
Abstract
Innate lymphoid cells (ILCs) are a lymphocyte population that is mostly resident at mucosal surfaces. They help to induce an appropriate immune response to the microbiome at homeostasis. In healthy people, the mucosal immune system works symbiotically with organisms that make up the microbiota. ILCs play a critical role in orchestrating this balance, as they can both influence and in turn be influenced by the microbiome. ILCs also are important regulators of the early response to infections by diverse types of pathogenic microbes at mucosal barriers. Their rapid responses initiate inflammatory programs, production of antimicrobial products and repair processes. This review will focus on the role of ILCs in response to the microbiota and to microbial infections of the lung and intestine.
Similar content being viewed by others

Introduction
Diverse microorganisms, including bacteria, viruses, and fungi, are associated with the mucosal barriers of the body.1 These barrier surfaces include the gastrointestinal and respiratory tracts, along with others, such as the skin and reproductive tract. They are sites of continual interactions between microbes, tissue cells and the immune system. The bacterial contents of these sites are among the best-defined microbial constituents at these surfaces, and they can have both beneficial and deleterious effects on the immune response and inflammatory disease.2 In the case of steady-state interactions with the microbiota, innate lymphoid cells (ILCs) have been reported to play important roles in regulating the immune response at mucosal barriers through the rapid production of cytokines and other mediators.3 Likewise, during infections, ILCs participate in the orchestration of the early response to clear pathogens at mucosal sites, thereby preventing systemic infection.
ILCs are cells that share a common progenitor with B and T lymphocytes, but they lack antigen receptors encoded by genes generated by RAG-dependent DNA rearrangements. This lack of receptor clonal diversity, coupled with their rapid responses, serves to categorize ILCs as part of the innate immune system. Studies suggest that ILCs arose approximately 500 million years ago in conjunction with adaptive immune B and T cells. They perhaps evolved to participate in the requirement for longer-lived and larger organisms to live in harmony with a more complex microbiota while providing defense against pathogens.4 ILCs have been reported to participate in the course of several diseases of mucosal tissues, including inflammatory bowel disease, asthma and colorectal cancer; these topics have been reviewed elsewhere.5,6,7,8 There also have been a number of recent, excellent reviews on ILC biology and function,9,10,11,12,13 including the role of these cells in general tissue homeostasis. Here we will focus specifically on the role of ILCs in influencing the response to the microbiome during homeostasis and during infection at two mucosal barriers, the lung and the intestine.
Subsets of ILCs
ILCs populate mucosal barriers, respond to cues from other cells and produce cytokines and other substances that mediate appropriate immune responses. While evidence for TLR function in ILCs in vitro has been reported,14,15 in vivo evidence for direct detection of microbes by ILCs, either by TLR signaling, or by other means, is sparse. Therefore, ILCs generally are not the first responders. Like other lymphocytes, ILCs rely on cytokine signaling through the common cytokine receptor γ chain (γc or CD132) for their development. Thus, ILCs were not decreased in Rag-deficient mice, but Il2rg-deficient mice displayed a more than 90% reduction.9,16 Similar to CD4+ T cells, functional subsets of ILCs have been identified, including ILC1, ILC2 and ILC3, with ILC1s similar to Th1 cells, ILC2s similar to Th2 cells and ILC3s similar to Th17 cells. For example, ILC1s were reported to be similar to Th1 cells in their dependence on the transcription factor T-bet17 and they exhibited the ability to secrete IFNγ. Likewise, ILC2s were similar to Th2 cells in their requirement for the transcription factor GATA-3 and they produced type 2 cytokines, prominently IL-5 and IL-13, but also IL-4 and IL-9.18,19 Finally, ILC3s showed similarities to Th17 cells and they depended on the transcription factor RORγt for their differentiation and they produced IL-22 as well as IL-17.19 Together, these three subsets may be classified as helper ILCs.
There is heterogeneity within the three principal ILC subsets. For example, ILC2s could be divided further into two subsets, natural ILC2s (nILC2) and inflammatory ILC2 (iILC2).20,21 nILC2s, which are Lin− ST2+ KLRG1int cells, were found in the lung during homeostatic conditions and responded to IL-33 stimulation. However, iILC2, which are Lin− ST2- KLRG1hi cells, are generally not found in the lungs, nor in most other peripheral tissues in naïve mice. iILC2s were only present after helminth infection or IL-25 exposure following recruitment from the intestine to the lung.20,21 Interestingly, some iILC2s exhibited the ability to produce IL17 and expressed intermediate amounts of the transcription factor that drives IL-17 expression, RORγt.20 Additionally, ILC3s can be separated based on their expression of CCR6 and natural cytotoxicity receptors (NCR) such as NKp46 in mouse and NKp44 in human. CCR6+ ILC3s are also known as lymphoid tissue inducer (LTi) cells and they have been demonstrated to aid in the development of lymph nodes and tertiary lymphoid follicles in peripheral tissues, including mucosal barriers.22 Maternally derived retinoic acid intake and fetal RA signaling control LTi cell differentiation before birth.23 RA has multiple basal functions in the intestine that influence interactions with the microbiome and the response to infections, including homing of lymphocytes to the mucosae, IgA synthesis, regulatory T-cell accumulation, and ILC subset skewing.24,25,26,27,28 There is a degree of plasticity between the three main ILC subsets. For example, RA stimulation in the presence of IL-1β and IL-23 has been reported to drive ILC1 plasticity and conversion to ILC3s29 but also, in other contexts, some ILC3 converted to ILC1.30,31
Natural killer (NK) cells may be classified as killer ILCs. They resemble cytotoxic CD8+ T cells in their ability to produce perforin and granzyme B,32 although NK cells are sometimes placed in a separate lymphocyte category from ILCs. With regard to cytokine production, many NK cells produce IFNγ, similar to ILC1s, CD8+ and Th1 CD4+ cells.33,34 Additionally, both NK cells and ILC1 express characteristic surface receptors, such as NK1.1 and NKp46. NK cell development is distinct from helper ILCs, however, because precursors of NK cells did not require IL-7 or the transcription factor GATA3.35 Instead, NK cell differentiation is distinguished by dependence on the transcription factor EOMES.36
ILCs were shown to be generated in both the fetal liver and bone marrow from the common lymphoid progenitor.37,38 Studies of parabiotic mice have demonstrated that ILCs were mostly tissue-resident cells,39 but the ILC subsets were differentially present in different sites. For example, the mouse lung contained a significant number of ILC2s,40 with lesser amounts of ILC1s and ILC3s at steady state.40 In the intestine, ILC1s were the major subset within the small intestine epithelium41,42 and ILC3s were the predominant ILC subset in the lamina propria of both the small and large intestine.42,43 In the intestine, ILC2s were more frequent in the colon lamina propria compared to the small intestine.42,43 Of note, these findings are specific to the steady state and alterations in the proportions of the ILC subsets occur following some infections. NK cells are the only ILC type found to a significant extent in the circulation, and they also are the most numerous ILC type in the lung and several other tissues.42
Essential functions of ILCs
To assess the phenotype in mice lacking only ILCs, investigators have relied on several techniques, most often comparison of Rag−/− mice lacking B and T cells to analyses of Rag−/−gc−/− mice also lacking ILCs in addition to the absence of B and T cells. Some studies have analyzed the effect of ILC depletion by treatment of Rag−/− mice with anti-Thy1 antibodies. These methods for deletion of all ILCs have limitations. For example, the microbiome is highly altered in Rag−/− mice and ILC populations in the intestinal epithelium of these mice were greatly expanded in number. Furthermore, anti-Thy1 antibodies could have effects even in T-cell-deficient Rag−/− mice on Thy1+ cell types besides ILCs, such as neuronal cells or fibroblasts. Therefore, while these methods have been used to provide much important information, and have supported suggestions that ILCs are critical for a variety of immune responses, studies in mice have been hampered by the absence of a means to specifically eliminate all ILCs, without effects on other cell types. Some methods have been developed, however, to analyze the function of particular ILC subsets. For example, Staggerer/RorαFlox-Cd127Cre mice, a strain with deletion of the gene encoding the transcription factor RORα mediated by Il7ra-driven Cre recombinase, were reported to be selectively deficient for ILC2s.44 Rora is highly transcribed in a few other IL-7 receptor-dependent lymphocyte types, however, such as iNKT cells. Similarly, deletion of the gene encoding the aryl hydrocarbon receptor by Cre recombinase controlled by the gene encoding RORγt (Rorc-Cre x Ahrf/f) specifically depleted ILC3s, although this also affected IL-22 secretion by CD4+ T cells.45
In humans, evidence for the essentiality of ILCs is still lacking. In fact, in severe combined immunodeficiency patients that received hematopoietic stem cell transplants, T lymphocytes were reconstituted but ILC reconstitution was absent or these cells were very limited in number. These patients showed no signs of disease after many years.46 It remains to be determined if a more thorough categorization of similar individuals is required to rule out illnesses dependent on the absence of ILCs, or if this represents a species difference in the relative importance of ILCs comparing laboratory mice to humans. We note that the immune system of mammals contains additional populations of innate-like lymphocytes, including γδ T cells, iNKT cells and mucosal-associated invariant T (MAIT) cells, and these lymphocytes all share aspects of their transcriptome and in some cases, show similar functions.47 Furthermore, one innate-like population may expand in the absence of another.48 Therefore, there could be a degree of redundancy between these cell populations, but this does not provide a cogent argument that ILCs are not important in any circumstance.
ILC interactions with the microbiome
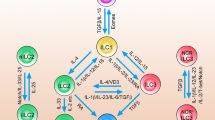
Homeostasis between the immune system and the microbiome is essential for both the lung and the intestine. A diverse and appropriate population of commensals aids in promoting the optimal immune response to pathogens, while also preventing a destructive inflammatory response to nonpathogenic microbes. In this section, as summarized in Fig. 1, we will describe how the different ILC subsets function to promote a barrier immune system in harmony with the microbiome.

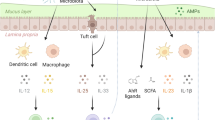
ILC subsets in the intestine are illustrated and for simplicity the role of cytokines in activating ILCs is emphasized, as opposed to other influences of ILC behavior. NK cells and ILC1s can be found mostly in the epithelium. Expression of IL-25R by ILC2s in the lamina propria allows them to respond to IL-25 from tuft cells. Activation of ILC2s leads to IL-5 and IL-13 production, which promotes goblet cell mucus production. Lamina propria ILC3s respond to IL-23 and IL-1β production by myeloid cells. Steady-state cytokine production by ILCs, especially IL-22, signals Paneth cells and epithelial cells to induce antimicrobial peptide (AMP) production and other responses, such as fucosylation of glycoproteins and glycolipids, both of which contribute to the composition of the microbiome.
