
Abstract
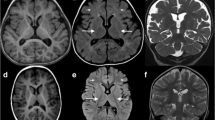
Congenital disorders of glycosylation (CDGs) are a genetically heterogeneous group of disorders caused by the defects in the synthesis and processing of glycoproteins. CDG is caused by mannosyl-oligosaccharide glucosidase (MOGS) deficiency, and is an extremely rare type, and only six patients have been reported. Here, we report a patient from China with facial dysmorphism, infantile spams, developmental delay, low vison, and abnormal liver function and low immunoglobulin. Brain MRI showed hypoplasia of the corpus callosum and slightly wide sulci at bilateral frontal parietal lobes. Compound heterozygous mutations of (c.1694G>A: R565Q and c.1619G>A: R540H) in exon 4 of MOGS gene (NM_006302.2) were identified by whole exome sequencing. Further investigation showed that the gene expression of MOGS in patients’ peripheral blood was decreased. We observed that two mutations were associated with lower protein expression of MOGS, cell growth, and cell cycle in transiently transfected Hela cells. We also noticed that cell cycle–related proteins, β-catenin, cyclin D1, and C-myc, were decreased in mutant cells. In conclusion, our study suggested whole exome sequencing, and genes associated with CDGs should be analyzed in patients with infantile spams and multiple system involvement, and mutant MOGS–impaired cell cycle progression. Our work broadens the mutation spectrum of MOGS gene.



Similar content being viewed by others

References
Van Scherpenzeel M, Willems E, Lefeber DJ (2016) Clinical diagnostics and therapy monitoring in the congenital disorders of glycosylation. Glycoconj J 33(3):345–358. https://doi.org/10.1007/s10719-015-9639-x
Freeze HH (2006) Genetic defects in the human glycome. Nat Rev Genet 7(7):537–551. https://doi.org/10.1038/nrg1894
Freeze HH, Eklund EA, Ng BG, Patterson MC (2012) Neurology of inherited glycosylation disorders. Lancet Neurol 11(5):453–466. https://doi.org/10.1016/S1474-4422(12)70040-6
Sadat MA, Moir S, Chun TW, Lusso P, Kaplan G, Wolfe L, Memoli MJ, He M, Vega H, Kim LJY, Huang YHN (2014) Glycosylation, hypogammaglobulinemia, and resistance to viral infections. Curr Opin Cell Biol 370(17):82–91. https://doi.org/10.1038/jid.2014.371
Correspondence P (2009) Conotruncal heart defects in three patients with congenital disorder of glycosylation type Ia (CDG Ia). J Med Genet 46(4):287–289. https://doi.org/10.1136/jmg.2008.057620
Marques-da-Silva D, Dos Reis Ferreira V, Monticelli M et al (2017) Liver involvement in congenital disorders of glycosylation (CDG). A systematic review of the literature. J Inherit Metab Dis 40(2):195–207. https://doi.org/10.1007/s10545-016-0012-4
Leroy JG (2006) Congenital disorders of N-glycosylation including diseases associated with O- as well as N-glycosylation defects. Pediatr Res 60(6):643–656. https://doi.org/10.1203/01.pdr.0000246802.57692.ea
Jaeken J, Hennet T, Matthijs G, Freeze HH (2009) CDG nomenclature: time for a change! Biochim Biophys Acta 1792:825–826. https://doi.org/10.1016/j.bbadis.2009.08.005
Freeze HH, Chong JX, Bamshad MJ, Ng BG (2014) Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am J Hum Genet 94(2):161–175. https://doi.org/10.1016/j.ajhg.2013.10.024
Kim YM, Seo GH, Jung E, Jang JH, Kim SZ, Lee BH (2018) Characteristic dysmorphic features in congenital disorders of glycosylation type IIb. J Hum Genet 63(3):383–386. https://doi.org/10.1038/s10038-017-0386-7
De Praeter CM, Gerwig GJ, Bause E et al (2000) A novel disorder caused by defective biosynthesis of N-linked oligosaccharides due to glucosidase I deficiency. Am J Hum Genet 66(MIM 266265):1744–1756. https://doi.org/10.1086/302948
