
Abstract
The microbiota colonizing the root endophytic compartment and surrounding rhizosphere soils contribute to plant growth and health. However, the key members of plant soil and endophytic microbial communities involved in inhibiting or assisting pathogen invasion remain elusive. By utilizing 16S high-throughput sequencing and a molecular ecological network (MEN) approach, we systematically studied the interactions within bacterial communities in plant endophytic compartments (stem and root) and the surrounding soil (bulk and rhizosphere) during bacterial wilt invasion. The endophytic communities were found to be strongly influenced by pathogen invasion according to analysis of microbial diversity and community structure and composition. Endophytic communities of the infected plants were primarily derived from soil communities, as assessed by the SourceTracker program, but with rare migration from soil communities to endophytic communities observed in healthy plants. Soil and endophytic microbiomes from infected plants showed modular topology and greater complexity in network analysis, and a higher number of interactions than those in healthy plants. Furthermore, interactions among microbial members revealed that pathogenic Ralstonia members were positively correlated with several bacterial genera, including Delftia, Stenotrophomonas, Bacillus, Clostridium XlVa, Fontibacillus, Acidovorax, Herminiimonas, and three unclassified bacterial genera, in infected plant roots. Our findings indicated that the pathogen invasion in the rhizosphere and endophytic compartments may be highly associated with bacteria that are normally not detrimental, and sometimes even beneficial, to plants.
Similar content being viewed by others

Intoduction
The soil microbial community can significantly impact plant growth, development, and resistance against soil-borne pathogens in agricultural ecological systems.1 Previous studies have revealed that the bulk soil is the main reservoir for microorganisms colonizing the rhizosphere.2 The plant drives the migration of the microorganisms by depositing specific root-excreted exudates at the soil–root interface.2,3 Meanwhile, soil microorganisms play essential roles in improving plant nutrient acquisition, enhancing stress tolerance, protection against soil-borne pathogens, and host immune regulation.4,5,6 The rhizosphere community is a subset of soil microbes that are subsequently filtered by niche utilization attributes and interactions with the host to inhabit the endophytic compartment.7 Meanwhile, a variety of microbes with diverse functions may migrate into plants and become transient endophytes, those consistently found within root and stem tissues are either candidate symbionts or stealthy pathogens;7,8 however, a mechanistic role for derivation of endophytic communities from soil communities has yet to be established.
Soil-borne bacterial plant pathogens attack crops and cause significant losses.9,10 Bacterial wilt caused by Ralstonia species is a devastating disease of Solanaceae crops (e.g., tobacco, tomato, and egg plants) with large-scale crop losses worldwide,11 and extensive efforts have been made to prevent and control this disease.12,13 Rhizosphere microbiota can function as a first line of defense against pathogen invasion4,14,15 with several studies revealing that endophytic microbes that colonize plants without inducing disease may also contribute to host resistance against pathogens.16,17 These endophytes suppress diseases via the induction of host resistance genes, competition, or the production of bioactive compounds.18 At present, the mechanistic role of plant endophytes during the infection period of pathogens is poorly understood.
With the development of high-throughput sequencing, related data-mining technologies have advanced greatly, including the SourceTracker program, a useful computational tool based on Bayesian approach that can be applied to estimate the proportions of taxa from certain environmental sources.19 SourceTracker has been widely used in different fields, such as tracking microbial contamination in aquatic systems,20 fecal pollution in recreational freshwater, and sources of airborne microorganisms in the indoor environment.21,22 Besides, the microbial interactions within certain habitat could be explored by the newly developed co-occurrence network approaches.23,24,25,26 Within network structure, the configuration and the distribution of links among interacting microbial members, can provide strong predictions on the function and stability of ecosystems, and recent modeling studies have linked these interaction network structures to community invasion resistance in plant soils and endophytic microbiota.27,28,29 Network analysis could also identify keystone microbial members or other microorganisms that may function in the defense against pathogen invasion.25,30,31,32,33 Therefore, network interactions both within the soil and endophytic communities and between the resident communities and invading pathogen are likely to be important for plant health and fitness.
The objectives of this study were to (i) investigate the characteristics of bacterial communities in soil, both bulk and rhizosphere, and within the endophytic compartments of roots and stems from healthy and wilt disease infected tobacco plants; (ii) track the source of microbial migration from soils communities to endophytic communities during pathogen invasion; and (iii) analyze the interactions between pathogen and other microbiota through network analysis. Through our results, we propose a possible road map that shows microbial source migration and thus reveals the core microbiota during plant wilt disease invasion.
Results
Bacterial community diversity and composition of healthy and infected samples
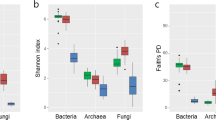
A total of 774,995 high-quality reads (bulk and rhizosphere soils, 405,438; endophyte roots and stems, 369,557) were obtained from 80 samples through high-throughput sequencing analysis. According to diversity indices, Chao1 (Fig. 1a) and Faith’s phylogenetic diversity (PD, Fig. 1b),34 the healthy and infected samples showed similar bacterial community diversity in both bulk and rhizosphere soils. Interestingly, the bacterial diversities of all infected root and stem samples were significantly higher than those of healthy root and stem samples (Student’s t-test, P < 0.01), which indicated more endophytic bacterial species in the infected plants than healthy ones. The principal coordinates analysis (PCoA) plot of microbial communities revealed a clear separation between endophytic and soil samples, and between healthy and infected endophytic samples (Supplementary Fig. 1).