ILC1s and NK cells
While NK cells and ILC1s play roles in the immune response to pathogens, by quickly producing IFNγ,34,41 less is known about their relationship with the microbiome. In mice and humans, these cells produce IFNγ in response to IL-12, IL-15 and/or IL-18.33,34 ILC1s may also be important, however, for preventing infection with opportunistic intestinal pathogens, such as Clostridium difficile. Rag1−/− mice were largely protected from Clostridium difficile infection, while Rag−/−gc−/− mice were susceptible.49 Furthermore, mice deficient for both Rag2 and the transcription factor T-bet developed a spontaneous severe colitis dependent on the commensal organism Helicobacter typhlonius.50 These data suggest that colonic, T-bet-dependent ILCs may play an active role in modulating the response to elements of the microbiome.
ILC2s
ILC2s drive a type 2 immune response in both lung and colon that is reported to maintain an appropriate inflammatory balance in both organs.51 ILC2s have been shown to be a major source of IL-13 in the lung at steady state,52 which is required for alternatively activated macrophage (M2) differentiation.53,54 M2 macrophages promoted tissue homeostasis, as opposed to the inflammation associated with excess M1 macrophages.55 In the intestine, IL-13, produced in part by ILC2s, contributed to goblet cell differentiation and mucus production,56,57 which are important for interactions with the microbiome and infectious agents. Consistent with this observation, mice deficient for IL-13R exhibited impaired mucus production.58 ILC2s were not only capable of producing cytokines typical of type 2 or Th2 immune responses, but they also produced amphiregulin (AREG) which binds to epithelial growth factor receptor (EGFR). These data indicate that ILC2s can induce tissue-healing responses that are important for limiting infection to barrier tissues.59
ILC2s are maintained in the tissues by IL-2 and IL-7,60,61 and they have the ability to respond to cytokines produced by other innate immune cells, such as IL-18, IL-25, IL-33 and TSLP.51,62 At steady state in mucosal barriers, epithelial cells produced amounts of IL-25 and IL-33 sufficient to drive basal levels of IL-5 and IL-13 production.51,63,64 In the lung, reports indicated that epithelial IL-33 induced the IL-13 production by ILC2s that led to M2 macrophage accumulation.53,54 IL-33 secreted by lung epithelial cells acted as an alarmin indicating epithelial damage. Subsequent AREG production by ILC2s in response to IL-33 promoted resolution of tissue damage and maintenance of the barrier.59,65 At homeostasis, IL-25 is produced by tuft cells in the intestine and lung. Tuft cells are an epithelial cell type that likely can sense different small molecules, including microbiome-derived succinate, through expression and activation of the succinate receptor.66,67 In the intestine, tuft cell-derived IL-25 activated ILC2s to secrete IL-1363 that contributed to a feedforward loop that further activated tuft cell IL-25 production. This loop allowed for an appropriate strength of the type 2 response.63 IL-18 derived from several cell types also has been shown to stimulate IL-13 production by ILC2s, especially in the lung and skin.62 Therefore, several cytokines can promote ILC2 activation under steady-state conditions, with the activation pathway somewhat tissue dependent.
There is also one report, however, suggesting that ILC2s directly sensed the microbiome. Human ILC2s expressed TLRs 1, 4 and 6. In culture, ILC2s were able to produce IL-5 and IL-13 in response to TLR stimulation.14 The in vivo relevance of this observation remains to be determined.
There is controversy regarding the question of dependence of intestinal ILC2s by the microbiome. Transcriptional programs of ILC2 residing in the small intestine lamina propria have been shown to be affected by colonization by commensal bacteria.19 One laboratory observed an increased number of ILC2s in the small intestine of germ-free mice,68 while a second report suggested there was no difference.62 Furthermore, in this latter study, germ-free mice had the same number of resident ILC2s in lung and other tissues as specific pathogen-free mice, suggesting that the number of ILC2s was independent of the microbiota outside of the intestine.62
ILC3s
The interaction of ILC3s with the microbiome has been explored most extensively in the intestine, where these cells are most numerous. The microbiome drives increased myeloid cell IL-23 and IL-1β at steady state, which played a prominent role in ILC3 activation.69,70 Epithelial cells were also shown to be capable of producing IL-1β.71 In response to IL-23 and IL-1β, ILC3s produced a variety of cytokines including IL-17, IL-22, IFNγ, and GM-CSF.72 Notably, there was a clear role for the microbiome in the induction of ILC3 cytokine production, with reduced cytokines by ILC3s from germ-free mice.24,73,74
The CCR6+ subset of ILC3s was a significant pool of cells capable of secreting IL-17 and IL-22 in the intestine of mice,75 but they also exhibited additional functions that could affect the microbiome. CCR6+ ILC3s highly expressed MHCII and exhibited antigen-presenting function.76 As a result, they influenced commensal bacteria-specific CD4+ T-cell responses through direct presentation of microbiota-derived antigens. CCR6+ ILC3s also expressed CD1d, allowing for antigen presentation to iNKT cells.77 ILC3 expression of MHC class II, and likely their ability to carry out antigen presentation, negatively regulated the interaction of T follicular helper (TFH) cells and B cells, which led to reduced mucosal IgA synthesis in the colon with corresponding effects on the microbiome.78 In Peyer’s patches and small intestine lamina propria, by contrast, both T-cell-dependent and T-cell-independent IgA at steady state were dependent on RORγt+ ILCs, via ILC expression of the TNF superfamily member lymphotoxin.78,79,80 In addition to IL-17 and IL-22, CCR6− ILC3s also could acquire expression of T-bet, which induced IFNγ and tumor necrosis factor (TNF) production.81 This T-bet expression occurred in response to IL-12 and IL-15 production in response to the microbiome.29
The function of ILC3s in the intestine was influenced by aryl hydrocarbon receptor signaling (AhR). AhR serves as a receptor for multiple ligands derived from endogenous, dietary or microbial sources. AhR stimulation in ILC3s improved cell survival and increased IL-22 production.82,83,84 IL-22 acted on receptors expressed by intestinal epithelial cells (IECs) to promote intestinal stem cell regeneration, production of mucus and antimicrobial peptides, enhanced tight junction formation and fucosylation of proteins and lipids.85,86,87,88,89 Notably, the antimicrobial peptides produced due to IL-22 stimulation prevented bacteria from associating with the mucus layer.90 IL-22 receptor signals promoted the addition of terminal fucose sugars to proteins and glycolipids on the surface of epithelial cells. These terminal fucose sugars have been shown to influence the composition of the commensal microbiota.86,91 Therefore, while the microbiota induced homeostatic production of IL-22 by ILC3s, these lymphocytes in turn influenced the microbiota, in part by secreting IL-22. This cross-talk is exemplified by the increased colonization by a Clostridiaceae family microbe, segmented filamentous bacteria (SFB), in mice deficient for ILC3s.92 SFB promoted homeostatic Th17 cell accumulation, which is noninflammatory, in the lamina propria of the ileum.93,94 Of note, AhR-deficient mice displayed impaired IL-22 production and increased SFB colonization, suggesting that AhR stimulation may be an important regulator of IL-22 function.95 Prostaglandin E2 also promoted ILC3 production of IL-22 at steady state.96 Blockade of prostaglandin E2 led to systemic infection and inflammation by commensal microbes, due to reduced ILC3 production of IL-22.96 ILC3s also were shown to express the rearranged during transfection (RET) tyrosine kinase receptor, which allowed them to respond to glial-derived neurotrophic factor (GDNF) family ligands to drive IL-22 synthesis.97 ILC3 production of GM-CSF and IL-2 also contributed to intestinal homeostasis.24,98 GM-CSF produced by ILC3s induced production of IL-10 and RA by intestinal macrophages and DCs, which promoted regulatory T-cell (Treg) accumulation in the large intestine.24,99,100 IL-2, also produced by ILC3s, was induced by production of IL-1β by intestinal macrophages. This IL-2 was important for maintaining Tregs in the small intestine.98 Tregs have been found to prevent an aberrant immune response to the normal microbiome; in their absence, severe colitis resulted.101 In conclusion, ILC3s can integrate several types of signals in order to influence the microbiome and the response of the immune system to commensal bacteria.
The effector functions of ILCs during infections
Once pathogens enter the body at mucosal sites, ILCs participate in the early defense response to clear pathogens in order to avoid systemic infections. ILC effector functions are primarily mediated through cytokine secretion during infection. NK cells have long been known to be potent first-line effector cells in helping control pathogen infections.102,103 In this section, we will include NK cells but we will emphasize the roles of ILC1, ILC2 and ILC3 in reviewing knowledge about ILC effector functions against four types of pathogenic microbes, including various types of intracellular pathogens, extracellular bacteria, helminths and fungi. Of note, in a number of publications, the essential function of ILCs has been demonstrated using immunocompromised mouse models of infection. Therefore, the susceptibility to these infections could be due to defects in the mucosal and/or systemic components of the immune system. Analyses of mucosal tissues, however, combined with studies of the early phases of the response, can help to identify an important ILC function in mucosal tissues, without necessarily excluding systemic effects. The overall findings on microbial infections discussed in this section have been summarized in Fig. 2 and Table 1.

ieILC1 intraepithelial ILC1.
Intracellular pathogens
Obligate intracellular pathogens are entirely dependent on host cells to supply them with energy sources. Obligate intracellular pathogens include all viruses, certain protozoa, such as Toxoplasma gondii, and some bacteria. By contrast, facultative intracellular bacteria prefer the intracellular environment of host cells for growth but are capable of surviving outside of host cells. For protection against intracellular pathogens at acute infection, IFNγ is an important cytokine (Fig. 3a). IFNγ aids in clearing infected cells through the activation of macrophages and other cell types. Therefore, IFNγ-producing ILC subsets, such as NK cells, ILC1s, and some ILC3s, can be important for early host defense against this type of pathogens.

For simplicity the role of cytokines in activating ILCs is emphasized. a Following infection by intracellular pathogens, such as viruses, protozoa, and facultative intracellular bacteria, myeloid-derived IL-12 activates NK cells, ILC1s and CCR6− ILC3s (NKp46+ ILC3s and NKp46− ILC3s) to stimulate the production of IFNγ. ILC-derived IFNγ recruits and activates phagocytes to clear pathogens. b Following helminth infection, increases in tuft cell-derived IL-25 and IEC-derived IL-33 drive IL-5 and IL-13 production by ILC2s. ILC2-derived IL-5 recruits and activates eosinophils, and IL-13 induces the production of mucus by goblet cells for clearance of helminths. c Upon infection by extracellular pathogens such as protozoa and bacteria, DC-derived IL-23 and IL-1β activate ILC3s to produce IL-22 and IL-17. ILC3-derived IL-22 and IL-17 induce the production of antimicrobial peptides for host defense. d Upon infection by fungi, DC-derived IL-23 activates ILC3s to produce IL-22 and IL-17. ILC3-derived IL-22 and IL-17 induce the production of antimicrobial peptides. iILC2s, which express intermediate amounts of RORγt, also can produce IL-17 to protect against fungal infection.
