
Abstract
Lithium-manganese orthosilicate powders were synthesized via a modified solvothermal method. Three different weight ratios of sucrose used as a carbon source were investigated for finding an effective carbon coating process. Synthesized materials were analyzed by structural and morphological (X-ray powder diffraction, scanning electron microscopy, N2 adsorption/desorption, thermogravimetry analysis) and electrochemical (chronopotentiometry, cyclic voltammetry) methods. By increasing the sucrose content, the electrochemical performance improvement of lithium-manganese orthosilicate powder was observed, such as an increase in its specific capacity (132%) and cyclability (5%) compared with uncoated sample. The enhancement in Li2MnSiO4 performance is related to an increase in its electrical conductivity and enlargement in the electrochemically active surface area due to carbon coating process.

Graphical Abstract
Similar content being viewed by others
Introduction
The interest in lithium-manganese orthosilicate (Li2MnSiO4, LMS) has rapidly increased since the first Dominko’s publication regarding the LMS compound synthesis by the modified Pechinni sol-gel process [1]. Li2MnSiO4 shows advantages toward commonly used positive electrode materials in Li-ion systems, such as a fast synthesis duration, low-priced substrates, and a high theoretical capacity of 333 mAh g−1. Such high capacity is related to the fact that one mole of Li2MnSiO4 can intercalate/deintercalate 2 moles of lithium ions per formula unit during galvanostatic charge/discharge processes, involving Mn4+/Mn3+ and Mn3+/Mn2+ redox couples [1, 2]. In addition, a wide range of working potential 1.5–4.8 V [3], low cost, and high safety of the LMS compounds arise a growing attractiveness of this material [4].
Unfortunately, the amorphization of the sample over the first charging causes a change in the crystallographic structure, and consequently a capacity drop and cyclic stability decline through following cycles. Other disadvantages of Li2MnSiO4 can be attributed to the differences in the structural stability of the delithiated phases, Jahn-Teller distortion of Mn3+ ions, Mn dissolution in the electrolyte, and low intrinsic conductivity in the range of 10−14 to 10−16 S cm−1 [5]. As a result, LMS exhibits reasonably good electrochemical performance only at elevated temperatures and low charge/discharge current rates. In order to improve the electrochemical parameters of lithium-manganese orthosilicate, carbon coatings were widely used with carbon sources in various forms, e.g., acetic acid [6, 7], adipic acid [8, 9], citric acid [1, 2, 10], and sucrose [10, 11]. Moreover, the carbon coating process can be executed during the synthesis (in situ) [10] or after the completed synthesis, but before calcination (ex situ) [10, 11].
Among a variety of synthesis methods, one of the most commonly used techniques to produce orthosilicates is solvo-/hydrothermal synthesis. These techniques are simple, clean, low-cost, and of low-energy consumption and are suitable to prepare high-quality materials in closed systems. In addition to the sol-gel methods, this technique is characterized by a very short synthesis time (several minutes for supercritical fluid techniques). Murukanahally successfully synthesized Li2MnSiO4 nanoparticles (4–5 nm in diameter), which showed good electrochemical performance and delivered 283/220 mAh g−1 after first and fiftieth discharge process, respectively. However, the mentioned LMS/C composite contained ~ 18 wt% of amorphous carbon as a carbon coat. Muraliganth et al. obtained Li2MnSiO4/C powder which delivered 210 and 250 mAh g−1 at C/20 current rate in room temperature and 55 °C, respectively. The electrode polarization is greatly reduced at 55 °C compared to room temperature experiments; however, the capacity fade of the examined lithium-manganese orthosilicate increased, and the material retained only 15% of its initial capacity after 20 cycles at elevated temperature [12]. Kuezma et al. examined the performance of Li2MnSiO4 at higher current rate of C/10 and lower potential window of 2.0–4.5 V. The electrochemical properties of LMS powder were like Muraliganth’s material, with a dramatic fall in the cyclability after fifth cycle [13]. The high discharge capacities in cited works are due to a high phase purity and monodispersed nanoparticles of lithium-manganese orthosilicate powders, which facilitate short reaction pathways for the lithium insertion/exertion process [14, 15].