a Diversity based on Chao1 index; b phylogenetic diversity calculated as Faith’s PD based on 97% similarity. Statistical analysis of the data was performed using Student’s t-test (*p < 0.05; **p < 0.01). HBS: bulk soils samples of healthy tobacco; IBS: bulk soil samples of wilt-infected tobacco; HRS: rhizosphere samples of healthy tobacco; IRS: rhizosphere samples of wilt-infected tobacco; HR: root samples of healthy tobacco; IR: root samples of wilt-infected tobacco; HS: stem samples of healthy tobacco; IS: stem samples of wilt-infected tobacco.
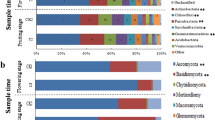
All OTUs were classified into 828 genera (soils, 730; endophytes, 242) belonging to 28 phyla (soils, 28; endophytes, 13). The top 10 most dominant OTUs (≥1.0% relative abundance) of soil and endophytic samples are shown in Fig. 2a, and 20 dominant genera (≥1.0% relative abundance) in endophytic compartments are shown in Fig. 2b. The bulk and rhizosphere soil communities were dominated by Arthrobacter, Acinetobacter, Massilia, Sphingomonas, Falsibacillus, Bradyrhizobium, Rhodanobacte, Sphingobium, Gaiella, and Terrabacter, but the relative abundances of these genera were reduced or absent in endophytic compartments. In addition, the relative abundances of some genera in soils and endophytic compartments were altered by pathogenic wilt invasion. The relative abundance of Ralstonia genus, which included many pathogenic species of plant wilt, was detected much higher in infected bulk and rhizosphere soils than healthy soils. Although we could not technically affirm all those OTUs assigned to Ralstonia are pathogenic by utilizing 16S high-throughput sequencing, this result still indicated those potential pathogens have been enriched from the soils close to the rhizosphere. Other genera, such as Chryseobacterium, Rhodococcus, Burkholderia, Noviherbaspirillum, Acinetobacter, Sphingomonas, Falsibacillus, and Bradyrhizobium showed a similar trend as pathogenic Ralstonia. In contrast, Massilia, Nocardioides, Sphingobium, Gaiella, and Conexibacte were relatively lower in infected soils than healthy soils. Within the endophytic compartments (Fig. 2b), the relative abundances of Ralstonia, Stenotrophomonas, Paenibacillus, Achromobacter, and Rhizobium of infected samples were increased compared to healthy samples. However, the relative abundances of Pseudomonas, Bacillus, and Falsibacillus, which are often considered to be plant-beneficial bacteria, showed a significant decrease as compared with healthy samples.

a Relative abundance of top 10 most dominant OTUs in healthy and infected bulk soils, rhizosphere, roots, and stems samples with 10 replicates. b Average relative abundance of bacterial genera showing significant difference among the healthy and infected root and stem samples. HBS1-10: 10 bulk soils samples of healthy tobacco; IBS1-10: 10 bulk soil samples of wilt-infected tobacco; HRS1-10: 10 rhizosphere samples of healthy tobacco; IRS1-10: 10 rhizosphere samples of wilt-infected tobacco; HR1-10: 10 root samples of healthy tobacco; IR1-10: 10 root samples of wilt-infected tobacco; HS1-10: 10 stem samples of healthy tobacco; IS1-10: 10 stem samples of wilt-infected tobacco.
SourceTracker analysis of bacterial community from soil to endophytic compartments
We utilized the SourceTracker program19 to study the proportion of endophytic bacterial communities derived from soils. According to the source apportionment results, there were differences in the sources of endophytic bacterial communities between infected and healthy plants (Supplementary Table 1). In healthy samples (Fig. 3), the majority of rhizosphere soil bacteria community members (74.98%) were derived from the bulk soil, but rare members of the endophytic communities of plant were derived from the soil bacteria community, indicating there is a clear boundary between interior and exterior of healthy plants. In infected samples (Fig. 3), the rhizosphere soil bacterial communities were mainly (71.4%) derived from the bulk soil, while the stems endophytic communities were mainly derived from the roots (94.9%). Importantly, more than half of endophytic root communities were derived from the bulk (50.1%) and rhizosphere (11.7%) soils, indicating most of endophytic microbial species in infected plant could be tracked back from the soils.

SourceTracker analysis results of healthy (left) and infected (right) samples.
Molecular ecological network analysis on soil and endophytic communities
Molecular ecological networks (MENs) analyses were performed to reveal the microbial interactions within soil and endophytic microbial communities, and their topological properties are shown in Supplementary Table 2. The average connectivity was used to assess network complexity and showed that the soil and endophytic compartments of infected plants were more complex than those of healthy plants (avgK: infected soils: 4.79 > healthy soils: 3.189; infected endophyte: 10.927 > healthy endophyte: 7.225). The average path lengths in infected and healthy soils were 4.035 and 5.048, respectively, and in infected and healthy endophytic compartments were 2.63 and 3.388, respectively; these values were very close to the logarithm of total number of network size and markedly different from other networks, therefore exhibiting the network properties of typical small world.35 These results suggested that all nodes were highly interlinked within the networks. The differences of topological properties were compared between the empirical and corresponding random networks for modularity analyses. Finally, the modularity value (M) for infected and healthy soils were 0.524 and 0.66 and for infected and healthy endophytic compartments were 0.427 and 0.514, respectively. These values were all higher than the M values in corresponding randomized networks, which implied all of the constructed MENs had modular architectures. Furthermore, the constructed random network results showed that network indices (e.g., average clustering coefficient, average path length, modularity) were all different between any two networks of infected and healthy sample groups (Supplementary Table 2).