Viral infections
Enteric viruses that target IECs, such as rotavirus and norovirus, are a leading cause of gastroenteritis worldwide. Rotavirus has a preferential tropism for the villous epithelium of the small intestine in humans and mice and is a leading cause of childhood gastroenteritis.104,105 IL-22-producing ILC3s were shown to act as an amplifier of IFNλ production by IECs during rotavirus infection. IL-22R signals, acting together with signals to the IFNλ receptor, induced optimal STAT1 activation.106 Murine norovirus (MNV) is typically asymptomatic, but it induced intestinal inflammation in genetically susceptible mice.107,108,109 In fact, MNV promoted healthy intestinal function and reversed intestinal abnormalities in germ-free mice.68 Interestingly, MNV improved the outcome following intestinal injury through induction of IL-22 by ILC3s in an IFNα R1- and CCR2-dependent manner.68,110 Human immunodeficiency virus (HIV), and its primate counterpart the simian immunodeficiency virus (SIV), target the gastrointestinal tract as a major site of viral transmission and replication.111,112,113 These lentiviral infections break down the integrity of the gastrointestinal mucosa and lead to alteration of gut microbiome and associated disease progression.114,115,116 HIV-1/SIV infection induced the rapid loss of ILC3s in the intestinal mucosa.117,118 Transcriptional profiling during acute infection revealed increased expression of genes linked with a strong IFN acute-phase response and evidence of gut barrier breakdown.119 These studies suggest that IL-22 from ILC3s has a protective role against some enteric viral infections in the intestine, either through inhibition of viral replication in IECs or possibly by induction of epithelial cell proliferation and epithelial regeneration after damage.
Viruses that target the respiratory tract, such as influenza virus, respiratory syncytial virus (RSV), vaccinia virus (VACV), reovirus, and adenovirus, cause common colds, bronchiolitis and pneumonia. Influenza viruses typically infect the host through oral or nasal uptake of aerosolized virus particles leading to mild to severe pneumonia.120 The role of NK cells during viral infection is usually protective.121 After infection through interactions with DCs and macrophages, NK cells were recruited to the lung from the blood and became activated to secrete a variety of effector cytokines, including IFNγ.122,123,124 Influenza virus infection also enhanced ILC1 activation, similar to NK cells, and ILC1-derived IFNγ contributed to antiviral immunity,125 likely through the activation of macrophages. Another report suggested, however, that the function of IFNγ in the lung during infection may be harmful by limiting protective ILC2 activity.126 Influenza virus infection led to increased lung ILC2s126 and ILC2-derived AREG in the lung helped limit immunopathology by restoring lung function and barrier integrity and by impairing remodeling of respiratory tissues after influenza virus-induced damage.65 Interestingly, during influenza virus infection, ILC2s have also been reported to convert into ILC1s, providing another example of ILC subset plasticity.127 Furthermore, ILC-derived IL-22 has been suggested to contribute to host protection during influenza infection.128,129 Respiratory syncytial virus (RSV) causes infections of the lungs and respiratory tract and is the most common cause of bronchiolitis and pneumonia in infants.130 A recent study demonstrated that an elevated ILC2 number in nasal aspirates correlated with infant RSV-induced disease severity.131 CD4+ T cells contributed to ILC2 activation during RSV infection, partly via IL-2 production in the lungs.132 Lung ILC2s regulated RSV-induced CD4+ T-cell activation and expansion in turn, via OX40/OX40L interactions.133
Vaccinia virus (VACV) initially replicates in airway epithelial cells before spreading to secondary sites of infection, mainly the draining lymph nodes, spleen, gastrointestinal tract, and reproductive organs.134 Recovery from a respiratory VACV infection required a controlled inflammatory response by both innate and adaptive immune cells. IFNγ production by CD8+ T cells was demonstrated to control virus dissemination,134 and NK cells were the primary producers of early IFNγ production during VACV infection.135 It is uncertain if IFNγ-producing helper ILC subsets contributed to early host defense against these viral infections.
Protozoan infections
Obligate intracellular protozoans that infect the small intestine epithelium are among the most common infections worldwide.136 The innate immune system of immunocompetent hosts typically clears intestinal protozoans during acute infection. However, in immunocompromised individuals, these pathogens can cause chronic, potentially life-threatening, infections. The role of NK cells and NK cell-derived IFNγ for control of T. gondii, Cryptosporidium and other protozoan infections has been well-described.103,137,138 Less is known, however, about the function of other ILC subsets during parasitic infections, and mechanistic studies have been mostly limited to T. gondii. T. gondii is a widespread protozoan that is capable of infecting all warm‐blooded vertebrates.139 Accordingly, T. gondii causes toxoplasmosis in humans, one of the most common infections worldwide.136 T. gondii infection is caused by several means including ingestion of undercooked, contaminated meat or water, through contact with cat feces that contain T. gondii, by transfusion of infected blood, by infected organ transplantation or by transmission from mother-to-fetus through the placenta.140 Following oral infection by T. gondii, the organism infects the gut mucosa by direct invasion of small intestine epithelial cells.141 T. gondii crosses the intestinal barrier and expands locally in the small intestine, particularly in the ileum, within hours.142 It can disseminate systemically to sites such as the mLN, liver and spleen within days and finally reaches the central nervous system.143,144 IFNγ has been shown to have a critical function for survival during T. gondii infection through the elimination of the parasite.145 During acute infection, TLR signaling in DCs led to IL-12 production that controlled the infection through initiating IFNγ production.146 Following exposure to IL-12, IFNγ was mainly produced by T cells, NK cells and ILC1s. For production of IFNγ by NK cells, IL-12 and additional cytokines, such as IL-1β and IL-18, were required during T. gondii infection.147,148,149 It has been shown, however, that parasite-infected NK cells were defective for host defense.150 Furthermore, it is uncertain if NK cell IFNγ is actually beneficial during infection, as it was reported that antibody-mediated NK cell depletion did not affect the survival of T. gondii-infected mice.151 Interestingly, ILC1s represent a primary IFNγ-secreting population during acute infection with T. gondii.17 ILCs may participate in immunopathology by other mechanisms besides excessive IFNγ production. For example, NK1+ cells activated by IL-15 secreted CCL3, which enhanced intestinal recruitment of inflammatory monocytes during T. gondii infection.152 Ahr−/− mice, which have a defect in ILC3s, were susceptible to T. gondii, but exhibited increased T-cell activation following infection, suggesting that ILC3s contributed to host defense against T. gondii infection through limiting immunopathology mediated by T-cell activation.153
The protozoan Giardia lamblia is an extracellular pathogen found worldwide that causes giardiasis, a disease that is associated with gastrointestinal malfunction.154 This infection mostly occurs via oral uptake of G. lamblia cysts in contaminated drinking water. Following oral infection, G. lamblia colonizes the small intestine where the organism attaches to the intestinal epithelium, replicates vegetatively and disrupts intestinal barrier function. A recent study has shown that G. lamblia infection increased the number of ILC3s in the small intestinal lamina propria and augmented IL-17A production by them.155 It is still not known, however, if this ILC3-derived IL-17A is important for host defense.
Facultative intracellular bacterial infections
Pathogenic facultative intracellular bacteria that target intestinal tissues, such as Yersinia enterocolitica (Y. enterocolitica) and Salmonella typhimurium (S. typhimurium), are common causes of food-borne infectious gastroenteritis. These bacterial infections can lead to severe systemic infections, especially in immunocompromised individuals. It is well known that IFNγ plays a critical role in intestinal immunity against them.156,157,158 While CD4+ T cells are important after the second week of bacterial infection, Y. enterocolitica is frequently cleared in 1−2 weeks.159,160 Furthermore, athymic (nude) mice, which lack T cells, are resistant to infection with L. monocytogenes and S. typhimurium.161,162 These studies suggest that innate immune cells are sufficient to mediate early host defense against these types of bacteria.
The role of ILCs in intestinal bacterial infection has been well demonstrated with several intracellular bacteria. Y. enterocolitica is a gram-negative, rod-shaped, pathogenic bacterium that infects the small intestine, especially the ileum. After oral uptake, Y. enterocolitica replicates in the small intestine, invades Peyer’s patches of the distal ileum, and disseminates to the spleen and liver.163 Recently, it has been shown that ILC populations, especially CCR6− ILC3s, including those that do or do not express NKp46, were a major source of IFNγ production for host protection against Y. enterocolitica.164,165 ILC1s and NK cells also released this cytokine after Y. enterocolitica infection, and likely contributed to host defense as well. The herpes virus entry mediator (HVEM), a member of the TNF receptor superfamily (also known as TNFRSF14), was expressed by all small intestine ILC subsets in mice and all ILCs in human peripheral blood.164,166 Interestingly, mice with an ILC3-specific deletion of the gene encoding HVEM exhibited reduced IFNγ production, higher bacterial burdens and increased mortality after Y. enterocolitica infection. LIGHT, a member of the TNF superfamily, was shown to serve as the ligand that signaled HVEM for the induction of protective IFNγ secretion from ILC3s. Thus, HVEM signaling mediated by LIGHT was shown to play a critical role in regulating ILC3-derived IFNγ production for protection following Y. enterocolitica infection.164 HVEM signaling in cells other than lymphocytes also has a critical role for host defense against other bacterial infections, such as Citrobacter rodentium, Streptococcus pneumoniae and Clostridium difficile.167,168
Salmonella typhimurium is a gram-negative, rod-shaped, pathogenic bacterium that can attach to IECs and attack them by producing toxins. Once S. typhimurium invades the gut mucosa, it can cause systemic infections. IFNγ plays a critical role in protection against S. typhimurium. IFNγ-controlled mucin release by goblet cells restricted pathogen growth in intestinal tissue by formation of a mucin gel.169 During salmonellosis, NKp46+ ILC3s were the main source of protective IFNγ production.81,170 It has been shown that Tbx21−/− mice, which lack several IFNγ-producing cell types, including NKp46+ ILC3s, had a significant defect in the ability to secrete mucus.81 IL-22 and lymphotoxin production by ILC3s contributed to host defense from S. typhimurium infection through induction by IECs of the fucosylation of their glycoproteins and glycolipids, which was mediated by the enzyme fucosyltransferase 2 (Fut2).170 The fucose group was metabolized by commensal bacteria into beneficial metabolites, such as short-chain fatty acids, that boosted barrier immunity. This process also led to reduced expression of the S. typhimurium virulence gene.86
The role of ILCs in the infection of lung tissue with facultative intracellular bacteria also has been demonstrated. Mycobacterium tuberculosis (Mtb) mainly affects the lungs and is among the ten leading causes of mortality worldwide.171 The importance of IFNγ-producing T cells for protection against Mtb has been demonstrated172 and NK cell-derived IFNγ was protective in T-cell-deficient mice.173 During Mtb infection, ILCs, especially ILC3s, accumulated in the lungs of humans and mice, and mice with specific deletion of ILC3s exhibited increased susceptibility to Mtb infection. Increased susceptibility in this model was observed in Il17−/−Il22−/− double knockout mice, implicating a role for these ILC3-derived cytokines. Furthermore, Mtb infection increased expression of the C-X-C motif chemokine receptor 5 (CXCR5) on circulating ILC3s and also increased plasma levels of its ligand, C-X-C motif chemokine ligand 13 (CXCL13). ILCs therefore located in lymphoid follicles and granuloma-like structures after Mtb infection.174 These data suggest a protective role for ILC3s in regulating early Mtb infection through signaling by CXCR5 and secretion of cytokines. The mechanism underlying the protective effects of IL-17 and IL-22 in this infection remains unknown. Previously, it has been proposed that IL-17-producing CD4+ T cells promoted chemokine expression in the lung and the recruitment of IFNγ-producing CD4+ T cells, which ultimately impaired bacterial growth.175 IL-17 and IL-22 derived from ILC3s could have a similar function.
