schlosberg C, Hiken JF, Saied L, Sanchez C, Giuntoli T, Bumb C, Cooper C, Kitchens RT, Lin A, Phommaly C, Davies SR, Zhang J, Kavuri MS, McEachern D, Dong YY, Ma C, Pluard T, Naughton M, Bose R, Suresh R, McDowell R, Michel L, Aft R, Gillanders W, DeSchryver K, Wilson RK, Wang S, Mills GB, Gonzalez-Angulo A, Edwards JR, Maher C, Perou CM, Mardis ER, Ellis MJ (2013) Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep 4:1116–1130
Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, Kalyana-Sundaram S, Wang R, Ning Y, Hodges L, Gursky A, Siddiqui J, Tomlins SA, Roychowdhury S, Pienta KJ, Kim SY, Roberts JS, Rae JM, Van Poznak CH, Hayes DF, Chugh R, Kunju LP, Talpaz M, Schott AF, Chinnaiyan AM (2013) Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet 45:1446–1451
Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA, Hudis C, Chen D, Taran T, Hortobagyi G, Greene G, Berger M, Baselga J, Chandarlapaty S (2013) ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 45:1439–1445
Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, Yelensky R, Brown M, Miller VA, Sarid D, Rizel S, Klein B, Rubinek T, Wolf I (2013) D538G mutation in estrogen receptor-alpha: a novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res 73:6856–6864
Jeselsohn R, Buchwalter G, De Angelis C, Brown M, Schiff R (2015) ESR1 mutations-a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol 12:573–583
Schiavon G, Hrebien S, Garcia-Murillas I, Cutts RJ, Pearson A, Tarazona N, Fenwick K, Kozarewa I, Lopez-Knowles E, Ribas R, Nerurkar A, Osin P, Chandarlapaty S, Martin LA, Dowsett M, Smith IE, Turner NC (2015) Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci Transl Med 7:313ra182
Chandarlapaty S, Chen D, He W, Sung P, Samoila A, You D, Bhatt T, Patel P, Voi M, Gnant M, Hortobagyi G, Baselga J, Moynahan ME (2016) Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: a secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol 2:1310–1315
Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, Ferrer-Lozano J, Perez-Fidalgo JA, Cristofanilli M, Gomez H, Arteaga CL, Giltnane J, Balko JM, Cronin MT, Jarosz M, Sun J, Hawryluk M, Lipson D, Otto G, Ross JS, Dvir A, Soussan-Gutman L, Wolf I, Rubinek T, Gilmore L, Schnitt S, Come SE, Pusztai L, Stephens P, Brown M, Miller VA (2014) Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res 20:1757–1767
Fanning SW, Mayne CG, Dharmarajan V, Carlson KE, Martin TA, Novick SJ, Toy W, Green B, Panchamukhi S, Katzenellenbogen BS, Tajkhorshid E, Griffin PR, Shen Y, Chandarlapaty S, Katzenellenbogen JA, Greene GL (2016) Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife 5:e12792
Weir HM, Bradbury RH, Lawson M, Rabow AA, Buttar D, Callis RJ, Curwen JO, de Almeida C, Ballard P, Hulse M, Donald CS, Feron LJ, Karoutchi G, MacFaul P, Moss T, Norman RA, Pearson SE, Tonge M, Davies G, Walker GE, Wilson Z, Rowlinson R, Powell S, Sadler C, Richmond G, Ladd B, Pazolli E, Mazzola AM, D'Cruz C, De Savi C (2016) AZD9496: an oral estrogen receptor inhibitor that blocks the growth of ER-positive and ESR1-mutant breast tumors in preclinical models. Cancer Res 76:3307–3318
Bahreini A, Li Z, Wang P, Levine KM, Tasdemir N, Cao L, Weir HM, Puhalla SL, Davidson NE, Stern AM, Chu D, Park BH, Lee AV, Oesterreich S (2017) Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res 19:60
Hill KK, Roemer SC, Churchill ME, Edwards DP (2012) Structural and functional analysis of domains of the progesterone receptor. Mol Cell Endocrinol 348:418–429
Daniel AR, Gaviglio AL, Knutson TP, Ostrander JH, D'Assoro AB, Ravindranathan P, Peng Y, Raj GV, Yee D, Lange CA (2015) Progesterone receptor-B enhances estrogen responsiveness of breast cancer cells via scaffolding PELP1- and estrogen receptor-containing transcription complexes. Oncogene 34:506–515
Mohammed H, Russell IA, Stark R, Rueda OM, Hickey TE, Tarulli GA, Serandour AA, Birrell SN, Bruna A, Saadi A, Menon S, Hadfield J, Pugh M, Raj GV, Brown GD, D'Santos C, Robinson JL, Silva G, Launchbury R, Perou CM, Stingl J, Caldas C, Tilley WD, Carroll JS (2015) Progesterone receptor modulates ERalpha action in breast cancer. Nature 523:313–317