To gain a deeper insight into the interactions among soil and endophytic microorganisms, the four networks were visualized and found to exhibit significantly different network structures (Fig. 4a–d). The network structures of soil and endophytic communities appeared to be significantly altered during tobacco wilt bacterial pathogen with the bacterial communities of infected plants (Fig. 4b, d) showing higher complexity and connectivity than those of healthy plants (Supplementary Table 2, Fig. 4a, c). The interactions among potential pathogenic Ralstonia and other bacterial members were observed in the networks of infected soils and endophytic compartments that were not found in the corresponding healthy networks. In addition, we also noted that there were more nodes (9 nodes) of potential pathogenic Ralstonia OTUs and more links (38 links) among potential Ralstonia and other bacterial OTUs in the network of infected endophytic compartments than the infected soils network (1 node and 12 links), indicating that the greater number of interactions among potential pathogenic Ralstonia and other organisms in infected endophytic compartments than in infected soils might play an important role in determining the migration of pathogens from soil into endophytic communities during tobacco bacterial wilt invasion.

a Bacterial networks of soil communities in healthy samples (bulk + rhizosphere soils). b Bacterial networks of soil communities in infected samples (bulk + rhizosphere soils). c Bacterial networks of endophytic communities in healthy samples (roots + stems). d Bacterial networks of endophytic communities in infected samples (roots + stems). e Networks of bacterial communities in infected stems. f Networks of bacterial communities in infected roots. Each node color represents a microbial species at phylum level. Ralstonia was labeled at the genus level. Blue links represent positive interactions between nodes and red links represent negative interactions.
Based on the above network structures, we performed further analysis on the endophytic network of infected roots and stems, in order to verify that which part played a more important role in the process of pathogenic wilt invasion. The topological properties of networks are shown in Supplementary Table 2 and the visualized individual networks of infected stems and roots are shown in Fig. 4e, f, respectively. The endophytic microbiota of healthy (Supplementary Fig. 2b) and infected roots (Fig. 4f) had more complex and highly connected bacterial community than those of healthy (Supplementary Fig. 2a) and infected stems (Fig. 4e). In addition, there were more nodes (8) of potential pathogenic Ralstonia OTUs and a greater number of links (69 in total) among Ralstonia and other microbial members in the network of infected roots than in infected stems (4 nodes, 6 links), which implied that the invasion by pathogenic Ralstonia was associated with the more varied interactions with other microbial members in pla"nt roots than in infected plant stems.
Network of interactions among potential pathogenic Ralstonia and other microbial members in endophytic roots
To further reveal which endophytic microorganisms may be important in aiding or inhibiting bacterial wilt outbreaks, subnetworks for the interactions among pathogenic Ralstonia and other microbial members were analyzed to identify “inferred” key organisms in the MENs of infected roots (Fig. 5). The network of interactions in infected roots revealed that potential pathogenic bacteria (Ralstonia) were negatively correlated with several bacterial genera, including Pigmentiphaga, Bosea, Variovorax, Sphingobacterium, and one unclassified bacteria (family: Enterobacteriaceae), but were positively correlated with other groups including Delftia, Stenotrophomonas, Bacillus, Clostridium XlVa, Fontibacillus, Acidovorax, Herminiimonas, and three unclassified bacterial genera (family: 2 Burkholderiaceae, Rhizobiales). These bacteria that correlated positively and negatively with potential pathogenic Ralstonia members, respectively, may play important roles in assisting and inhibiting bacterial wilt infections. Our observations show pathogen invasion might be aided by positively correlated native microbial members who may assist in colonization and/or enriched through a mutualistic relationship in tobacco roots during the infection process of wilt disease.

Each node is labeled at the genus level and unclassified OTUs are labeled with their family information. Blue links represent positive interactions between nodes and red links represent negative interactions.
Discussion
Several members of the genus Ralstonia, especially R. solanacearum, are well-known and important phytopathogens due to their ability to cause wilt symptoms and economic losses in many cultivated members the Solanaceous family of plants.9,10 Meanwhile, the diversity of resident microbes could also affect the antagonistic and/or facilitative interactions between plants and pathogens.27,36 Our results showed that the community diversity of infected roots and stems were higher than in healthy samples according to both Chao1 and PD indices (Fig. 1), which could be explained by the fact that the plant’s defense system was disrupted after bacterial wilt invasion, allowing more organisms from soil microbial communities to enter the plant. This result was also consistent with previous research that endophytes are believed to play important roles in priming host defenses against pathogen invasion37 and high diversity might increase community invasion resistance due to interactive effects on community stability.38
Through the species classification, we found the OTUs assigned to potential pathogenic Ralstonia (we could not technically affirm all those OTUs assigned to Ralstonia are pathogenic by just using 16S sequences) were rarely observed in all healthy samples (bulk soils, rhizosphere, stems and roots), but showed fairly high abundances in the infected plant samples, consistent with field observations of plant wilt. Correspondingly, the relative abundances of some bacteria were clearly altered after bacterial wilt infection. There was a decline in the relative abundances of Arthrobacter, Massilia, Nocardioides, Sphingobium, Gaiella, and Conexibacter in infected bulk and rhizosphere soils (Fig. 2a). These compositional changes could be a consequence of pathogen invasion. For example, Arthrobacter (49.7% and 19.1% reduction in infected bulk and rhizosphere soil, respectively) is known to have pathogen suppression potential for Fusarium wilt.39 In the endophytic compartments, the relative abundances of Lactococcus, Pseudomonas, Bacillus, Falsibacillus, and Leuconostoc, often considered to be plant-beneficial microbes,40,41,42,43,44,45,46 