In several other infections of mice with facultative intracellular bacteria, ILCs have been shown to be activated early to secrete cytokines such as IFNγ, including infections with Campylobacter jejuni and Shigella flexneri.176,177 While these data suggest that ILC responses could be essential for host defense against these microbes, this remains to be demonstrated.
Helminth infections
Helminths are invertebrates with elongated, flat or round bodies and are comprised of three taxonomic groups: roundworms (nematodes), tapeworms (cestodes), and flukes (trematodes). They can live as parasites in animals and plants, or as free-living organisms in aquatic and terrestrial environments. Infection by helminths is generally prevalent in developing countries and can cause morbidity and mortality in immunocompromised or malnourished individuals. To study helminth infections, Nippostrongylus brasiliensis (N. brasiliensis), which is a gastrointestinal roundworm that infects rodents, is widely used. Third-stage larvae (L3) of N. brasiliensis infect mice through the skin and migrate to the lungs, where they are coughed up, swallowed and reach the small intestine. Intestinal worms develop into mature adults that produce eggs. They are expelled in C57BL/6 mice by days 10−15 post infection (p.i.).178 Production of the Th2 cytokines IL-13 and IL-5 was shown to be required for worm expulsion, through the induction of goblet cell hyperplasia, which is important for mucin secretion, and activation and recruitment of eosinophils to the sites of infection (Fig. 3b).179 Interestingly, Rag1−/− mice were protected from N. brasiliensis infection and had similar levels of IL-13 and IL-5 compared to C57BL/6 mice.180 Furthermore, adoptive transfer of IL-13+ ILC2s rescued impaired worm expulsion, suggesting that ILC2 function is sufficient for worm clearance.180,181
Infection with a different helminth, Heligmosomoides polygyrus (H. polygyrus), was controlled by IL-4-dependent immunity. H. polygyrus is a naturally occurring intestinal roundworm of rodents. Following H. polygyrus infection, ILC2s produced significant amounts of IL-4, and this contributed to subsequent ILC2 expansion and Th2 cell differentiation.182 ILC2s also expanded in vivo in response to the type-2-inducing cytokines IL-25 and IL-33.61,180,181,183 IL-25 was constitutively produced by tuft cells, and tuft cell numbers were markedly increased in the small intestine following helminth infections with N. brasiliensis or H. polygyrus.63,67,184 After infection, tuft cell-derived IL-25 stimulated ILC2s to secrete increased IL-13, which acted on epithelial stem cells to promote differentiation of tuft and goblet cells. In this way, a tuft cell-ILC2-epithelial response positive feedback circuit is generated that can drive small intestinal remodeling during parasite infection.67 ILC2s activated by IL-25 also promoted antigen-specific Th2 and Th9 function that contributed to the control of infection with the roundworm Trichinella spiralis. This infection is caused by consuming undercooked or raw meat.185 Lack of IL-22, in contrast, impaired worm expulsion upon N. brasiliensis infection and infection with a different helminth, the roundworm Trichuris muris, despite normal levels of type 2 cytokine production. This was due to reduced goblet cell hyperplasia, suggesting that ILC3-derived IL-22 also contributed to host defense against helminths.186
ILC2 function is negatively and positively regulated during infections by several molecules, including transcription factors and cytokines known to regulate cytokine production by other immune cell types. For example, the transcription factor AhR, important for IL-22 production, is highly expressed by intestinal ILC2s. AhR expression by ILC2s, however, suppressed the normal cellular functions of these cells through inhibition of expression of the IL-33 receptor ST2, IL-5, IL-13 and AREG, in a cell-intrinsic manner.187 AhR deficiency therefore enhanced ILC2 function and led to enhanced protective immunity against intestinal H. polygyrus infection.187 Loss of the Th1 and ILC1 transcription factor T-bet also led to expansion and increased activity of ILC2s and enhanced protection from the roundworm Trichinella spiralis.188 By contrast, loss of ILC-intrinsic Arginase 1 (Arg1) prevented ILC2 responses after N. brasiliensis infection.189 It was shown that Arg1 is critical for regulating ILC2 responses through its effects on cellular metabolism, by controlling arginine catabolism, polyamine biosynthesis and aerobic glycolysis.189 ILC2s preferentially use fatty acids (FAs) to maintain their function during helminth infection and it has been proposed that enhanced FA usage and FA-dependent IL-13 production by ILC2s could represent a host adaptation to maintain barrier immunity under dietary restriction.190
In the lung, infection with the roundworm Strongyloides venezuelensis (S. venezuelensis) increased the number of alveolar epithelial type II cells (ATII) and the level of IL-33. ATII-derived IL-33 induced accumulation of ILC2s, which produced IL-5 and IL-13, leading to lung eosinophilic inflammation and worm expulsion.191 Interestingly, S. venezuelensis-exposed mice were significantly more resistant to infection by N. brasiliensis and this resistance also was dependent on ILC2s, suggesting that mice acquired a type of immune memory or trained immunity by ILC2s following S. venezuelensis infection.192 Infection with the fluke Schistosoma mansoni (S. mansoni) elicited the expression of TSLP and IL-33, which may have activated ILC2s,193 although ILC2 activation was not tested. Using a mouse model of coinfection with Mtb and S. mansoni, however, IL-25 and ILC2s induced pulmonary fibrosis that occurred independently of T-cell-mediated antigen-specific responses.194 In summary, ILCs have been shown to provide protection from infection of mice with a variety of helminths, in most cases by eliciting and stimulating type 2 immunity.
Extracellular bacterial infections
Extracellular bacterial pathogens may adhere to epithelial cell surfaces and some secrete potent toxins leading to infectious diseases. One of the best studied examples of such a pathogen in the intestine is Citrobacter rodentium (C. rodentium), a gram-negative, enteric murine pathogen that shares an infection strategy and virulence factors with the human diarrheagenic pathogens enteropathogenic E. coli and enterohemorrhagic E. coli (EPEC and EHEC). C. rodentium mainly colonizes the cecum and proximal colon and causes acute, robust colitis, colonic crypt hyperplasia and dysbiosis.195 C. rodentium were able to colonize the colon by days 7−14 p.i., but bacterial levels in the cecum and colon virtually were cleared by days 21−28 p.i. in wild-type mice.196 While T cells and B cells were required for clearance of C. rodentium, ILCs were essential for early host defense. Mice deficient for the bZip transcription factor Nfil3, which have a loss of NK cells, ILC1s, and ILC3s, were highly susceptible to C. rodentium infection.197 The ILC3 subset of ILCs was essential in the absence of T cells, but partially redundant when T cells were present.45 Considering ILC3 subsets, NKp46+ ILC3s were not required in fully immune competent mice, suggesting that LTi and/or NKp46− ILC3s provided protection.45
IL-22 was shown to be important for protection from C. rodentium198 and IL-22-deficient mice therefore exhibited increased intestinal epithelial damage, systemic bacterial burden and mortality (Fig. 3c).53,74,198,199,200 This cytokine played crucial roles in host defense against C. rodentium and other extracellular pathogens by maintaining the epithelial barrier through multiple functions, including induction of antimicrobial peptide expression, stimulation of epithelial cell proliferation, and modification of the microbiome. ILC3s, stimulated by IL-23, were the main source of IL-22 early, while CD4+ T cells were the major source of IL-22 later in C. rodentium infection.201,202 IL-22 was indispensable for the induction of antimicrobial agents by colonic epithelial cells, such as RegIIIγ and RegIIIβ, following C. rodentium infection. Exogenous RegIIIγ improved the survival of IL-22-deficient mice, suggesting that IL-22-mediated production of antimicrobial proteins was essential.52
Several factors have been shown to regulate the differentiation and localization of intestinal ILC3s with consequences for the host response to C. rodentium. IL-22-producing ILC3s were observed to be AhR-dependent; as a consequence, AhR was critical for the clearance of C. rodentium.42,82,187 Mice with epithelial-specific overexpression of Cyp1a, which depletes AhR ligands, also displayed loss of IL-22+ ILC3s and increased susceptibility to C. rodentium infection.203 The transcription factor Ikaros is a binding partner of AhR in ILC3s. Ikaros negatively regulated the number of ILC3s in a cell-intrinsic manner, through zinc finger-dependent inhibition of the transcriptional activity of AhR. Therefore, in mice deficient for Ikaros expression, ILC3s exhibited an enhanced protective activity for C. rodentium.204 Recently, GPR183, a G protein receptor that binds oxysterols, was shown to be required for ILC3-mediated protection against C. rodentium by influencing the localization of ILC3s in the lamina propria.205
Clostridium difficile (C. difficile) is a spore-forming, gram-positive, toxin-producing anaerobic bacterium that overgrows in hospitalized patients after prolonged use of antibiotics. It causes infectious diarrhea and pseudomembranous colitis.206 The pathogenicity of C. difficile is mediated by two clostridial toxins, toxin A (TcdA) and B (TcdB), which disrupt the cytoskeletal structure and the tight junctions of epithelial cells.207 Antibiotic-treated IL-22-deficient mice were susceptible to C. difficile infection,208 providing a model system for studying pathogenesis by these bacteria. ILCs were the predominant source of IL-22 and IFNγ following C. difficile infection.49 Loss of ILC1s in mice lacking T-bet (Tbx21−/− mice) that are also Rag protein deficient, or ILC1-derived IFNγ led to increased susceptibility to C. difficile.49 In the absence of IL-22, enterobacterial pathobionts translocated from the intestinal lumen into peripheral organs following C. difficile infection.208 Thus, these studies showed that ILC1-derived IFNγ and ILC3-derived IL-22 have a cooperative role for host defense against C. difficile in the intestine and peripheral organs.