Most of the Li2MnSiO4/C composites obtained through solvo- and hydrothermal reactions contain relatively huge amounts of amorphous carbon. Kempaiah et al. synthesized LMS/C compound through solvothermal process with 13.3 wt% amorphous carbon [14]. Composite obtained by Devaraju et al. contained 18 wt% of carbonaceous modifications [16]. Muraliganth et al. prepared Li2MnSiO4/C composite by solvothermal reaction with 12 wt% of amorphous carbon through sucrose reduction [12]. Such high amounts of electrochemically inactive species in the electrode lead to limited gravimetric capacity of the cell.
In this work, we examined the effect of the sucrose additive on final Li2MnSiO4/C material. Sucrose was used as a carbon coating source in this modified solvothermal synthesis. The amorphous carbon in prepared composites was on low level (1–3 wt%), demonstrating its positive effect on structural, morphological, and electrochemical properties of this new LMS positive electrode material for lithium-ion cells.
Experimental
Synthesis of Li2MnSiO4 by Solvothermal Method
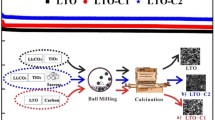
Li2MnSiO4/C composite was synthesized by a solvothermal method. The schematic of the synthesis steps is shown in Fig. 1. Firstly, 0.0368 mol Si(OC2H5)4 (TEOS, Sigma-Aldrich) was dissolved in 40 ml of ethyl alcohol (EtOH, POCH) and 25 ml of deionized water and then mixed with 0.015 mol of citric acid (Sigma-Aldrich) and either 0.001, 0.002, or 0.003 mol of sucrose (Sigma-Aldrich) for SLMS1, SLMS2, or SLMS3 samples, respectively. Then, 0.0386 mol of ethylene glycol (POCH) were added to the so-prepared solutions (solution A). The mixtures were then sonicated on ultrasonic scrubber for 30 min and heated to 30 °C to combine the reagents. 0.0372 mol of Mn(C2H3O2)2·4H2O (Sigma-Aldrich) and 0.0761 mol of Li(C2H3O2)·2H2O (Sigma-Aldrich) were separately dissolved (solutions B and C) in identical solvent mixtures: 40-ml EtOH and 25 ml of deionized water. Then solutions B and C were both added dropwise to the previously prepared solutions (A). The final solution, containing hydrolyzed SiO2 from TEOS, was moved to the reactor and heated to 150 °C for 24 h under 30 bar pressure. The resulting gels were heat-treated in the tubular oven for 17 h at 700 °C in a reducing atmosphere of Ar/5 wt% H2 to form a carbon-coated Li2MnSiO4.

Schematic representation of the synthesis procedure
Structural and Morphological Analyses
X-ray diffraction (XRD) measurements were carried out in the classical Bragg-Brentano geometry. X-ray tube C was used in the optical system of the 1-degree slit and Ni filter (1:20). The copper (Cu) lamp was charged with 40 mA and 40 kV. Measurements were done in the 2θ range of 10°–120° with a step of 0.008°.
Merlin scanning electron microscope (Zeiss) was used together with a Quantax 400 energy dispersive X-ray spectroscope (Bruker) in SEM and EDX measurements. Electron beam energy used for X-ray excitation was 3 keV. The sample image was obtained from the resultant of two in-lens and secondary electron detectors in order to obtain the best contrast.
Thermal stability of carbon products was studied by thermogravimetry (TGA) using a TA Q50 instrument. The measurements were performed under oxidizing atmosphere (N2:O2 95:5 vol%) with the heating rate of 10 °C min−1.
The Micromeritics® ASAP 2060 apparatus, at 77.349 K in the range of 0.01 to 0.995 relative N2 pressure p·(p0)−1 equipment, was used to evaluate adsorption/desorption of N2. ASAP software was used for data processing. The surface area was obtained using the BET method, whereas a distribution and pore volume were calculated using the BJH method for desorption curves and H-K for microporous systems. Moreover, ASAP software was used to estimate average grain size of LMS powders, assuming powder density of 4.0 g cm−3.
Electrochemistry
The orthosilicate powders were used for electrochemical tests. Firstly, the sample (active material) was homogenized with Vulcan XC72R carbon (conductive additive) for 15 min in an agate mortar. As-prepared blend was mixed with 5 wt% PVDF solution (dissolved in N-methyl-2-pyrrolidone) and stirred on a magnetic stirrer to unify the slurry. This mixture was then applied on to the aluminum foil with automatic applicator and, in order to evaporate the residual solvent, dried in vacuum at 120 °C overnight. Electrodes were cut from the layer, compressed at 200 bar, weighed, and re-dried in a vacuum. Such prepared samples were transported to the glove box (MBraun Unilab MB-20-G).