showed significant decrease compared to the healthy samples. These decreases suggest that the normal endophytic taxa were either actively excluded by the host immune system or outcompeted by more-successful colonizers.47,48 The genera Lactococcus, Enterococcus, and Leuconostoc are recognized as lactic acid bacteria,40 with the ability to act as plant growth promoting bacteria, inhibiting wilt of tomato caused by Ralstonia solanacearum.49 Members of the genera Bacillus, Enterococcus, and Falsibacillus are widely recognized as the biocontrol agents with the ability to secrete antibiotics or other antimicrobial proteins,41,42,43,44,45,46 and have been applied to prevent and control bacterial diseases of alfalfa, tobacco, and cucumber.50,51,52 Meanwhile, the relative abundances of Stenotrophomonas, Paenibacillus, Achromobacter, Rhizobium, Clostridium, Delftia, Acidovorax, and Microbacterium in infected samples showed a marked increase compared to healthy samples, suggesting they may be involved in the process of pathogen invasion and have mutualistic relationships with pathogenic members of Ralstonia,53 or perhaps they are opportunists, able to take advantage of potential ecological niches opened by pathogen invasion.47 These changes in microbiome composition and structure indicate a change in the root exudates caused by pathogen invasion or a sophisticated plant immune system,54,55,56 which drives either differential recruitment of beneficial microbes and/or differential exclusion to enable wilt resistance in plant roots and stems.12
Soil microorganisms likely affect plant immune defense and pathogen migration, therefore understanding how plant endophytes interact with the soil microorganisms may provide a “road map” to define the pathogen invasion process. The SourceTracker program has been used to estimate the proportions of contaminants in a given community that come from potential source environments19 and has been used to analyze the relationship between human-associated microbial communities and home surfaces.57 In this study, we utilized this program to track the source of plant rhizospheric and endophytic microbial communities during the process of pathogenic wilt invasion. A previous study had shown that R. solancearum invaded plants via the roots, multiplied, and then aggressively colonize the xylem elements in the vascular system, blocking water transport such that infected plants wilt and die.58 Our results showed different sources for microbiota in infected plants, in which the bacteria communities in the rhizosphere soils were mainly (71.4%) derived from the bulk soil. This is consistent with the previous studies that the bulk soil was the main source of microbial species richness in plant rhizosphere.2,3,59 However, root endophytic communities were mainly derived from the bulk soils (50.1%) rather than from the rhizosphere soil (11.7%) (Fig. 3). It was concluded that pathogen invasion may begin in the bulk soils, transfer to the plant roots, and in turn infect plant stems.
In the recent years, visualization of interactomes from diverse organisms has led to great progress in network biology.60,61 While several studies have established a positive correlation between community diversity and invasion resistance, it is less clear how interactions between members within resident communities are involved in this process.62 From the perspective of resource utilization and competition, plants and soil microbes can have direct co-evolutionary relationships, such as those between plants and pathogens.63,64 It is becoming more evident that pathogenic and mutualistic–symbiotic organisms influence plant microbial community diversity and succession.65,66 In this study, we performed network analyses on soil and endophytic bacterial community interactomes of infected and healthy plants, and revealed their topological features (Supplementary Table 2, Fig. 4a–d). The soil (Fig. 4b) and endophytic microbiota (Fig. 4d) of infected plants exhibited more complex, and highly connected bacterial communities than the respective communities of healthy plants, (Fig. 4a, c). In this sense, by changing soil community structure, invasive pathogenic microbes could generate positive feedback that enhances both their own competitiveness and subsequent interactions with their neighbors. Crucially, highly connected and modular microbiota could prime the plant immune system for accelerated activation of defense against the pathogen.54,55,67
In addition, we found that there were more nodes (9 nodes) of pathogenic Ralstonia members and a greater number of links (38 links) among Ralstonia and other microbial members in the network of infected endophytic compartments (Fig. 4d) than in the infected soil network (1 node, 12 links) (Fig. 4b). A greater number of Ralstonia nodes (8) and links (69) between Ralstonia and other microbial members were observed in the infected root network (Fig. 4f) as compared to infected stems (4 nodes, 6 links) (Fig. 4e). Based on these network topological data and source tracking analyses results (Fig. 3), we predicted that the endophytic microbiota played important role in the suppression of plant pathogens and that, from the perspective of microbial interactions and source tracking, plant roots were the critical migration site during the process of tobacco bacterial wilt disease.
Network analysis also revealed the relationships between pathogen and other associated bacteria species (Fig. 5). The highly connected and anomalously correlated nodes are either targets or helpers of diverse pathogens.68 Microbes that positively interact with the pathogenic Ralstonia members were the preferred helpers for pathogen attack in tobacco bacterial wilt disease.68 We identified previously unknown bacteria (Delftia, Stenotrophomonas, Bacillus, Clostridium XlVa, Fontibacillus, Acidovorax, Herminiimonas, and three unclassified bacterial genera (family: 2 Burkholderiaceae, Rhizobiales)) that may have a positive effect on wilt disease invasion, and were enriched in infected roots. For instance, species belonging to the Rhizobiales are intriguing and extensively researched for including both bacteria with the ability to fix nitrogen when in symbiosis with leguminous plants and pathogenic bacteria to plants,69 could colonize both below- and above-ground tissues of tobacco using a dynamic invasion process that involves both epiphytic and endophytic life styles.70 These non-detrimental microbial members could closely collaborate with pathogens in the endophytic root compartment. This is consistent with our source tracking analyses results that the root was the key compartment for microbial community assembly from soil into endophytic communities during tobacco bacterial wilt invasion.