Gram-positive Streptococcus pneumoniae (S. pneumoniae) and gram-negative Klebsiella pneumoniae (K. pneumoniae) are extracellular commensal bacteria in humans. However, they also can act as opportunistic pathogens that cause acute or chronic infections, especially in immunocompromised hosts. S. pneumoniae is the most common cause of community-acquired pneumonia. It is a commensal bacterium of the human upper respiratory tract, but under specific circumstances, it can migrate to the lower respiratory tract, causing pneumonia, otitis media, septicemia and meningitis.209,210 S. pneumoniae induced IL-17 and IL-22 production in the lung, especially by CCR6+ ILC3s.211 IL-22-deficient mice were susceptible to S. pneumoniae infection, which could be rescued by systemic administration of recombinant IL-22.212 Most lung ILC3s coexpressed RORγt and CCR6. The CCR6 ligand, CCL20, was strongly upregulated in the lung during infection, suggesting that recruitment of ILC3s to the lung might be due to increased CCL20.211 Flagellin, the agonist for Toll-like receptor 5 (TLR5), enhanced the production of IL-17 and IL-22 by ILC3s via activation of DCs, thereby contributing to protection from S. pneumoniae.211,213 In contrast, ILC2-derived IL-13 was detrimental in S. pneumoniae infection due to the persistence of M2 type alveolar macrophages, which were less efficient in triggering inflammatory responses compared to M1 macrophages.53 It has been shown that IL-22 production by NKp46+ ILCs was increased during K. pneumoniae infection and that IL-22 promoted host defense.214 The importance of IL-22 in this infection is controversial, however, as another study reported that IL-17A produced by ILC3s contributed to K. pneumoniae through enhancement of the antimicrobial activity of inflammatory monocytes by IL-17A.215 IFNγ production by NK cells also contributed to protection from K. pneumoniae by promoting macrophage activation.178
Fungal infections
Many kinds of fungi colonize the human body, but they usually do not cause infection in healthy individuals. However, fungi can become opportunistic pathogens and cause various fungal diseases in immunocompromised individuals, ranging from mild mucosal infections to severe systemic infections. Examples include Candida albicans (C. albicans), which can be found in the oral cavity, intestine and skin. Similarly, Cryptococcus neoformans (C. neoformans) and Aspergillus fumigatus (A. fumigatus) can be found in the lung. These three fungi are often present without causing disease, but they cause many cases of opportunistic mycoses that are associated with a high rate of mortality.216
The mucosal epithelium is the first line of defense against Candida species. Upon recognition of the invading Candida species, epithelial cells secreted antimicrobial peptides, such as β-defensins, to inhibit fungal growth.217,218 It is well known that IL-17 is required for protective antifungal immunity at mucosal surfaces.219 IL-17 induced expression of antimicrobial peptides, which have direct antifungal activity toward Candida.220 IL-22 can act with IL-17 to enhance the expression of these antimicrobial peptides.221 IL-17 also was shown to have a critical role in the prevention of invasive fungal infections by activating neutrophils to kill fungi.222 Interestingly, athymic mice that lack T cells were resistant to C. albicans, indicating that innate immune cells have an essential function.161 Indeed, ILCs were the primary source of IL-17 during oropharyngeal C. albicans infection223 (Fig. 3d). IL-17A and IL-17F were produced upon C. albicans infection in an IL-23-dependent manner.223 ILC2s, which express intermediate amounts of RORγt, contributed to protection against C. albicans infection through production of IL-17.20
In the lungs of neutropenic mice with A. fumigatus infection, NK cells were the major population of cells capable of generating IFNγ and NK cell-derived IFNγ also was essential to host defense against A. fumigatus.224,225 During infection with C. neoformans, IL-33/ST2 signaling contributed to the expansion of ILC2s and their production of IL-13 that likely facilitated C. neoformans growth and dissemination, although type 2 immune responses from CD4+ T cells also played a role in this example of infection-induced immunopathology. However, ILC2s were not able to induce type 2 inflammation in pulmonary cryptococcosis in the absence of adaptive lymphocytes.226,227
Conclusions
ILCs are the most recently discovered lymphocyte population, but already numerous studies have demonstrated that they can act as sentinels of the mucosal immune system, which contribute to an appropriate immune response to the microbiome while providing active protection following acute infection. For example, at steady state, ILC2s and ILC3s respond to diverse cues and produce cytokines to maintain and integrity of the mucosal tissues. Infection-augmented IL-4, IL-5 and IL-13 production by ILC2s plays a critical role in the clearance of helminths. Also, ILC3 production of IL-17, IL-22, and other cytokines such as IFNγ directly contribute to host defense against extracellular bacterial and fungal infections. Finally, during viral, bacterial and protozoal infections, NK cells and ILC1s become activated and serve as early producers of IFNγ to drive pathogen clearance. It is likely that the full range of signals inducing tissue-resident ILC activation, the types of responses that can be elicited by ILCs, and the full extent of the plasticity between ILC subsets remain to be fully characterized. NK cells have been studied much longer, and it is known that primed NK cells could be recruited into a secondary immune response following viral infection.228 This phenomenon, sometimes referred to as “innate memory”, suggests that ILCs might exhibit similar changes after stimulation. In fact, ILC2s stimulated with IL-33 during an allergy response acquired a memory-like phenotype because, compared to naïve ILC2s, they produced even higher levels of cytokines after a subsequent IL-33 stimulation.229 Additional research will be required to characterize the long-term responses of ILCs to acute activation signals and to identify the mucosal infections in humans in which the responses by these cells are most significant for host protection.
References
Cianci, R., Pagliari, D., Piccirillo, C. A., Fritz, J. H. & Gambassi, G. The Microbiota and immune system crosstalk in health and disease. Mediators Inflamm. 2018, 2912539 (2018).
Sun, X. & Jia, Z. Microbiome modulates intestinal homeostasis against inflammatory diseases. Vet. Immunol. Immunopathol. 205, 97–105 (2018).
Britanova, L. & Diefenbach, A. Interplay of innate lymphoid cells and the microbiota. Immunol. Rev. 279, 36–51 (2017).
Vivier, E., van de Pavert, S. A., Cooper, M. D. & Belz, G. T. The evolution of innate lymphoid cells. Nat. Immunol. 17, 790–794 (2016).
Yu, S., Kim, H. Y., Chang, Y. J., DeKruyff, R. H. & Umetsu, D. T. Innate lymphoid cells and asthma. J. Allergy Clin. Immunol. 133, 943–950 (2014).
Flores-Borja, F. et al. Crosstalk between innate lymphoid cells and other immune cells in the tumor microenvironment. J. Immunol. Res. 2016, 7803091 (2016).
Panda, S. K. & Colonna, M. Innate lymphoid cells: a potential link between microbiota and immune responses against cancer. Semin. Immunol. 41, 101271 (2019).
Peters, C. P., Mjosberg, J. M., Bernink, J. H. & Spits, H. Innate lymphoid cells in inflammatory bowel diseases. Immunol. Lett. 172, 124–131 (2016).
Colonna, M. Innate lymphoid cells: diversity, plasticity, and unique functions in immunity. Immunity 48, 1104–1117 (2018).
Vivier, E. et al. Innate lymphoid cells: 10 years on. Cell 174, 1054–1066 (2018).
Kotas, M. E. & Locksley, R. M. Why innate lymphoid cells? Immunity 48, 1081–1090 (2018).
Artis, D. & Spits, H. The biology of innate lymphoid cells. Nature 517, 293–301 (2015).
Sonnenberg, G. F. & Hepworth, M. R. Functional interactions between innate lymphoid cells and adaptive immunity. Nat. Rev. Immunol. 19, 599–613 (2019).
Maggi, L. et al. Human circulating group 2 innate lymphoid cells can express CD154 and promote IgE production. J. Allergy Clin. Immunol. 139, 964–976 e964 (2017).
Crellin, N. K. et al. Regulation of cytokine secretion in human CD127(+) LTi-like innate lymphoid cells by Toll-like receptor 2. Immunity 33, 752–764 (2010).
Kim, M. Y. et al. OX40 ligand and CD30 ligand are expressed on adult but not neonatal CD4+CD3- inducer cells: evidence that IL-7 signals regulate CD30 ligand but not OX40 ligand expression. J. Immunol. 174, 6686–6691 (2005).
Klose, C. S. N. et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 157, 340–356 (2014).
Rauber, S. et al. Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat. Med. 23, 938–944 (2017).
Gury-BenAri, M. et al. The spectrum and regulatory landscape of intestinal innate lymphoid cells are shaped by the microbiome. Cell 166, 1231–1246 e1213 (2016).
Huang, Y. et al. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat. Immunol. 16, 161–169 (2015).
Huang, Y. et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science 359, 114–119 (2018).
Tsuji, M. et al. Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity 29, 261–271 (2008).
van de Pavert, S. A. et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature 508, 123–127 (2014).
Mortha, A. et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 343, 1249288 (2014).
Larange, A. & Cheroutre, H. Retinoic acid and retinoic acid receptors as pleiotropic modulators of the immune system. Annu. Rev. Immunol. 34, 369–394 (2016).
Seo, G. Y. et al. Retinoic acid, acting as a highly specific IgA isotype switch factor, cooperates with TGF-beta1 to enhance the overall IgA response. J. Leukoc. Biol. 94, 325–335 (2013).
Seo, G. Y. et al. Retinoic acid acts as a selective human IgA switch factor. Hum. Immunol. 75, 923–929 (2014).
Kim, M. H., Taparowsky, E. J. & Kim, C. H. Retinoic acid differentially regulates the migration of innate lymphoid cell subsets to the gut. Immunity 43, 107–119 (2015).
Bernink, J. H. et al. Interleukin-12 and -23 control plasticity of CD127(+) group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity 43, 146–160 (2015).
Bernink, J. H. et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 14, 221–229 (2013).
Vonarbourg, C. et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity 33, 736–751 (2010).
Argentati, K., Bartozzi, B., Bernardini, G., Di Stasio, G. & Provinciali, M. Induction of natural killer cell activity and perforin and granzyme B gene expression following continuous culture of short pulse with interleukin-12 in young and old mice. Eur. Cytokine Netw. 11, 59–66 (2000).
Simoni, Y. et al. Human innate lymphoid cell subsets possess tissue-type based heterogeneity in phenotype and frequency. Immunity 46, 148–161 (2017).
Weizman, O. E. et al. ILC1 confer early host protection at initial sites of viral infection. Cell 171, 795–808 e712 (2017).
Yagi, R. et al. The transcription factor GATA3 is critical for the development of all IL-7Ralpha-expressing innate lymphoid cells. Immunity 40, 378–388 (2014).
Gordon, S. M. et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity 36, 55–67 (2012).
Harly, C., Cam, M., Kaye, J. & Bhandoola, A. Development and differentiation of early innate lymphoid progenitors. J. Exp. Med. 215, 249–262 (2018).