The electrochemical performance of Li2MnSiO4 powders was tested in a Swagelok®-type three-electrode system. Li2MnSiO4 electrodes were used as the working electrodes, whereas metallic lithium acts as the counter and reference electrodes. Trilayer separators (polypropylene (PP) polyethylene (PE) polypropylene (PP)) of 25 μm thickness (Celgard® 2325) were immersed in 1 M LiPF6 dissolved in ethylene carbonate and dimethyl carbonate (1:1 wt./wt., BASF). Galvanostatic charge/discharge tests and cyclic voltammetry (CV) were tested on multichannel battery tester Sollich Atlas 1361 and Solartron SI 1287 Electrochemical Interface. Chronopotentiometry (CP) charge/discharge experiments were made in the potential range of 1.5 and 4.8 V (vs. Li+/Li0) for 20 cycles at 0.1 C current rate (where C corresponds to 333 mA g−1) at room temperature. Similarly, in case of CV measurements, the working electrode was polarized between 1.5 and 4.8 V (vs. Li+/Li0) at a scan rate of 0.1 mV s−1 for 2 cycles.
Results and Discussion
Structure and Morphology of Li2MnSiO4/C Composites
Structural and Morphological Analyses of SLMS Samples
Figure 2 presents XRD patterns of Li2MnSiO4/C powders. The main phase in all analyzed SLMS samples was identified as lithium-manganese orthosilicate (ICSD 98-026-2720; Pnma space group) as expected [17]. The additional reflexes originating from impurities, such as MnO, Li2SiO3, and Mn2SiO4, were present in the examined powders. The dominant impurity phase in SLMS1 powder consisted of Li2SiO3, while MnO and Mn2SiO4 were mostly present in SLMS2 and SLMS3 composites. The lowest amount of the impurity phases were characterized in the SLMS3 sample. This suggests that a sucrose addition reduces the amount of contaminations obtained during solvothermal synthesis process, due to separation of ions in the xerogel and gas evolution processes during heat treatment [18, 19]. The crystallite size of SLMS samples was estimated from the Scherrer’s equation. The calculated crystallite sizes were found to be 40 nm, 33 nm, and 33 nm for SLMS1, SLMS2, and SLMS3, respectively.

XRD patterns of all Li2MnSiO4 powders. All detected phases are marked using different colors, namely, Li2MnSiO4 (green), Mn2SiO4 (blue), MnO (purple), and Li2SiO3 (orange). Cu lamp charged with 40 mA and 40 kV; measurement done in the range of 10° to 120° with a step of 0.008°
The SEM images of Li2MnSiO4 powders are shown in Fig. 3. A uniform distribution of material grains in the range of 10 to 100 nm were in all the synthesized samples. Some agglomeration and a creation of cubic and pellet-like structures (up to few hundred nanometers in diameter) were also visible on the recorded SEM images.

SEM images of SLMS1 (a, b), SLMS2 (c, d), and SLMS3 (e, f) powders. Beam energy was 3–5 keV; spectrum acquisition was ca. 120 s
Active Surface Area and Pore Distribution Studies
Figure 4 shows N2 adsorption/desorption isotherms and a pore distribution of Li2MnSiO4 powders. The adsorption curves presented a course similar to Brauner’s type-II isotherms, while at the same time the desorption isotherm did not overlap with adsorption one. The IUPAC type-H3 of hysteresis loop suggested a slit-shape geometry of pores present in analyzed powders [20]. BET-specific surface area (SSA) calculations showed an increase in Li2MnSiO4 powders SSA with an increased amount of carbon source added into the solvothermal reactor. Calculated BET-specific surface area for LMS powders was found to be 22.15 ± 0.06, 45.89 ± 0.11, and 48.75 ± 0.15 m2 g−1 for SLMS1, SLMS2, and SLMS3 powders, respectively. One can see that a small carbon addition can improve the SSA by a factor of over two.