Taken together, we infer that infection by pathogenic Ralstonia members may be highly associated with positive interactions between them and non-detrimental bacteria including Delftia, Stenotrophomonas, Bacillus, Clostridium XlVa, Fontibacillus, Acidovorax, Herminiimonas, and three unclassified bacterial genera (family: 2 Burkholderiaceae, Rhizobiales), and that these non-detrimental bacteria could obtain benefits from promoting pathogen, which might lead to the migration of many additional bacterial genera into plant root and stems from bulk soils, eventually causing an outbreak of tobacco bacterial wilt disease. This discovery will provide potential ideas and a theoretical basis for controlling tobacco bacterial wilt disease. Further work is needed to confirm these findings.
Methods
Sample collection and processing
A total of 80 samples were collected from five different tobacco field sites located in the Chenzhou Tobacco-growing region of Hunan province (general locations are shown in Supplementary Fig. 3). The same tobacco cultivar Yunyan 87 was cultivated at all sites included both healthy and severely infected (grade 5–9 infection).71 The samples included 10 bulk soils samples of healthy tobacco (HBS), 10 bulk soil samples of wilt-infected tobacco (IBS), 10 rhizosphere samples of healthy tobacco (HRS), 10 rhizosphere samples of wilt-infected tobacco (IRS), 10 root samples of healthy tobacco (HR), 10 root samples of wilt-infected tobacco (IR), 10 stem samples of healthy tobacco (HS), 10 stem samples of wilt-infected tobacco (IS). Each sample was a composite formed by mixing together five sub-samples from the same plant. The samples were collected from each field using checkerboard sampling method on June 2016 (tobacco was at its mature stage).
Bulk soil samples close to the plant root but not adhere to the root were collected by shaking off plant root. After shaking off bulk soils, the adhering rhizosphere soil samples were collected in PBS (0.1% Tween 80) with a brush. After stirring for 5 min, the resulting suspension was then poured into a sterile centrifuge tube, this process was repeated a further two times. The suspensions were mixed and centrifuged; the resulting sediment pellets were stored at −80 °C prior to DNA extraction. The roots and stems were washed with 75% ethanol, 2.5% sodium hypochlorite, and sterile water, respectively. Subsequently, the roots and stems were cut into small pieces and ground into homogenate using a mortar with addition of PBS, then washed into a centrifuge tube and let stand for 30 min. The homogenate solution was centrifuged and the supernatant was removed, and the resulting cell pellets (endophytic samples) were stored at −80 °C prior to DNA extraction.
DNA extraction and amplicon sequencing
A total of 80 samples were sequenced following the procedure below. Total DNA was extracted using the FastDNATM SPIN kit (MP Biomedicals). DNA concentration and quality were assessed by a NanoDrop Spectrophotometer (Nano-100, Aosheng Instrument Co. Ltd). To amplify the V5-V6 region of 16S rRNA gene, we used the 799F (5′-AACMGGATTAGATACCCKG-3′)/1115R (5′-AGGGTTGCGCTCGTTG-3′) primers to avoid amplifying chloroplast DNA.72 The 12 bp barcodes have been added into the 5′-ends of both forward and reverse primers to distinguish every samples in high-throughput sequencing. The PCR amplification was performed in a 50 μl reaction system with 1.5 μl dNTP mixture, 0.5 μl Taq DNA Enzyme (TaKaRa, Beijing, China), 5 μl 10× PCR buffer, 1.5 μl of both 10 uM forward and reverse primers, 20–30 ng of DNA template. The thermal cycle operations were defined as follows: 94 °C for 1 min, 30 cycles of 94 °C for 20 s, 57 °C for 25 s, and 68 °C for 45 s, then extension at 72 °C for 10 min, and finally stored at 4 °C.
Detection and purification of positive PCR amplicons were conducted by agarose gel electrophoresis and E.Z.N.A.TM Gel Extraction Kit (Omega BioTek, Norcross, USA). The purified amplicons were quantified by using a NanoDrop Spectrophotometer, and the optical density of the gel was analyzed with the Gel Image Analysis System (Taxon-1600). Subsequently, we established a standard regression model including DNA concentration and optical density to obtain the required volume of 150 ng DNA based on their net optical reference. The amplicons were pooled together and the mixed samples were used to prepare the sequencing library with VAHTS™ Nano DNA Library Prep Kit for Illumina according to the MiSeq Reagent Kit Preparation Guide (Illumina). The samples were sequenced using a Miseq sequencing machine (Illumina) at Central South University, Changsha, China.
Sequence data preprocessing and bioinformatics approaches
Preprocessing of sequence data was performed with a series of bioinformatics tools integrated into an in-house pipeline (http://mem.rcees.ac.cn:8080). All reads with less than two mismatches were sorted and assigned to different samples according to barcodes. The forward and reverse primers sequences were trimmed off. Paired-end reads of adequate length, with at least 30 bp overlap, were combined by FLASH program73 to obtain full-length sequences with an average length of 222 bp. Unqualified sequences were filter out by the Btrim program with a threshold of quality value >20 and window size of 5.74 Sequences with ambiguous bases were discarded, only targeted sequences with a length of 290~310 bp passed strict quality filtering. Next, UPARSE75 was used to remove chimeras and generate operational taxonomy units (OTUs) at a similarity of 97%. A large table where the columns contain 80 samples and the rows represented OTUs was created as OTU table, and the total read counts were resampling with the lowest sequences (9256 sequences for soil samples and 7539 sequences for endophytic samples) that were used for downstream analysis. The rarefaction curves of microbial communities for all samples are shown in Supplementary Fig. 4.