Constantinides, M. G., McDonald, B. D., Verhoef, P. A. & Bendelac, A. A committed precursor to innate lymphoid cells. Nature 508, 397–401 (2014).
Gasteiger, G., Fan, X., Dikiy, S., Lee, S. Y. & Rudensky, A. Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 350, 981–985 (2015).
Spencer, S. P. et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science 343, 432–437 (2014).
Fuchs, A. et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity 38, 769–781 (2013).
Kim, C. H., Hashimoto-Hill, S. & Kim, M. Migration and tissue tropism of innate lymphoid cells. Trends Immunol. 37, 68–79 (2016).
Luci, C. et al. Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat. Immunol. 10, 75–82 (2009).
Oliphant, C. J. et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity 41, 283–295 (2014).
Song, C. et al. Unique and redundant functions of NKp46+ ILC3s in models of intestinal inflammation. J. Exp. Med. 212, 1869–1882 (2015).
Vely, F. et al. Evidence of innate lymphoid cell redundancy in humans. Nat. Immunol. 17, 1291–1299 (2016).
Michelet, X. et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol. 19, 1330–1340 (2018).
Matsuda, J. L. et al. Homeostasis of V alpha 14i NKT cells. Nat. Immunol. 3, 966–974 (2002).
Abt, M. C. et al. Innate immune defenses mediated by two ILC subsets are critical for protection against acute Clostridium difficile infection. Cell Host Microbe 18, 27–37 (2015).
Powell, N. et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity 37, 674–684 (2012).
Van Dyken, S. J. et al. A tissue checkpoint regulates type 2 immunity. Nat. Immunol. 17, 1381–1387 (2016).
Halim, T. Y. et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40, 425–435 (2014).
Saluzzo, S. et al. First-breath-induced type 2 pathways shape the lung immune environment. Cell Rep. 18, 1893–1905 (2017).
de Kleer, I. M. et al. Perinatal activation of the interleukin-33 pathway promotes type 2 immunity in the developing lung. Immunity 45, 1285–1298 (2016).
Gundra, U. M. et al. Alternatively activated macrophages derived from monocytes and tissue macrophages are phenotypically and functionally distinct. Blood 123, e110–e122 (2014).
Kondo, M., Tamaoki, J., Takeyama, K., Nakata, J. & Nagai, A. Interleukin-13 induces goblet cell differentiation in primary cell culture from Guinea pig tracheal epithelium. Am. J. Respir. Cell Mol. Biol. 27, 536–541 (2002).
Tukler Henriksson, J., Coursey, T. G., Corry, D. B., De Paiva, C. S. & Pflugfelder, S. C. IL-13 stimulates proliferation and expression of mucin and immunomodulatory genes in cultured conjunctival goblet cells. Invest. Ophthalmol. Vis. Sci. 56, 4186–4197 (2015).
Ramalingam, T. R. et al. Unique functions of the type II interleukin 4 receptor identified in mice lacking the interleukin 13 receptor alpha1 chain. Nat. Immunol. 9, 25–33 (2008).
Monticelli, L. A. et al. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc. Natl. Acad. Sci. USA 112, 10762–10767 (2015).
Roediger, B. et al. IL-2 is a critical regulator of group 2 innate lymphoid cell function during pulmonary inflammation. J. Allergy Clin. Immunol. 136, 1653–1663 e1657 (2015).
Neill, D. R. et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370 (2010).
Ricardo-Gonzalez, R. R. et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat. Immunol. 19, 1093–1099 (2018).
von Moltke, J., Ji, M., Liang, H. E. & Locksley, R. M. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 529, 221–225 (2016).
Divekar, R. & Kita, H. Recent advances in epithelium-derived cytokines (IL-33, IL-25, and thymic stromal lymphopoietin) and allergic inflammation. Curr. Opin. Allergy Clin. Immunol. 15, 98–103 (2015).
Monticelli, L. A. et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 12, 1045–1054 (2011).
Nadjsombati, M. S. et al. Detection of succinate by intestinal tuft cells triggers a type 2 innate immune circuit. Immunity 49, 33–41 e37 (2018).
Schneider, C. et al. A metabolite-triggered tuft cell-ILC2 circuit drives small intestinal remodeling. Cell 174, 271–284 e214 (2018).
Kernbauer, E., Ding, Y. & Cadwell, K. An enteric virus can replace the beneficial function of commensal bacteria. Nature 516, 94–98 (2014).
Shaw, M. H., Kamada, N., Kim, Y. G. & Nunez, G. Microbiota-induced IL-1beta, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J. Exp. Med. 209, 251–258 (2012).
Buonocore, S. et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464, 1371–1375 (2010).
Peeters, P. M., Wouters, E. F. & Reynaert, N. L. Immune homeostasis in epithelial cells: evidence and role of inflammasome signaling reviewed. J. Immunol. Res. 2015, 828264 (2015).
Melo-Gonzalez, F. & Hepworth, M. R. Functional and phenotypic heterogeneity of group 3 innate lymphoid cells. Immunology 150, 265–275 (2017).
Sanos, S. L. et al. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat. Immunol. 10, 83–91 (2009).
Satoh-Takayama, N. et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 29, 958–970 (2008).
Longman, R. S. et al. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J. Exp. Med. 211, 1571–1583 (2014).
Hepworth, M. R. et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4(+) T cells. Science 348, 1031–1035 (2015).
Saez de Guinoa, J. et al. CD1d-mediated activation of group 3 innate lymphoid cells drives IL-22 production. EMBO Rep. 18, 39–47 (2017).
Melo-Gonzalez, F. et al. Antigen-presenting ILC3 regulate T cell-dependent IgA responses to colonic mucosal bacteria. J. Exp. Med. 216, 728–742 (2019).
Kruglov, A. A. et al. Nonredundant function of soluble LTalpha3 produced by innate lymphoid cells in intestinal homeostasis. Science 342, 1243–1246 (2013).
Reboldi, A. et al. IgA production requires B cell interaction with subepithelial dendritic cells in Peyer’s patches. Science 352, aaf4822 (2016).
Klose, C. S. et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature 494, 261–265 (2013).
Kiss, E. A. et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 334, 1561–1565 (2011).
Qiu, J. et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 36, 92–104 (2012).
Lee, J. S. et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat. Immunol. 13, 144–151 (2011).
Gronke, K. et al. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature 566, 249–253 (2019).
Pickard, J. M. et al. Rapid fucosylation of intestinal epithelium sustains host-commensal symbiosis in sickness. Nature 514, 638–641 (2014).
Wang, Y., Mumm, J. B., Herbst, R., Kolbeck, R. & Wang, Y. IL-22 increases permeability of intestinal epithelial tight junctions by enhancing claudin-2 expression. J. Immunol. 199, 3316–3325 (2017).
Sugimoto, K. et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 118, 534–544 (2008).
Dixon, B. R., Radin, J. N., Piazuelo, M. B., Contreras, D. C. & Algood, H. M. IL-17a and IL-22 induce expression of antimicrobials in gastrointestinal epithelial cells and may contribute to epithelial cell defense against Helicobacter pylori. PLoS ONE 11, e0148514 (2016).
Vaishnava, S. et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 334, 255–258 (2011).
Pickard, J. M. & Chervonsky, A. V. Intestinal fucose as a mediator of host-microbe symbiosis. J. Immunol. 194, 5588–5593 (2015).
Guo, X. et al. Innate lymphoid cells control early colonization resistance against intestinal pathogens through ID2-dependent regulation of the microbiota. Immunity 42, 731–743 (2015).
Ivanov, I. I. et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009).
Omenetti, S. et al. The intestine harbors functionally distinct homeostatic tissue-resident and inflammatory Th17 cells. Immunity 51, 77–89 e76 (2019).
Qiu, J. et al. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity 39, 386–399 (2013).
Duffin, R. et al. Prostaglandin E(2) constrains systemic inflammation through an innate lymphoid cell-IL-22 axis. Science 351, 1333–1338 (2016).
Ibiza, S. et al. Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature 535, 440–443 (2016).
Zhou, L. et al. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature 568, 405–409 (2019).
Hoshi, N. et al. MyD88 signalling in colonic mononuclear phagocytes drives colitis in IL-10-deficient mice. Nat. Commun. 3, 1120 (2012).
Vicente-Suarez, I. et al. Unique lamina propria stromal cells imprint the functional phenotype of mucosal dendritic cells. Mucosal Immunol. 8, 141–151 (2015).
Liu, H., Hu, B., Xu, D. & Liew, F. Y. CD4+CD25+ regulatory T cells cure murine colitis: the role of IL-10, TGF-beta, and CTLA4. J. Immunol. 171, 5012–5017 (2003).
Horowitz, A., Stegmann, K. A. & Riley, E. M. Activation of natural killer cells during microbial infections. Front. Immunol. 2, 88 (2011).
Ivanova, D. L. et al. Innate lymphoid cells in protection, pathology, and adaptive immunity during Apicomplexan infection. Front. Immunol. 10, 196 (2019).
Ward, R. L. Mechanisms of protection against rotavirus in humans and mice. J. Infect. Dis. 174(Suppl 1), S51–S58 (1996).
Bartlett, A. V. 3rd, Bednarz-Prashad, A. J., DuPont, H. L. & Pickering, L. K. Rotavirus gastroenteritis. Annu. Rev. Med. 38, 399–415 (1987).
Hernandez, P. P. et al. Interferon-lambda and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nat. Immunol. 16, 698–707 (2015).
Cadwell, K. et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 141, 1135–1145 (2010).
Basic, M. et al. Norovirus triggered microbiota-driven mucosal inflammation in interleukin 10-deficient mice. Inflamm. Bowel Dis. 20, 431–443 (2014).
Seamons, A. et al. Obstructive lymphangitis precedes colitis in murine norovirus-infected Stat1-deficient mice. Am. J. Pathol. 188, 1536–1554 (2018).
Neil, J. A. et al. IFN-I and IL-22 mediate protective effects of intestinal viral infection. Nat. Microbiol. 4, 1737–1749 (2019).
Heise, C., Vogel, P., Miller, C. J., Halsted, C. H. & Dandekar, S. Simian immunodeficiency virus infection of the gastrointestinal tract of rhesus macaques. Functional, pathological, and morphological changes. Am. J. Pathol. 142, 1759–1771 (1993).
Heise, C., Miller, C. J., Lackner, A. & Dandekar, S. Primary acute simian immunodeficiency virus infection of intestinal lymphoid tissue is associated with gastrointestinal dysfunction. J. Infect. Dis. 169, 1116–1120 (1994).
Veazey, R. S. et al. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science 280, 427–431 (1998).
Dandekar, S., George, M. D. & Baumler, A. J. Th17 cells, HIV and the gut mucosal barrier. Curr. Opin. HIV AIDS 5, 173–178 (2010).
