
Abstract
Improvement in traits of agronomic importance is the top breeding priority of crop improvement programs. Majority of these agronomic traits show complex quantitative inheritance. Identification of quantitative trait loci (QTLs) followed by fine mapping QTLs and cloning of candidate genes/QTLs is central to trait analysis. Advances in genomic technologies revolutionized our understanding of genetics of complex traits, and genomic regions associated with traits were employed in marker-assisted breeding or cloning of QTLs/genes. Next-generation sequencing (NGS) technologies have enabled genome-wide methodologies for the development of ultra-high-density genetic linkage maps in different crops, thus allowing placement of candidate loci within few kbs in genomes. In this review, we compare the marker systems used for fine mapping and QTL cloning in the pre- and post-NGS era. We then discuss how different NGS platforms in combination with advanced experimental designs have improved trait analysis and fine mapping. We opine that efficient genotyping/sequencing assays may circumvent the need for cumbersome procedures that were earlier used for fine mapping. A deeper understanding of the trait architectures of agricultural significance will be crucial to accelerate crop improvement.
Similar content being viewed by others


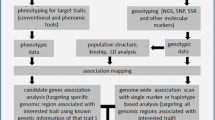
Introduction
Genetic improvement in crop plants is a continuous process of developing improved cultivars to meet the ever-increasing human demand for food, nutrition and energy. Natural variations available for important agronomic traits were utilized in crop improvement activities across the world over the century. Mendel’s experiments on genetic inheritance in the nineteenth century constituted the scientific basis for understanding genetics of plant traits and crop improvement through systematic plant breeding. Majority of the agriculturally important traits are complex or quantitative in nature (Abe et al. 2012). Traditionally, these traits were examined using morphological data and statistical analysis based on mean, variance and covariance of relatives. However, these studies achieved limited success because of low level of polymorphism and strong influence by environment. The discovery of molecular marker technology in 1980s made a major breakthrough in understanding the genetics of complex traits. Concurrent refinements in statistical packages enabled construction of genetic linkage maps based on genotypic data for various mapping populations, thus paving the way to discover quantitative trait loci (QTLs) controlling important quantitative traits.
DNA markers associated with the QTL region were used for making rapid and accurate selections and for introgressing traits in many crop species (Kulwal et al. 2012; Varshney 2016). The QTL regions identified by standard mapping procedure often extend to several centiMorgans (cMs) on genetic map (equivalent to several Mbs on physical map) and might contain a large number of genes (Varshney et al. 2014). Therefore, it is very difficult to pinpoint causative locus responsible for a specific trait. Moreover, introgression of such broad QTL regions based on flanking markers may carry undesirable genes (linkage drag), thereby affecting the performance of improved cultivars carrying the introgressed genomic segments. Therefore, genetic resolution of the mapping procedures should be enhanced to allow QTL placement within the shortest possible genomic region (marker interval) using innovative strategies. This process of refining the QTL region is called as fine mapping.
Three factors, viz. population size (Dinka et al. 2007), phenotyping (Cobb et al. 2013) and number of markers, mainly determine the success of QTL dissection, fine mapping and further cloning of the QTLs. Conventional fine mapping process involves screening of a large number of individuals with the DNA markers flanking the target QTLs, followed by phenotyping of the selected recombinant plants and progeny testing. The fine-mapped region obtained through this process is positioned on the physical map, and candidate genes are then identified. In recent years, the discovery of single-nucleotide polymorphism (SNP) markers in combination with evolving sequencing technologies has led a remarkable improvement in fine mapping procedures. SNPs are universal and the most abundant class of genetic variation among the individuals of a given species. High amenability of SNP markers to automation has broken the dominance of medium-throughput simple sequence repeat (SSR) markers that dominated crop research and breeding during the last two decades. Furthermore, massively parallel or high-throughput NGS technologies dramatically reduced per sample genotyping/sequencing cost and increased throughput (Varshney et al. 2009a). As a result, whole-genome sequences are available for majority of the important crop species (Michael and Jackson 2013, https://www.ncbi.nlm.nih.gov/assembly/organism/2759/all/). With the latest SNP genotyping platforms in place, it is now possible to genotype tens of thousands of samples in a short span of time. In this review, we discuss and critically appraise the efforts to fine-map QTLs, cloning QTLs/genes and identification of candidate/causative genes in the pre- and post-NGS era.
Genotyping tools and approaches for fine mapping in the pre-NGS era
In the early 1990s, DNA-based markers like restriction fragment length polymorphism (RFLP), amplified fragment length polymorphism (AFLP), SSR markers were used for trait dissection. Tomato (Solanum lycopersicum) is the first model crop plant species where the use of RFLP markers and QTL identification were reported (Paterson et al. 1988). In the pre-NGS era, SSR markers were most extensively used in genetic and plant breeding studies owing to their several advantages over other marker systems including higher polymorphism rate, genome-wide distribution and amenability to automation (Gupta and Varshney 2000).
In the pre-NGS era, QTL cloning involved two broad steps: Firstly, QTL region underlying the trait of interest is identified by using a limited number of DNA markers. Later, the identified QTL region is refined by mapping the QTL-flanking DNA markers onto their physical positions in order to identify respective BAC (bacterial artificial chromosome) clone on the physical map. The clones thus identified were used for sequencing and developing DNA markers within the QTL region (Fig. 1). A limited number of studies have reported fine mapping using SNP markers obtained from BAC/YAC clones in the pre-NGS era (Fridman et al. 2000; Kamolsukyunyong et al. 2001; Wang et al. 2009). Such studies could reach gene level of refinement, demonstrating the potential of SNP markers in refining the broad QTL regions. Although SNPs are more advantageous than other DNA-based makers, lack of high-throughput SNP discovery and genotyping methods in pre- NGS era greatly constrained their widespread utilization for fine mapping studies.