Statistical analysis
Student’s t-test was conducted to test statistical significance of differences between two groups. We calculated two measurements of alpha-diversity to assess the diversity of soil and endophytic microbial communities. PD was calculated according to Faith’s approach via Picante package in R (v.3.2.5).34 The Chao1 value76 was calculated using Mothur software.77 Unweighted PCoA based on UniFrac distance matrix78 was used to examine difference in microbial community structures.
SourceTracker analysis
We created an implementation of SourceTracker19 within an in-house pipeline (http://mem.rcees.ac.cn:8080) which consisted of relevant bioinformatics tools. The SourceTracker analysis was constructed as follows: based on OTUs data (filter OTUs present in less than 1% of the samples from the OTU table), estimated the proportion of rhizosphere communities from bulk soil communities, root endophyte communities from rhizosphere communities, and stem endophyte communities from root endophyte communities. The percentage value was derived from the statistical average of the results of SourceTracker.
Random matrix theory-based molecular ecology networks
To elucidate microbial interactions in soil and endophytic communities during wilt disease invasion, we constructed phylogenetic MENs via a Random Matrix Theory (RMT)-based approach in molecular ecological network analysis pipeline (MENA, http://ieg2.ou.edu/MENA/).23,24,79 This has been described previously in detail,23 and will only be summarized here. First, only OTUs that were present in more than eight samples were included in the analysis. Threshold values ranging from 0.01 to 0.99 with 0.01 intervals were applied to the Spearman rank correlation matrix. The optimal threshold value was estimated when the nearest-neighbor spacing distribution followed the Poisson distribution well, which is associated with characteristic nonrandom properties in a complex system.79,80 Furthermore, the appropriate identical threshold value was selected to generate networks for comparing the different networks under the same conditions.23 The empirical networks of soil and endophytic communities were all analyzed by above methods, and the random networks were generated by rewiring the positions of all links of MENs with the same numbers of nodes and links in corresponding empirical networks (Supplementary Table 2). The constructed networks of soil and endophytic communities in healthy samples and infected samples, and the sub-network of specific interactions between the pathogen and other microbial members were visualized by Cytoscape 3.3.0.81
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
16S rRNA gene sequencing data of all samples were submitted to the NCBI SRA database (https://www.ncbi.nlm.nih.gov/) under the accession PRJNA540089.
References
Mendes, L. W., Raaijmakers, J. M., Hollander, M. D., Mendes, R. & Tsai, S. M. Influence of resistance breeding in common bean on rhizosphere microbiome composition and function. ISME J. 12, 212–224 (2017).
Jones, D. L., Nguyen, C. & Finlay, R. D. Carbon flow in the rhizosphere: carbon trading at the soil–root interface. Plant Soil 321, 5–33 (2009).
Mendes, R., Garbeva, P. & Raaijmakers, J. M. The rhizosphere microbiome: significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 37, 634–663 (2013).
Mendes, R. et al. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 332, 1097–1100 (2011).
Mendes, L. W., Kuramae, E. E., Navarrete, A. A., Van Veen, J. A. & Tsai, S. M. Taxonomical and functional microbial community selection in soybean rhizosphere. ISME J. 8, 1577–1587 (2014).
Pérez-Jaramillo, J. E., Mendes, R. & Raaijmakers, J. M. Impact of plant domestication on rhizosphere microbiome assembly and functions. Plant Mol. Biol. 90, 635–644 (2016).
Schulz, B., Boyle, C. & Sieber, T. Microbial Root Endophytes (Springer Dordrecht, Dordrecht, Netherlands, 2006).
Hallmann, J., Quadt-Hallmann, A., Mahaffee, W. F. & Kloepper, J. W. Bacterial endophytes in agricultural crops. Can. J. Microb. 43, 895–914 (1997).
Hayward, A. C. Biology and epidemiology of bacterial wilt caused by Pseudomonas solanacearum. Annu. Rev. Phytopathol. 29, 65–87 (1991).
Chen, Y. et al. Biocontrol of tomato wilt disease by Bacillus subtilis isolates from natural environments depends on conserved genes mediating biofilm formation. Environ. Microbiol. 15, 848–864 (2013).
Jiang, G. F. et al. Bacterial wilt in China: history, current status, and future perspectives. Front. Plant Sci. 8, 1549 (2017).
Kwak, M. J. et al. Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. 36, 1100–1109 (2018).
Janvier, C. et al. Soil health through soil disease suppression: which strategy from descriptors to indicators? Soil Biol. Biochem. 39, 1–23 (2007).
Chapelle, E., Mendes, R., Bakker, P. A. H. & Raaijmakers, J. M. Fungal invasion of the rhizosphere microbiome. ISME J. 10, 265–268 (2016).
Mendes, L. W., Raaijmakers, J. M., de Hollander, M., Mendes, R. & Tsai, S. M. Influence of resistance breeding in common bean on rhizosphere microbiome composition and function. ISME J. 12, 212–224 (2018).
Wilson, D. Endophyte: the evolution of a term, and clarification of its use and definition. Oikos 73, 274–276 (1995).
Johnston-Monje, D. & Raizada, M. N. Conservation and diversity of seed associated endophytes in zea across boundaries of evolution, ethnography and ecology. PLoS ONE 6, e20396 (2011).
Mousa, W. K. et al. Root-hair endophyte stacking in finger millet creates a physicochemical barrier to trap the fungal pathogen Fusarium graminearum. Nat. Microbiol. 1, e16167 (2016).
Knights, D. et al. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 8, 761–763 (2011).
Liu, G. et al. Assessing the origin of bacteria in tap water and distribution system in an unchlorinated drinking water system by SourceTracker using microbial community fingerprints. Water Res. 138, 86–96 (2018).
Staley, C. et al. Application of sourceTracker for accurate identification of fecal pollution in recreational freshwater: a double-blinded study. Environ. Sci. Technol. 52, 4207–4217 (2018).