N2 adsorption/desorption isotherms (a, c, e) and pore distribution (b, d, f) for HLMS1 (a, b), HLMS2 (c, d), and HLMS3 (e, f) powders
Pore distribution in examined powders displayed a similar trend in every analyzed sample. One can see four dominant pore ranges: 0.6–0.7, 0.9–1.0, 3–4, and 20–40 nm. Pore volumes calculated during adsorption/desorption experiments increased in value as the sucrose content in the reactor increased. The highest volumes were found to be 0.2489 and 0.0145 cm3 g−1 for SLMS3 powder and pore diameter of 1.7–300 and < 1.7 nm, respectively. Moreover, SLMS3 composite showed lowest average grain size calculated from adsorption/desorption analysis, reaching 31 nm in diameter. This value is comparable to the XRD findings and combined with SEM observations suggests most homogenous distribution of Li2MnSiO4 particles. The results of N2 adsorption/desorption experiments are also listed in Table 1.
Carbon Content Analysis Through TGA Experiments
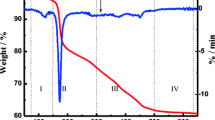
Thermogravimetric analysis is a very common technique used for a determination of carbon content in the sample. Generally, when the sample is exposed to oxygen (air) atmosphere during the TGA analysis, the carbon decomposition starts above 200 °C and up to 400 °C [21]. Figure 5 shows that the carbon content in our Li2MnSiO4 composite is close to 1 wt% for SLMS1, about 1.75 wt% for SLMS2, and above 3 wt% for SLMS3. At around 550 °C, the weight gain corresponded to the increase of oxygen mass derived from the crystal transformation of MnO impurity phase into Mn2O3.

TGA of Li2MnSiO4 powders synthesized by a modified solvothermal method with sucrose as a carbon source
Electrochemistry
Galvanostatic Charge/Discharge Tests
The results of galvanostatic charge/discharge tests for Li2MnSiO4 powders are presented in Fig. 6 a to e and Table 2. Capacities delivered during the first charge/discharge cycle were 77/60, 115/90, and 166/139 mAh g−1 for SLMS1, SLMS2, and SLMS3 powders, respectively. Increasingly, the sucrose content resulted in an enhancement of LMS electrochemical activity. The capacity increase during the first charge/discharge cycle can be assigned to the enhanced electronic conductivity and electron transport through Li2MnSiO4 electrodes. This is because of an improved electrochemically active surface area of LMS grains [22]. During a continuous charge/discharge cycling, one can see a rapid drop of specific capacity in lithium-manganese orthosilicate powders. This phenomenon can be related to the phase amorphization during the first charge process [23, 24] and the continuous crystal degradation due to the Jahn-Teller effect of Mn ions and manganese dissolution into the electrolyte [23]. After twenty charge/discharge cycles, the examined powders delivered capacities of 23/23, 46/40, and 63/60 mAh g−1 and thus retained 38%, 49%, and 43% of their initial capacity for SLMS1, SLMS2, and SLMS3 powders, respectively. As concluded from SEM and N2 adsorption/desorption experiments, increasing the amount of sucrose in the reactant mixture results in reduced grain size of synthesized powders. Therefore, by reduction of the Li2MnSiO4 particle size, the diffusion pathways for Li+ ions can be shortened. This would enhance the lithium ion transport properties, increase the contact area with the electrolyte, and improve the electrical conductance of the electrode through the conductive carbon network.

Charge/discharge curves (a–c) of Li2MnSiO4 powders (solid line, 1st cycle; dashed line, 2nd cycle; shortly dashed line, 5th cycle; dashed dot line, 10th cycle; dashed dot dot line, 20th cycle). Cyclability of Li2MnSiO4 powders during charge (d) and discharge (e) processes (charge/discharge current, C/10; potential range, from 1.5 to 4.8 V vs. Li+ /Li0). Cyclic voltamperometry curves (a) of Li2MnSiO4 powders (scan rate, 0.1 mV s−1; potential range, from 1.5 to 4.8 V vs. Li+ /Li0)
One can see that even though SLMS3 powder shows the best electrochemical performance (the highest specific capacity), its cyclability is lower than SLMS2 compound. This phenomenon can be explained by longer periods of time, during which SLMS3 compound was present in the potentials above 4.5 V (vs. Li+/Li0). Since the conventional electrolyte mixture of 1 M LiPF6 in EC/DMC starts to decompose above 4.5 V (vs. Li+/Li0) [23, 25], the cell with SLMS3 powder could produce more electrolyte decomposition products. As it was exposed to those potentials for the extended time (due to its improved capacity reached at such high potentials), which in turn caused highest degree of electrolyte decomposition [25], it induced a faster electrode degradation. However, this trend requires a further examination.