Kane MS, Davids M, Adams C, Wolfe LA, Cheung HW, Gropman A, Huang Y, Ng BG, Freeze HH, Adams DR, Gahl WA, Boerkoel CF (2016) Mitotic intragenic recombination: a mechanism of survival for several congenital disorders of glycosylation. Am J Hum Genet 98(2):339–346. https://doi.org/10.1016/j.ajhg.2015.12.007
Li M, Xu Y, Wang Y, Yang X-A, Jin D (2019) Compound heterozygous variants in MOGS inducing congenital disorders of glycosylation (CDG) IIb. J Hum Genet 64(3):265–268. https://doi.org/10.1038/s10038-018-0552-6
Monticelli M, Ferro T, Jaeken J, dos Reis Ferreira V, Videira PA (2016) Immunological aspects of congenital disorders of glycosylation (CDG): a review. J Inherit Metab Dis 39(6):765–780. https://doi.org/10.1007/s10545-016-9954-9
Scott K, Gadomski T, Kozicz T, Morava E (2014) Congenital disorders of glycosylation: new defects and still counting. J Inherit Metab Dis 37(4):609–617. https://doi.org/10.1007/s10545-014-9720-9
Barbosa EA, Fontes NC, Santos SCL et al (2019) Relative quantification of plasma N-glycans in type II congenital disorder of glycosylation patients by mass spectrometry. Clin Chim Acta 492(February):102–113. https://doi.org/10.1016/j.cca.2019.02.013
Bruneel A, Cholet S, Drouin-garraud V et al (2018) Complementarity of electrophoretic, mass spectrometric and gene sequencing techniques for the diagnosis and characterization of congenital disorders of glycosylation. Electrophoresis 39(24):3123–3132. https://doi.org/10.1002/elps.201800021
Jones MA, Rhodenizer D, da Silva C, Huff IJ, Keong L, Bean LJH, Coffee B, Collins C, Tanner AK, He M, Hegde MR (2013) Molecular diagnostic testing for congenital disorders of glycosylation (CDG): detection rate for single gene testing and next generation sequencing panel testing. Mol Genet Metab 110(1–2):78–85. https://doi.org/10.1016/j.ymgme.2013.05.012
Apweiler R (1999) On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta 1473(1):4–8
Kawauchi T, Shikanai M, Kosodo Y (2013) Extra-cell cycle regulatory functions of cyclin-dependent kinases (CDK) and CDK inhibitor proteins contribute to brain development and neurological disorders. Genes Cells:176–194. https://doi.org/10.1111/gtc.12029
Masclef L, Dehennaut V, Mortuaire M, Schulz C, Boyce M (2019) Cyclin D1 stability is partly controlled by O –GlcNAcylation. Front Endocrinol (Lausanne) 10(February):1–12. https://doi.org/10.3389/fendo.2019.00106
Stichelen SO, Dehennaut V, Buzy A et al (2014) O-GlcNAcylation stabilizes b-catenin through direct competition with phosphorylation at threonine. FASEB J 41:3325–3338. https://doi.org/10.1096/fj.13-243535
Gallo GL, Valko A, Aramburu SI, Etchegaray E, Völker C, Parodi AJ, D’Alessio C (2018) Abrogation of glucosidase I-mediated glycoprotein deglucosylation results in a sick phenotype in fission yeasts: model for the human MOGS-CDG disorder. J Biol Chem 293:19957–19973. https://doi.org/10.1074/jbc.RA118.004844
Funding
This research was supported in part by the National Key Research and Development Program of China (No. 2016YFC1306202), Wuhan Yellow Crane (Medicine and Healthcare) (2016-01), and Hubei Health and Family Planning Commission (WJ2015MB247).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interests
The authors declare that they have no competing interests.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Peiwei Zhao, Peng, X., Luo, S. et al. Identification and characterization of novel mutations in MOGS in a Chinese patient with infantile spams. Neurogenetics 21, 97–104 (2020). https://doi.org/10.1007/s10048-019-00590-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-019-00590-5