Prussin, A. J. & Marr, L. C. Sources of airborne microorganisms in the built environment. Microbiome 3, 78 (2015).
Deng, Y. et al. Molecular ecological network analyses. BMC Bioinformatics 13, 113 (2012).
Zhou, J. Z., Deng, Y., Luo, F., He, Z. L. & Yang, Y. F. Phylogenetic molecular ecological network of soil microbial communities in response to elevated CO2. mBio 2, e00122-00111 (2011).
Banerjee, S. et al. Determinants of bacterial communities in Canadian agroforestry systems. Environ. Microbiol. 18, 1805–1816 (2016).
PÕlme, S. et al. Host preference and network properties in biotrophic plant-fungal associations. N. Phytol. 217, 1230–1239 (2018).
Thebault, E. & Fontaine, C. Stability of ecological communities and the architecture of mutualistic and trophic networks. Science 329, 853–856 (2010).
Wei, Z. et al. Trophic network architecture of root-associated bacterial communities determines pathogen invasion and plant health. Nat. Commun. 6, e8413 (2015).
Wei, Z. et al. Ralstonia solanacearum pathogen disrupts bacterial rhizosphere microbiome during an invasion. Soil Biol. Biochem. 118, 8–17 (2018).
Berry, D. & Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 5, 219 (2014).
Layeghifard, M., Hwang, D. M. & Guttman, D. S. Disentangling interactions in the microbiome: a network perspective. Trends Microbiol. 25, 217–228 (2017).
Lurgi, M., Galiana, N., López, B. C., Joppa, L. N. & Montoya, J. M. Network complexity and species traits mediate the effects of biological invasions on dynamic food webs. Front. Ecol. Evol. 2, 1–11 (2014).
Galiana, N., Lurgi, M., Montoya, J. éM. & pez, B. C. L. ó Invasions cause biodiversity loss and community simplification in vertebrate food webs. Oikos 123, 721–728 (2014).
Faith, D. P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 61, 1–10 (1992).
Watts, D. J. & Strogatz, S. H. Collective dynamics of ‘small-world’ networks. Nature 393, 440–442 (1998).
Kefi, S. et al. More than a meal… integrating non-feeding interactions into food webs. Ecol. Lett. 15, 291–300 (2012).
Tan, R. X. & Zou, W. X. Endophytes: a rich source of functional metabolites. Nat. Prod. Rep. 18, 448–459 (2001).
McCann, K. S. The diversity-stability debate. Nature 405, 228–233 (2000).
Siegel-Hertz, K. et al. Comparative microbiome analysis of a Fusarium wilt suppressive soil and a Fusarium wilt conducive soil from the chateaurenard region. Front. Microbiol. 9, 568 (2018).
Ercolini, D., Moschetti, G., Blaiotta, G. & Coppola, S. Behavior of variable V3 region from 16S rDNA of lactic acid bacteria in denaturing gradient gel electrophoresis. Curr. Microbiol. 42, 199–202 (2001).
Schisler, D. A., Slininger, P. J., Behle, R. W. & Jackson, M. A. Formulation of Bacillus spp. for biological bontrol of plant diseases. Phytopathology 94, 1267–1271 (2004).
Ongena, M. & Jacques, P. Bacillus lipopeptides: versatile weapons for plant disease biocontrol. Trends Microbiol. 16, 115–125 (2008).
Ahimou, F., Jacques, P. & Deleu, M. Surfactin and iturin A effects on Bacillus subtilis surface hydrophobicity. Enzym. Microb. Technol. 27, 749–754 (2000).
Moyne, A. L., Cleveland, T. E. & Tuzun, S. Molecular characterization and analysis of the operon encoding the antifungal lipopeptide bacillomycin D. FEMS Microbiol. Lett. 234, 43–49 (2004).
Rosier, A., Bishnoi, U., Lakshmanan, V., Sherrier, D. J. & Bais, H. P. A perspective on inter-kingdom signaling in plant-beneficial microbe interactions. Plant Mol. Biol. 90, 537–548 (2016).
Al-Mughrabi, K. I. Biological control of Fusarium dry rot and other potato tuber diseases using Pseudomonas fluorescens and Enterobacter cloacae. Biol. Control 53, 280–284 (2010).
Lundberg, D. S. et al. Defining the core Arabidopsis thaliana root microbiome. Nature 488, 86–90 (2012).
Velásquez, A. C., Oney, M., Huot, B., Xu, S. & He, S. Y. Diverse mechanisms of resistance to Pseudomonas syringae in a thousand natural accessions of Arabidopsis thaliana. N. Phytol. 214, 1673–1687 (2017).
Harshini, R., Yasodha, P., Sabarinathan, K. G., Ambethgar, V. & David, P. M. M. Diversity of epiphytic lactic acid bacteria (lab) on insect oviposition sites. Int. J. Curr. Microbiol. Appl. Sci. 7, 607–621 (2018).
Handelsman, J., Raffel, S., Mester, Eh, Wunderlich, L. & Grau, C. R. Biological control of damping-off of alfalfa seedlings with Bacillus cereus UW85. Appl. Environ. Microbiol. 56, 713–718 (1990).
Fravel, D. R. & Spurr, H. W. Biocontrol of tobacco brown-spot disease by Bacillus cereus subsp. Mycoides in a controlled environment. Phytopathology 29, 930–932 (1977).
Cao, Y. et al. Bacillus subtilis SQR 9 can control Fusarium wilt in cucumber by colonizing plant roots. Biol. Fert. Soils 47, 495–506 (2011).
van Elsas, J. D. et al. Microbial diversity determines the invasion of soil by a bacterial pathogen. Proc. Natl Acad. Sci. USA 109, 1159–1164 (2012).