Estes, J. D. et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. 6, e1001052 (2010).
Crakes, K. R. & Jiang, G. Gut microbiome alterations during HIV/SIV infection: implications for HIV cure. Front. Microbiol. 10, 1104 (2019).
Xu, H. et al. IL-17-producing innate lymphoid cells are restricted to mucosal tissues and are depleted in SIV-infected macaques. Mucosal Immunol. 5, 658–669 (2012).
Li, H. et al. Hypercytotoxicity and rapid loss of NKp44+ innate lymphoid cells during acute SIV infection. PLoS Pathog. 10, e1004551 (2014).
Kloverpris, H. N. et al. Innate lymphoid cells are depleted irreversibly during acute HIV-1 infection in the absence of viral suppression. Immunity 44, 391–405 (2016).
Taubenberger, J. K. & Morens, D. M. The pathology of influenza virus infections. Annu. Rev. Pathol. 3, 499–522 (2008).
Hammer, Q., Ruckert, T. & Romagnani, C. Natural killer cell specificity for viral infections. Nat. Immunol. 19, 800–808 (2018).
Pommerenke, C. et al. Global transcriptome analysis in influenza-infected mouse lungs reveals the kinetics of innate and adaptive host immune responses. PLoS ONE 7, e41169 (2012).
Ennis, F. A. et al. Interferon induction and increased natural killer-cell activity in influenza infections in man. Lancet 2, 891–893 (1981).
Weiss, I. D. et al. IFN-gamma treatment at early stages of influenza virus infection protects mice from death in a NK cell-dependent manner. J. Interferon Cytokine Res. 30, 439–449 (2010).
Vashist, N. et al. Influenza-activated ILC1s contribute to antiviral immunity partially influenced by differential GITR expression. Front. Immunol. 9, 505 (2018).
Califano, D. et al. IFN-gamma increases susceptibility to influenza A infection through suppression of group II innate lymphoid cells. Mucosal Immunol. 11, 209–219 (2018).
Silver, J. S. et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat. Immunol. 17, 626–635 (2016).
Guo, H. & Topham, D. J. Interleukin-22 (IL-22) production by pulmonary Natural Killer cells and the potential role of IL-22 during primary influenza virus infection. J. Virol. 84, 7750–7759 (2010).
Kumar, P., Thakar, M. S., Ouyang, W. & Malarkannan, S. IL-22 from conventional NK cells is epithelial regenerative and inflammation protective during influenza infection. Mucosal Immunol. 6, 69–82 (2013).
Piedimonte, G. & Perez, M. K. Respiratory syncytial virus infection and bronchiolitis. Pediatr. Rev. 35, 519–530 (2014).
Vu, L. D. et al. Elevated levels of type 2 respiratory innate lymphoid cells in human infants with severe RSV bronchiolitis. Am. J. Respir. Crit. Care Med. 200, 1414–1423 (2019).
Han, X. et al. Essential role of CD4(+) T cells for the activation of group 2 innate lymphoid cells during respiratory syncytial virus infection in mice. Immunotherapy 11, 1303–1313 (2019).
Wu, J. et al. Critical role of OX40/OX40L in ILC2-mediated activation of CD4(+)T cells during respiratory syncytial virus infection in mice. Int. Immunopharmacol. 76, 105784 (2019).
Buller, R. M. & Palumbo, G. J. Poxvirus pathogenesis. Microbiol. Rev. 55, 80–122 (1991).
Abboud, G. et al. Natural killer cells and innate interferon gamma participate in the host defense against respiratory vaccinia virus infection. J. Virol. 90, 129–141 (2016).
Yarovinsky, F. Innate immunity to Toxoplasma gondii infection. Nat. Rev. Immunol. 14, 109–121 (2014).
Gigley, J. P. The diverse role of NK cells in immunity to Toxoplasma gondii Infection. PLoS Pathog. 12, e1005396 (2016).
Mead, J. R. & You, X. Susceptibility differences to Cryptosporidium parvum infection in two strains of gamma interferon knockout mice. J. Parasitol. 84, 1045–1048 (1998).
Tenter, A. M., Heckeroth, A. R. & Weiss, L. M. Toxoplasma gondii: from animals to humans. Int. J. Parasitol. 30, 1217–1258 (2000).
Hill, D. & Dubey, J. P. Toxoplasma gondii: transmission, diagnosis and prevention. Clin. Microbiol. Infect. 8, 634–640 (2002).
Dubey, J. P., Speer, C. A., Shen, S. K., Kwok, O. C. & Blixt, J. A. Oocyst-induced murine toxoplasmosis: life cycle, pathogenicity, and stage conversion in mice fed Toxoplasma gondii oocysts. J. Parasitol. 83, 870–882 (1997).
Dubey, J. P. Bradyzoite-induced murine toxoplasmosis: stage conversion, pathogenesis, and tissue cyst formation in mice fed bradyzoites of different strains of Toxoplasma gondii. J. Eukaryot. Microbiol. 44, 592–602 (1997).
Hand, T. W. et al. Acute gastrointestinal infection induces long-lived microbiota-specific T cell responses. Science 337, 1553–1556 (2012).
Konradt, C. et al. Endothelial cells are a replicative niche for entry of Toxoplasma gondii to the central nervous system. Nat. Microbiol. 1, 16001 (2016).
Sturge, C. R. & Yarovinsky, F. Complex immune cell interplay in the gamma interferon response during Toxoplasma gondii infection. Infect. Immun. 82, 3090–3097 (2014).
Mashayekhi, M. et al. CD8alpha(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity 35, 249–259 (2011).
Gazzinelli, R. T., Hieny, S., Wynn, T. A., Wolf, S. & Sher, A. Interleukin 12 is required for the T-lymphocyte-independent induction of interferon gamma by an intracellular parasite and induces resistance in T-cell-deficient hosts. Proc. Natl. Acad. Sci. USA 90, 6115–6119 (1993).
Cai, G., Kastelein, R. & Hunter, C. A. Interleukin-18 (IL-18) enhances innate IL-12-mediated resistance to Toxoplasma gondii. Infect. Immun. 68, 6932–6938 (2000).
Hunter, C. A., Chizzonite, R. & Remington, J. S. IL-1 beta is required for IL-12 to induce production of IFN-gamma by NK cells. A role for IL-1 beta in the T cell-independent mechanism of resistance against intracellular pathogens. J. Immunol. 155, 4347–4354 (1995).
Petit-Jentreau, L., Glover, C. & Coombes, J. L. Parasitized Natural Killer cells do not facilitate the spread of Toxoplasma gondii to the brain. Parasite Immunol. 40, e12522 (2018).
Hunter, C. A., Subauste, C. S., Van Cleave, V. H. & Remington, J. S. Production of gamma interferon by natural killer cells from Toxoplasma gondii-infected SCID mice: regulation by interleukin-10, interleukin-12, and tumor necrosis factor alpha. Infect. Immun. 62, 2818–2824 (1994).
Schulthess, J. et al. Interleukin-15-dependent NKp46+ innate lymphoid cells control intestinal inflammation by recruiting inflammatory monocytes. Immunity 37, 108–121 (2012).
Wagage, S. et al. The group 3 innate lymphoid cell defect in aryl hydrocarbon receptor deficient mice is associated with T cell hyperactivation during intestinal infection. PLoS ONE 10, e0128335 (2015).
Kraft, M. R., Klotz, C., Bucker, R., Schulzke, J. D. & Aebischer, T. Giardia’s epithelial cell interaction in vitro: mimicking asymptomatic infection? Front. Cell Infect. Microbiol. 7, 421 (2017).
Lee, H. Y., Park, E. A., Lee, K. J., Lee, K. H. & Park, S. J. Increased innate lymphoid cell 3 and IL-17 production in mouse lamina propria stimulated with Giardia lamblia. Korean J. Parasitol. 57, 225–232 (2019).
Autenrieth, I. B., Kempf, V., Sprinz, T., Preger, S. & Schnell, A. Defense mechanisms in Peyer’s patches and mesenteric lymph nodes against Yersinia enterocolitica involve integrins and cytokines. Infect. Immun. 64, 1357–1368 (1996).
Bao, S., Beagley, K. W., France, M. P., Shen, J. & Husband, A. J. Interferon-gamma plays a critical role in intestinal immunity against Salmonella typhimurium infection. Immunology 99, 464–472 (2000).
Harty, J. T. & Bevan, M. J. Specific immunity to Listeria monocytogenes in the absence of IFN gamma. Immunity 3, 109–117 (1995).
Cover, T. L. & Aber, R. C. Yersinia enterocolitica. N. Engl. J. Med. 321, 16–24 (1989).
Bottone, E. J. Yersinia enterocolitica: the charisma continues. Clin. Microbiol. Rev. 10, 257–276 (1997).
Nickol, A. D. & Bonventre, P. F. Anomalous high native resistance to athymic mice to bacterial pathogens. Infect. Immun. 18, 636–645 (1977).
Emmerling, P., Finger, H. & Bockemuhl, J. Listeria monocytogenes infection in nude mice. Infect. Immun. 12, 437–439 (1975).
Trulzsch, K., Oellerich, M. F. & Heesemann, J. Invasion and dissemination of Yersinia enterocolitica in the mouse infection model. Adv. Exp. Med. Biol. 603, 279–285 (2007).
Seo, G. Y. et al. LIGHT-HVEM signaling in innate lymphoid cell subsets protects against enteric bacterial infection. Cell Host Microbe 24, 249–260 e244 (2018).
Sorobetea, D. & Brodsky, I. E. HVEM LIGHTs the way for ILC3s. Cell Host Microbe 24, 187–188 (2018).
Bjorklund, A. K. et al. The heterogeneity of human CD127(+) innate lymphoid cells revealed by single-cell RNA sequencing. Nat. Immunol. 17, 451–460 (2016).
Shui, J. W. et al. HVEM signalling at mucosal barriers provides host defence against pathogenic bacteria. Nature 488, 222–225 (2012).
Sadighi Akha, A. A. et al. Interleukin-22 and CD160 play additive roles in the host mucosal response to Clostridium difficile infection in mice. Immunology 144, 587–597 (2015).
Songhet, P. et al. Stromal IFN-gammaR-signaling modulates goblet cell function during Salmonella Typhimurium infection. PLoS ONE 6, e22459 (2011).
Goto, Y. et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science 345, 1254009 (2014).
Yadav, J., Verma, S., Choudhary, D., Jaiwal, P. K. & Jaiwal, R. Tuberculosis: current status, diagnosis, treatment and development of novel vaccines. Curr. Pharm. Biotechnol. 20, 446–458 (2019).
Flynn, J. L. & Chan, J. Immunology of tuberculosis. Annu. Rev. Immunol. 19, 93–129 (2001).
Feng, C. G. et al. NK cell-derived IFN-gamma differentially regulates innate resistance and neutrophil response in T cell-deficient hosts infected with Mycobacterium tuberculosis. J. Immunol. 177, 7086–7093 (2006).