Singhal H, Greene ME, Tarulli G, Zarnke AL, Bourgo RJ, Laine M, Chang YF, Ma S, Dembo AG, Raj GV, Hickey TE, Tilley WD, Greene GL (2016) Genomic agonism and phenotypic antagonism between estrogen and progesterone receptors in breast cancer. Sci Adv 2:e1501924
Carroll JS, Hickey TE, Tarulli GA, Williams M, Tilley WD (2017) Deciphering the divergent roles of progestogens in breast cancer. Nat Rev Cancer 17:54–64
Fowler AM, Chan SR, Sharp TL, Fettig NM, Zhou D, Dence CS, Carlson KE, Jeyakumar M, Katzenellenbogen JA, Schreiber RD, Welch MJ (2012) Small-animal PET of steroid hormone receptors predicts tumor response to endocrine therapy using a preclinical model of breast cancer. J Nucl Med 53:1119–1126
Chan SR, Fowler AM, Allen JA, Zhou D, Dence CS, Sharp TL, Fettig NM, Dehdashti F, Katzenellenbogen JA (2015) Longitudinal noninvasive imaging of progesterone receptor as a predictive biomarker of tumor responsiveness to estrogen deprivation therapy. Clin Cancer Res 21:1063–1070
Salem K, Kumar M, Yan Y, Jeffery JJ, Kloepping KC, Michel CJ, Powers GL, Mahajan AM, Fowler AM (2019) Sensitivity and isoform specificity of (18)F-fluorofuranylnorprogesterone for measuring progesterone receptor protein response to estradiol challenge in breast cancer. J Nucl Med 60:220–226
Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, Fitzgibbons PL, Francis G, Goldstein NS, Hayes M, Hicks DG, Lester S, Love R, Mangu PB, McShane L, Miller K, Osborne CK, Paik S, Perlmutter J, Rhodes A, Sasano H, Schwartz JN, Sweep FC, Taube S, Torlakovic EE, Valenstein P, Viale G, Visscher D, Wheeler T, Williams RB, Wittliff JL, Wolff AC (2010) American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer (unabridged version). Arch Pathol Lab Med 134:e48–e72
Jiang H, Lei R, Ding SW, Zhu S (2014) Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 15:182
Magoc T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-bio.GN]
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303
Cingolani P, Platts A, Wang l L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6:80–92
Sherry ST, Ward M, Sirotkin K (1999) dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res 9:677–679
Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MDM, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ, Ding L (2013) Mutational landscape and significance across 12 major cancer types. Nature 502:333–339
Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, Staudt LM (2016) Toward a shared vision for cancer genomic data. N Engl J Med 375:1109–1112
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401–404
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6:pl1
Choi Y, Chan AP (2015) PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31:2745–2747
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249
Van Antwerp DJ, Verma IM (1996) Signal-induced degradation of I(kappa)B(alpha): association with NF-kappaB and the PEST sequence in I(kappa)B(alpha) are not required. Mol Cell Biol 16:6037–6045
Davaadelger B, Murphy AR, Clare SE, Lee O, Khan SA, Kim JJ (2018) Mechanism of telapristone acetate (CDB4124) on progesterone receptor action in breast cancer cells. Endocrinology 159:3581–3595
Strahle U, Klock G, Schutz G (1987) A DNA sequence of 15 base pairs is sufficient to mediate both glucocorticoid and progesterone induction of gene expression. Proc Natl Acad Sci U S A 84:7871–7875
MacGregor GR, Caskey CT (1989) Construction of plasmids that express E. coli beta-galactosidase in mammalian cells. Nucleic Acids Res 17:2365
Dustin D, Gu G, Fuqua SAW (2019) ESR1 mutations in breast cancer. Cancer 125:3714–3728
Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB (2002) Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem 277:5209–5218
Ghali RM, Al-Mutawa MA, Ebrahim BH, Jrah HH, Zaied S, Bhiri H, Hmila F, Mahjoub T, Almawi WY (2018) Progesterone receptor (PGR) gene variants associated with breast cancer and associated features: a case-control study. Pathol Oncol Res