Jones, J. D. G. & Dang, J. L. The plant immune system. Nature 444, 323–329 (2006).
Peter, N. D. & John, P. R. Plant immunity: towards an integrated view of plant-pathogen interactions. Nat. Rev. Genet. 11, 539–548 (2010).
Wu, K. et al. Root exudates from two tobacco cultivars affect colonization of Ralstonia solanacearum and the disease index. Eur. J. Plant Pathol. 141, 667–677 (2015).
Lax, S. et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science 345, 1048–1052 (2014).
Peeters, N., Guidot, A., Vailleau, F. & Valls, M. Ralstonia solancearum, a widespread bacterial plant pathogen in the post-genomic era. Mol. Plant Pathol. 14, 651–662 (2013).
Lareen, A., Burton, F. & Schäfer, P. Plant root-microbe communication in shaping root microbiomes. Plant Mol. Biol. 90, 575–587 (2016).
Mccormack, M. E., Lopez, J. A., Crocker, T. H. & Mukhtar, M. S. Making the right connections: Network biology and plant immune system dynamics. Curr. Opin. Plant Biol. 5, 2–12 (2016).
Cho, D. Y., Kim, Y. A. & Przytycka, T. M. Chapter 5: Network biology approach to complex diseases. PLoS Comput. Biol. 8, e1002820 (2012).
Li, M. et al. Facilitation promotes invasions in plant-associated microbial communities. Ecol. Lett. 22, 149–158 (2019).
Barberán, A., Bates, S. T., Casamayor, E. O. & Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 6, 343–351 (2012).
Keymer, D. P. & Lankau, R. Disruption of plant-soil-microbial relationships influences plant growth. J. Ecol. 105, 816–827 (2017).
Kardol, P. & Wardle, D. A. How understanding aboveground-belowground linkages can assist restoration ecology. Trends Ecol. Evol. 25, 670–679 (2010).
Philippot, L., Raaijmakers, J. M., Lemanceau, P. & van der Putten, W. H. Going back to the roots: the microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 11, 789–799 (2013).
Van, derEnt et al. Priming of plant innate immunity by rhizobacteria and beta-aminobutyric acid: differences and similarities in regulation. N. Phytol. 183, 419–431 (2009).
Ahmed, H. et al. Network biology discovers pathogen contact points in host protein-protein interactomes. Nat. Commun. 9, 2312 (2018).
Carvalho, F. M., Souza, R. C., Barcellos, F. G., Hungria, M. & Vasconcelos, A. T. R. Genomic and evolutionary comparisons of diazotrophic and pathogenic bacteria of the order Rhizobiales. BMC Microbiol. 10, e37 (2010).
Ji, K. X. et al. Movement of rhizobia inside tobacco and lifestyle alternation from endophytes to free-living rhizobia on leaves. J. Microbiol. Biotechnol. 20, 238–244 (2010).
Chinese Standards (GB). Grade and investigation method of tobacco diseases and insect pests (GB/T 23222-2008, 2008).
Chelius, M. & Triplett, E. The diversity of Archaea and bacteria in association with the roots of Zea mays L. Micro. Ecol. 41, 252–263 (2001).
Kong, Y. Btrim: a fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics 98, 152–153 (2011).
Mago, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011).
Edgar, R. C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998 (2013).
Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 11, 265–270 (1984).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009).
Lozupone, C. & Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235 (2005).
Zhou, J. Z. et al. Functional molecular ecological networks. mBio 1, e00169-10 (2010).
Luo, F. et al. Constructing gene co-expression networks and predicting functions of unknown genes by random matrix theory. BMC Bioinformatics 8, 299 (2007).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Acknowledgements
We thank Dr. James Walter Voordeckers for carefully editing the grammar of the manuscript and for some valuable suggestions for this paper twice. This project was supported by the National Key Research and Development Program (2018YFD0200800), Key Research Program of Frontier Sciences, Chinese Academy of Sciences (CAS, QYZDB-SSW-DQC026), the project of Key Laboratory of Environmental Biotechnology, Chinese Academy of Sciences (CAS, KF2018008), the Foundation for Tobacco Science of Chenzhou Tobacco Company of Hunan Province (201743100024128).
Author information
Authors and Affiliations
Contributions
Q.H., L.T. and S.G. performed the main experiments and analyzed data; Y.D. and Q.H. planned and designed the research, wrote the manuscript with substantial input from Y.X., X.X., W.Z., K.F. and Z.W.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hu, Q., Tan, L., Gu, S. et al. Network analysis infers the wilt pathogen invasion associated with non-detrimental bacteria. npj Biofilms Microbiomes 6, 8 (2020). https://doi.org/10.1038/s41522-020-0117-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41522-020-0117-2
This article is cited by
-
Analysis of endophytic bacterial diversity in seeds of different genotypes of cotton and the suppression of Verticillium wilt pathogen infection by a synthetic microbial community
BMC Plant Biology (2024)
-
The beneficial endophytic microbes enhanced tobacco defense system to resist bacterial wilt disease
Chemical and Biological Technologies in Agriculture (2024)
-
Xanthomonas infection and ozone stress distinctly influence the microbial community structure and interactions in the pepper phyllosphere
ISME Communications (2023)
-
Plant and soil-associated microbiome dynamics determine the fate of bacterial wilt pathogen Ralstonia solanacearum
Planta (2023)
-
Identifying the effects of cropping with different pear cultivars on microbial community composition and networks in orchard soils
Environmental Science and Pollution Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