Ardain, A. et al. Group 3 innate lymphoid cells mediate early protective immunity against tuberculosis. Nature 570, 528–532 (2019).
Khader, S. A. et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat. Immunol. 8, 369–377 (2007).
Malik, A., Sharma, D., St Charles, J., Dybas, L. A. & Mansfield, L. S. Contrasting immune responses mediate Campylobacter jejuni-induced colitis and autoimmunity. Mucosal Immunol. 7, 802–817 (2014).
Way, S. S., Borczuk, A. C., Dominitz, R. & Goldberg, M. B. An essential role for gamma interferon in innate resistance to Shigella flexneri infection. Infect. Immun. 66, 1342–1348 (1998).
Ivin, M. et al. Natural killer cell-intrinsic type I IFN signaling controls Klebsiella pneumoniae growth during lung infection. PLoS Pathog. 13, e1006696 (2017).
Kopf, M. et al. IL-5-deficient mice have a developmental defect in CD5+ B-1 cells and lack eosinophilia but have normal antibody and cytotoxic T cell responses. Immunity 4, 15–24 (1996).
Moro, K. et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463, 540–544 (2010).
Price, A. E. et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. USA 107, 11489–11494 (2010).
Pelly, V. S. et al. IL-4-producing ILC2s are required for the differentiation of TH2 cells following Heligmosomoides polygyrus infection. Mucosal Immunol. 9, 1407–1417 (2016).
Saenz, S. A. et al. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature 464, 1362–1366 (2010).
Gerbe, F. et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 529, 226–230 (2016).
Angkasekwinai, P. et al. ILC2s activated by IL-25 promote antigen-specific Th2 and Th9 functions that contribute to the control of Trichinella spiralis infection. PLoS ONE 12, e0184684 (2017).
Turner, J. E., Stockinger, B. & Helmby, H. IL-22 mediates goblet cell hyperplasia and worm expulsion in intestinal helminth infection. PLoS Pathog. 9, e1003698 (2013).
Li, S. et al. Aryl hydrocarbon receptor signaling cell intrinsically inhibits intestinal Group 2 innate lymphoid cell function. Immunity 49, 915–928 e915 (2018).
Garrido-Mesa, N. et al. T-bet controls intestinal mucosa immune responses via repression of type 2 innate lymphoid cell function. Mucosal Immunol. 12, 51–63 (2019).
Monticelli, L. A. et al. Arginase 1 is an innate lymphoid-cell-intrinsic metabolic checkpoint controlling type 2 inflammation. Nat. Immunol. 17, 656–665 (2016).
Wilhelm, C. et al. Critical role of fatty acid metabolism in ILC2-mediated barrier protection during malnutrition and helminth infection. J. Exp. Med. 213, 1409–1418 (2016).
Yasuda, K. et al. Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc. Natl. Acad. Sci. USA 109, 3451–3456 (2012).
Yasuda, K., Adachi, T., Koida, A. & Nakanishi, K. Nematode-infected mice acquire resistance to subsequent infection with unrelated nematode by inducing highly responsive group 2 innate lymphoid cells in the lung. Front. Immunol. 9, 2132 (2018).
Bourke, C. D. et al. Epidermal keratinocytes initiate wound healing and pro-inflammatory immune responses following percutaneous schistosome infection. Int. J. Parasitol. 45, 215–224 (2015).
Hams, E. et al. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 111, 367–372 (2014).
Collins, J. W. et al. Citrobacter rodentium: infection, inflammation and the microbiota. Nat. Rev. Microbiol. 12, 612–623 (2014).
Mundy, R., MacDonald, T. T., Dougan, G., Frankel, G. & Wiles, S. Citrobacter rodentium of mice and man. Cell. Microbiol. 7, 1697–1706 (2005).
Geiger, T. L. et al. Nfil3 is crucial for development of innate lymphoid cells and host protection against intestinal pathogens. J. Exp. Med. 211, 1723–1731 (2014).
Zheng, Y. et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14, 282–289 (2008).
Cella, M. et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 457, 722–725 (2009).
Sonnenberg, G. F., Monticelli, L. A., Elloso, M. M., Fouser, L. A. & Artis, D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity 34, 122–134 (2011).
Basu, R. et al. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity 37, 1061–1075 (2012).
Ahlfors, H. et al. IL-22 fate reporter reveals origin and control of IL-22 production in homeostasis and infection. J. Immunol. 193, 4602–4613 (2014).
Schiering, C. et al. Feedback control of AHR signalling regulates intestinal immunity. Nature 542, 242–245 (2017).
Li, S. et al. Ikaros inhibits group 3 innate lymphoid cell development and function by suppressing the aryl hydrocarbon receptor pathway. Immunity 45, 185–197 (2016).
Chu, C. et al. Anti-microbial functions of Group 3 innate lymphoid cells in gut-associated lymphoid tissues are regulated by G-protein-coupled receptor 183. Cell Rep. 23, 3750–3758 (2018).
Carroll, K. C. & Bartlett, J. G. Biology of Clostridium difficile: implications for epidemiology and diagnosis. Annu. Rev. Microbiol. 65, 501–521 (2011).
Di Bella, S., Ascenzi, P., Siarakas, S., Petrosillo, N. & di Masi, A. Clostridium difficile Toxins A and B: insights into pathogenic properties and extraintestinal effects. Toxins (Basel) 8, 134 (2016).
Hasegawa, M. et al. Interleukin-22 regulates the complement system to promote resistance against pathobionts after pathogen-induced intestinal damage. Immunity 41, 620–632 (2014).
van der Poll, T. & Opal, S. M. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet 374, 1543–1556 (2009).
Tuomanen, E. I., Austrian, R. & Masure, H. R. Pathogenesis of pneumococcal infection. N. Engl. J. Med. 332, 1280–1284 (1995).
Van Maele, L. et al. Activation of Type 3 innate lymphoid cells and interleukin 22 secretion in the lungs during Streptococcus pneumoniae infection. J. Infect. Dis. 210, 493–503 (2014).
Trevejo-Nunez, G., Elsegeiny, W., Conboy, P., Chen, K. & Kolls, J. K. Critical role of IL-22/IL22-RA1 signaling in Pneumococcal pneumonia. J. Immunol. 197, 1877–1883 (2016).
Kinnebrew, M. A. et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity 36, 276–287 (2012).
Xu, X. et al. Conventional NK cells can produce IL-22 and promote host defense in Klebsiella pneumoniae pneumonia. J. Immunol. 192, 1778–1786 (2014).
Xiong, H. et al. Innate lymphocyte/Ly6C(hi) monocyte crosstalk promotes Klebsiella pneumoniae clearance. Cell 165, 679–689 (2016).
Brown, G. D. et al. Hidden killers: human fungal infections. Sci. Transl. Med. 4, 165rv113 (2012).
Abiko, Y. et al. Upregulation of human beta-defensin 2 peptide expression in oral lichen planus, leukoplakia and candidiasis. an immunohistochemical study. Pathol. Res. Pr. 198, 537–542 (2002).
Bahri, R., Curt, S., Saidane-Mosbahi, D. & Rouabhia, M. Normal human gingival epithelial cells sense C. parapsilosis by toll-like receptors and module its pathogenesis through antimicrobial peptides and proinflammatory cytokines. Mediators Inflamm. 2010, 940383 (2010).
Huang, W., Na, L., Fidel, P. L. & Schwarzenberger, P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J. Infect. Dis. 190, 624–631 (2004).
Gorr, S. U. Antimicrobial peptides of the oral cavity. Periodontol 2000 51, 152–180 (2009).
Liang, S. C. et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 203, 2271–2279 (2006).
Gazendam, R. P., van de Geer, A., Roos, D., van den Berg, T. K. & Kuijpers, T. W. How neutrophils kill fungi. Immunol. Rev. 273, 299–311 (2016).
Gladiator, A., Wangler, N., Trautwein-Weidner, K. & LeibundGut-Landmann, S. Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J. Immunol. 190, 521–525 (2013).
Park, S. J., Hughes, M. A., Burdick, M., Strieter, R. M. & Mehrad, B. Early NK cell-derived IFN-{gamma} is essential to host defense in neutropenic invasive aspergillosis. J. Immunol. 182, 4306–4312 (2009).
Bouzani, M. et al. Human NK cells display important antifungal activity against Aspergillus fumigatus, which is directly mediated by IFN-gamma release. J. Immunol. 187, 1369–1376 (2011).
Piehler, D. et al. The IL-33 receptor (ST2) regulates early IL-13 production in fungus-induced allergic airway inflammation. Mucosal Immunol. 9, 937–949 (2016).
Flaczyk, A. et al. IL-33 signaling regulates innate and adaptive immunity to Cryptococcus neoformans. J. Immunol. 191, 2503–2513 (2013).
Sun, J. C., Beilke, J. N. & Lanier, L. L. Adaptive immune features of natural killer cells. Nature 457, 557–561 (2009).
Martinez-Gonzalez, I. et al. Allergen-experienced group 2 innate lymphoid cells acquire memory-like properties and enhance allergic lung inflammation. Immunity 45, 198–208 (2016).
De Luca, A. et al. IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunol. 3, 361–373 (2010).
Abe, K. et al. Th1-Th2 cytokine kinetics in the bronchoalveolar lavage fluid of mice infected with Cryptococcus neoformans of different virulences. Microbiol. Immunol. 44, 849–855 (2000).
Acknowledgements
This work was supported by grants from the National Institutes of Health P01 DK46763, R01 AI61516 and MIST U01 AI125955 to M.K.; F32 AI140581 to D.A.G.
Author information
Authors and Affiliations
Contributions
G.-Y.S., D.A.G., and M.K. planned the manuscript. G.-Y.S. D.A.G. wrote the initial manuscript draft and reviewed the manuscript. M.K. wrote and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Seo, GY., Giles, D.A. & Kronenberg, M. The role of innate lymphoid cells in response to microbes at mucosal surfaces. Mucosal Immunol 13, 399–412 (2020). https://doi.org/10.1038/s41385-020-0265-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-020-0265-y
This article is cited by
-
Mucosal T-cell responses to chronic viral infections: Implications for vaccine design
Cellular & Molecular Immunology (2024)
-
Unconventional immune cells in the gut mucosal barrier: regulation by symbiotic microbiota
Experimental & Molecular Medicine (2023)
-
Microbiota-dependent regulation of costimulatory and coinhibitory pathways via innate immune sensors and implications for immunotherapy
Experimental & Molecular Medicine (2023)
-
The Efficacy of Citrus maxima Peels Aqueous Extract Against Cryptosporidiosis in Immunecompromised Mice
Acta Parasitologica (2021)
-
Gut Innate Immunity and HIV Pathogenesis
Current HIV/AIDS Reports (2021)