Jahandoost S, Farhanghian P, Abbasi S (2017) The effects of sex protein receptors and sex steroid hormone gene polymorphisms on breast cancer risk. J Natl Med Assoc 109:126–138
Rowe SM, Coughlan SJ, McKenna NJ, Garrett E, Kieback DG, Carney DN, Headon DR (1995) Ovarian carcinoma-associated TaqI restriction fragment length polymorphism in intron G of the progesterone receptor gene is due to an Alu sequence insertion. Cancer Res 55:2743–2745
McKenna NJ, Kieback DG, Carney DN, Fanning M, McLinden J, Headon DR (1995) A germline TaqI restriction fragment length polymorphism in the progesterone receptor gene in ovarian carcinoma. Br J Cancer 71:451–455
Fuqua SA, Hill SM, Chamness GC, Benedix MG, Greene GL, O'Malley BW, McGuire WL (1991) Progesterone receptor gene restriction fragment length polymorphisms in human breast tumors. J Natl Cancer Inst 83:1157–1160
Pooley KA, Healey CS, Smith PL, Pharoah PD, Thompson D, Tee L, West J, Jordan C, Easton DF, Ponder BA, Dunning AM (2006) Association of the progesterone receptor gene with breast cancer risk: a single-nucleotide polymorphism tagging approach. Cancer Epidemiol Biomark Prev 15:675–682
Johnatty SE, Spurdle AB, Beesley J, Chen X, Hopper JL, Duffy DL, Chenevix-Trench G (2008) Progesterone receptor polymorphisms and risk of breast cancer: results from two Australian breast cancer studies. Breast Cancer Res Treat 109:91–99
Zhang B, Beeghly-Fadiel A, Long J, Zheng W (2011) Genetic variants associated with breast-cancer risk: comprehensive research synopsis, meta-analysis, and epidemiological evidence. Lancet Oncol 12:477–488
Gaudet MM, Milne RL, Cox A, Camp NJ, Goode EL, Humphreys MK, Dunning AM, Morrison J, Giles GG, Severi G, Baglietto L, English DR, Couch FJ, Olson JE, Wang X, Chang-Claude J, Flesch-Janys D, Abbas S, Salazar R, Mannermaa A, Kataja V, Kosma VM, Lindblom A, Margolin S, Heikkinen T, Kampjarvi K, Aaltonen K, Nevanlinna H, Bogdanova N, Coinac I, Schurmann P, Dork T, Bartram CR, Schmutzler RK, Tchatchou S, Burwinkel B, Brauch H, Torres D, Hamann U, Justenhoven C, Ribas G, Arias JI, Benitez J, Bojesen SE, Nordestgaard BG, Flyger HL, Peto J, Fletcher O, Johnson N, Dos Santos SI, Fasching PA, Beckmann MW, Strick R, Ekici AB, Broeks A, Schmidt MK, van Leeuwen FE, Van't Veer LJ, Southey MC, Hopper JL, Apicella C, Haiman CA, Henderson BE, Le Marchand L, Kolonel LN, Kristensen V, Grenaker Alnaes G, Hunter DJ, Kraft P, Cox DG, Hankinson SE, Seynaeve C, Vreeswijk MP, Tollenaar RA, Devilee P, Chanock S, Lissowska J, Brinton L, Peplonska B, Czene K, Hall P, Li Y, Liu J, Balasubramanian S, Rafii S, Reed MW, Pooley KA, Conroy D, Baynes C, Kang D, Yoo KY, Noh DY, Ahn SH, Shen CY, Wang HC, Yu JC, Wu PE, Anton-Culver H, Ziogoas A, Egan K, Newcomb P, Titus-Ernstoff L, Trentham Dietz A, Sigurdson AJ, Alexander BH, Bhatti P, Allen-Brady K, Cannon-Albright LA, Wong J, Chenevix-Trench G, Spurdle AB, Beesley J, Pharoah PD, Easton DF, Garcia-Closas M (2009) Five polymorphisms and breast cancer risk: results from the Breast Cancer Association Consortium. Cancer Epidemiol Biomark Prev 18:1610–1616
Wang-Gohrke S, Chang-Claude J, Becher H, Kieback DG, Runnebaum IB (2000) Progesterone receptor gene polymorphism is associated with decreased risk for breast cancer by age 50. Cancer Res 60:2348–2350
De Vivo I, Huggins GS, Hankinson SE, Lescault PJ, Boezen M, Colditz GA, Hunter DJ (2002) A functional polymorphism in the promoter of the progesterone receptor gene associated with endometrial cancer risk. Proc Natl Acad Sci U S A 99:12263–12268
Romano A, Delvoux B, Fischer DC, Groothuis P (2007) The PROGINS polymorphism of the human progesterone receptor diminishes the response to progesterone. J Mol Endocrinol 38:331–350
Agoulnik IU, Tong XW, Fischer DC, Korner K, Atkinson NE, Edwards DP, Headon DR, Weigel NL, Kieback DG (2004) A germline variation in the progesterone receptor gene increases transcriptional activity and may modify ovarian cancer risk. J Clin Endocrinol Metab 89:6340–6347