The diminishing of the synthetic method to the nanoscale results in the shortening of the lithium ion diffusion pathway and thus accelerates the intercalation/deintercalation process. Table 3 shows the results of solvo- and hydrothermal syntheses. Increasing the working temperature of the cell causes a reduction in viscosity which causes a faster diffusion of lithium ions. With the increase of the working temperature of the system, we also accelerate aging of the cell and increase the rate of electrolyte decomposition. The carbon additives such as PEDOT, carbon black, sucrose, citric acid, etc. improve the poor conductivity of the material based on orthosilicates. The improvement of the cell’s capacity has been achieved by increasing the working temperature of the cell [12, 14, 15]. The temperature increase caused faster aging of the battery, which may be the result of faster electrolyte degradation. The best results, due to the carbon content, were achieved between 13 and 18 wt% [13, 15, 26]. Too little amount of conductive carbon additive does not allow good conductivity to be obtained from all grains, while too much carbon content forms agglomerates from amorphous carbon, which inhibits lithium ion diffusion. In our work, it has been presented how a small addition of sucrose (1–3%) to the synthesis improves the electrochemical parameters of the cell in contrast to other works where the carbon is added after the gel is formed, i.e., only on the outer coating.
Cyclic Voltammetry
Figure 6 f presents cyclic voltammograms of Li2MnSiO4 powders. A large oxidation peak at ca. 4.5 V (vs. Li+/Li0) during the first oxidation process shifted to lower potential values and then to lower current densities in subsequent cycle. These phenomena might be correlated to lithium-manganese orthosilicate phase amorphization happening during first charging process. After the initial LMS amorphization, the shape of voltammograms changed exposing two redox pairs present at 2.9/3.2 and 4.0/4.3 V (vs. Li+/Li0) and related to Mn2+/Mn3+ and Mn3+/Mn4+ redox processes. Moreover, when the electrode potential approached to 4.6 V (vs. Li+/Li0), there was an increase in the current density flowing through the electrochemical cell, which can be related to electrolyte decomposition reactions at high potentials as suggested before. In the case of SLMS3 sample, one can see one additional reduction process, present below 1.8 V (vs. Li+/Li0), which could be related to unreacted contaminations in SLMS3 powder, which started to be visible on voltammograms due to enhanced conductivity through the electrode (as the amorphous carbon obtained from the sucrose reduction started to provide electrical connection between current collector, orthosilicate particles, and impurities present in the sample). The current density values for Li2MnSiO4/C composites increased together with the amount of sucrose added into the reactor chamber, suggesting enhanced kinetics of lithium insertion/extraction reaction into/out of lithium-manganese orthosilicate grains. This effect supports findings from galvanostatic charge/discharge test and is related to the enhanced conductivity of LMS electrodes, suggesting an increase in their electrochemically active surface area [10, 26,27,28,29].
Conclusions
In our work, we synthesized and characterized a series of lithium-manganese orthosilicate powders with a various carbon content. Use of the modified solvothermal method with sucrose as a carbon source led to a good quality Li2MnSiO4 powder with a small amount of impurities and enhanced electrochemical performance in lithium-ion cell. The increased amount of the carbon source used in solvothermal process significantly changed the LMS material properties such as improved electrochemical properties of LMS/C compounds, reduced grain size, and increased their specific surface area. A small particle size and large specific surface area, altogether with internal carbon coating, increased lithium-manganese orthosilicate electrochemically active surface area, which was responsible for an enhancement in LMS electrochemical performance. It is noteworthy that despite the improvement in Li2MnSiO4 properties, we observed a decrease in cycling stability related to a prolonged electrolyte decomposition at high potentials. Nonetheless, our work shows that modified solvothermal process with sucrose as a carbon source is an effective, fast, and cheap approach to manufacture a new generation of electrode materials for lithium-ion batteries.