Stenzig J, Schweikert A, Piasecki A, Hoppner G, Eschenhagen T, Rau T (2012) Progesterone receptor variants associated with the PROGINS haplotype exhibit functional properties similar to those of wild-type progesterone receptor. Pharmacogenet Genomics 22:629–641
Hovland AR, Powell RL, Takimoto GS, Tung L, Horwitz KB (1998) An N-terminal inhibitory function, IF, suppresses transcription by the A-isoform but not the B-isoform of human progesterone receptors. J Biol Chem 273:5455–5460
Sartorius CA, Melville MY, Hovland AR, Tung L, Takimoto GS, Horwitz KB (1994) A third transactivation function (AF3) of human progesterone receptors located in the unique N-terminal segment of the B-isoform. Mol Endocrinol 8:1347–1360
Abdel-Hafiz HA, Horwitz KB (2014) Post-translational modifications of the progesterone receptors. J Steroid Biochem Mol Biol 140:80–89
Leehy KA, Regan Anderson TM, Daniel AR, Lange CA, Ostrander JH (2016) Modifications to glucocorticoid and progesterone receptors alter cell fate in breast cancer. J Mol Endocrinol 56:R99–r114
Tung L, Shen T, Abel MG, Powell RL, Takimoto GS, Sartorius CA, Horwitz KB (2001) Mapping the unique activation function 3 in the progesterone B-receptor upstream segment. Two LXXLL motifs and a tryptophan residue are required for activity. J Biol Chem 276:39843–39851
Daniel AR, Gaviglio AL, Czaplicki LM, Hillard CJ, Housa D, Lange CA (2010) The progesterone receptor hinge region regulates the kinetics of transcriptional responses through acetylation, phosphorylation, and nuclear retention. Mol Endocrinol 24:2126–2138
Letz M, Bringmann P, Mann M, Mueller-Fahrnow A, Reipert D, Scholz P, Wurtz JM, Egner U (1999) Investigation of the binding interactions of progesterone using muteins of the human progesterone receptor ligand binding domain designed on the basis of a three-dimensional protein model. Biochim Biophys Acta 1429:391–400
Williams SP, Sigler PB (1998) Atomic structure of progesterone complexed with its receptor. Nature 393:392–396
Cui X, Schiff R, Arpino G, Osborne CK, Lee AV (2005) Biology of progesterone receptor loss in breast cancer and its implications for endocrine therapy. J Clin Oncol 23:7721–7735
Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, Tsimberidou AM, Vnencak-Jones CL, Wolff DJ, Younes A, Nikiforova MN (2017) Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 19:4–23
Acknowledgments
The authors thank the University of Wisconsin Biotechnology Center DNA Sequencing Facility for providing library preparation and next generation sequencing services. We also thank the UW Translational Research Initiatives in Pathology Lab for technical assistance and the University of Wisconsin Carbone Cancer Center Support Grant P30 CA014520. Also, we thank Derek M. Pavelec, PhD and the University of Wisconsin Carbone Cancer Center Computational Informatics Shared Resource for assistance in sequencing data analysis. We also thank the University of Wisconsin Optical Imaging Core for confocal microscopy and image collection. Lastly, we thank J. Julie Kim, PhD (Northwestern University) for generously providing the T47D PR KO cell line.
Funding
This work was supported by the University of Wisconsin Paul P. Carbone Cancer Center Young Investigator Award, the University of Wisconsin Institute of Clinical and Translational Research KL2 Scholar Award, 1KL2TR002374, the Clinical and Translational Science Award program, grant 1UL1TR002373, the University of Wisconsin School of Medicine and Public Health Shapiro Summer Research Program, and the University of Wisconsin Department of Radiology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(DOCX 45 kb)
Rights and permissions
About this article

Cite this article
Fowler, A.M., Salem, K., DeGrave, M. et al. Progesterone Receptor Gene Variants in Metastatic Estrogen Receptor Positive Breast Cancer. HORM CANC 11, 63–75 (2020). https://doi.org/10.1007/s12672-020-00377-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-020-00377-3