References
R. Dominko, M. Bele, M. Gaberšček, A. Meden, M. Remškar, J. Jamnik, Electrochem. Commun. 8, 217 (2006)
R. Dominko, M. Bele, A. Kokalj, M. Gaberscek, J. Jamnik, J. Power Sources 174, 457 (2007)
Y.X. Li, Z.L. Gong, Y. Yang, J. Power Sources 174, 528 (2007)
M. Armand, C. Michot, N. Ravet, M. Simoneau, P. Hovington, US Patent 6, 085,015 (2000)
R. Dominko, J. Power Sources 184, 462 (2008)
I. Belharouak, A. Abouimrane, K. Amine, J. Phys. Chem. C 113, 20733 (2009)
H. Duncan, A. Kondamreddy, P.H.J. Mercier, Y. Le Page, Y. Abu-Lebdeh, M. Couillard, P.S. Whitfield, I.J. Davidson, Chem. Mater. 23, 5446 (2011)
V. Aravindan, K. Karthikeyan, K.S. Kang, W.S. Yoon, W.S. Kim, Y.S. Lee, J. Mater. Chem. 21, 2470 (2011)
V. Aravindan, K. Karthikeyan, S. Ravi, S. Amaresh, W.S. Kim, Y.S. Lee, J. Mater. Chem. 20, 7340 (2010)
M. Boczar, M. Krajewski, M. Ratynski, B. Hamankiewicz, A. Czerwinski, Int. J. Electrochem. Sci. 13, 11636 (2018)
W. Liu, Y. Xu, R. Yang, Rare Metals 29, 511 (2010)
T. Muraliganth, K.R. Stroukoff, A. Manthiram, Chem. Mater. 22, 5754 (2010)
M. Kuezma, S. Devaraj, P. Balaya, J. Mater. Chem. 22, 21279 (2012)
D.M. Kempaiah, D. Rangappa, I. Honma, Chem. Commun. 48, 2698 (2012)
D. Rangappa, K.D. Murukanahally, T. Tomai, A. Unemoto, I. Honma, Nano Lett. 12, 1146 (2012)
M.K. Devaraju, T. Tomai, A. Unemoto, I. Honma, RSC Adv. 3, 608 (2013)
R.J. Gummow, N. Sharma, V.K. Peterson, Y. He, J. Solid State Chem. 188, 32 (2012)
G. Luo, J. He, X. Song, X. Huang, X. Yu, Y. Fang, D. Chen, J. Alloys Compd. 621, 268 (2015)
Y.J. Hao, Q.Y. Lai, J.Z. Lu, D.Q. Liu, X.Y. Ji, J. Alloys Compd. 439, 330 (2007)
K.S.W. Sing, Pure Appl. Chem. 54, 2201 (1982)
L.C. de Oliveira, C.A. Ribeiro, A.H. Rosa, W.G. Botero, J.C. Rocha, L.P. Cruz Romão, A. dos Santos, J. Braz. Chem. Soc. 20, 1135 (2009)
A. Kokalj, R. Dominko, G. Mali, A. Meden, M. Gaberscek, J. Jamnik, Chem. Mater. 19, 3633 (2007)
Q. Cheng, W. He, X. Zhang, M. Li, L. Wang, J. Mater. Chem. A 5, 10772 (2017)
C.A.J. Fisher, N. Kuganathan, M.S. Islam, J. Mater. Chem. A 1, 4207 (2013)
A. Guéguen, D. Streich, M. He, M. Mendez, F.F. Chesneau, P. Novák, E.J. Berg, J. Electrochem. Soc. 163, A1095 (2016)
M. Molenda, R. Dziembaj, M. Drozdek, E. Podstawka, L.M. Proniewicz, Solid State Ionics 179, 197 (2008)
M. Krajewski, B. Hamankiewicz, A. Czerwiński, Electrochim. Acta 219, 277 (2016)
M. Krajewski, B. Hamankiewicz, M. Michalska, M. Andrzejczuk, L. Lipinska, A. Czerwinski, RSC Adv. 7, 52151 (2017)
M. Ratyński, B. Hamankiewicz, M. Krajewski, M. Boczar, A. Czerwiński, RSC Adv. 8, 22546 (2018)
Funding
This work was supported by The Polish National Centre of Science through the research grant UMO2014/15/B/ST5/02118.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Boczar, M., Krajewski, M., Hamankiewicz, B. et al. Structure, Morphology, and Electrochemical Properties of Carbon-Coated Lithium-Manganese Orthosilicate with Sucrose as a Carbon Source. Electrocatalysis 11, 329–337 (2020). https://doi.org/10.1007/s12678-020-00590-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12678-020-00590-2