Comparison of marker development, map and QTL resolution during the pre- and post-NGS era. In the pre-NGS era, QTL resolution is low (10–30 cM), while in the post-NGS era the QTL resolution is high (0.5–10 cM)
Genotyping technologies and approaches for fine mapping in the post-NGS era
Increasing adoption of NGS-based assays for population genotyping has facilitated high-density linkage mapping in various crop species (Varshney et al. 2019a). Timeline for fine mapping in both the pre- and post-NGS era is compared in Fig. 2. Most importantly, the NGS technology has allowed parallelization of sequencing process, thereby generating thousands to millions of DNA sequences in a single run and reducing the sequencing cost over 1000-folds since its invention (Park and Kim 2016). New NGS-based protocols have emerged such as reduced representation libraries (RRLs), restriction-site-associated DNA sequencing (RAD), genotyping-by-sequencing (GBS), whole-genome resequencing (WGRS) and skim GBS that are capable of identifying and mapping massive number of SNPs in thousands of samples in one go (Varshney et al. 2019a). A dramatic reduction in sequencing cost has motivated researchers to obtain deeper view of the target genomic region by sequencing entire mapping populations. Additionally, one can generate new markers with lesser efforts and time using the available genome sequence for an identified QTL region using flanking marker information, which was not the case in the pre-NGS era (Fig. 2).

Diagrammatic illustration of duration required for fine mapping during the pre- and post-NGS era. Development of NGS-based markers has nearly reduced half the time span taken for fine mapping using non-NGS-based markers
The NGS technologies have offered more benefits to crops that lack whole-genome sequence information, as these technologies generate large-scale DNA markers by sequencing the entire population under study. For example, building a genetic linkage map with moderate density (1000 loci) in the pre-NGS era demanded considerable time and the effort of few technicians in comparison with the current NGS assays that allow highly saturated genetic maps (100,000 loci) to be constructed within few months with modest technical efforts (Yang et al. 2015). Based on the use or non-use of restriction enzymes, the NGS methods can be grouped into two categories: (i) whole-genome sequencing (including WGRS, RNA sequencing, exome capture) that does not employ restriction enzymes, (ii) reduced representation sequencing (such as RAD-seq, GBS, etc.) that uses restriction enzymes for reducing genome complexity (Fig. 3). As these techniques are adequately reviewed elsewhere, they are not discussed in detail in this review (Davey et al. 2011; Garg and Jain 2013; Goodwin et al. 2016).

Various techniques followed for fine mapping during the pre- and post-NGS era and their impact on fine mapping
Role of NGS in accelerating high-resolution mapping and gene discovery
Several new approaches have led to the fast gene discovery through high-resolution mapping using NGS technologies. We list few important such methods in this section.
High-density SNP arrays for faster mapping The SNP arrays or high-density genotyping based on resequencing is being used to generate large-scale marker profiles for a number of individuals (Rasheed et al. 2017; Pandey et al. 2017a; Roorkiwal et al. 2018; Saxena et al. 2018). The SNP chip is preferred over other high-density genotyping platforms as the SNP data it generates are less computationally demanding (Yuan et al. 2019). A high-quality SNP chip array, CottonSNP80K, was developed especially for intraspecific genotyping in cotton, and eight SNPs were found to be associated with salt stresses (Cai et al. 2017). In wheat, a high-throughput genotyping array (TaBW280K) containing 280,226 SNPs was developed and deployed for assessing the germplasm diversity as well as high-density linkage mapping (Rimbert et al. 2018). Similarly, SNP chip array containing 6,000 SNPs was developed in castor from WGRS dataset of 14 diverse lines (Senthilvel et al. 2019). This SNP chip array was validated on 314 inbred castor lines and can be applied in genome-wide analysis.
Bulked segregant RNA Seq (BSR-Seq) for gene discovery Whole transcriptome sequencing of contrasting bulks is referred to as BSR-Seq. The technique is particularly important for crops with large and complex genomes like wheat where resequencing still remains cost-ineffective (Liu et al. 2012a, b). Also, BSR-seq is suitable for fine mapping in crops that still lack a reference genome sequence. Technically similar to QTL-Seq, BSR-Seq relies on sequencing RNAs (cDNA) from extreme bulks for the trait of interest. It facilitates identification of the target region and development of markers near or within the gene of interest. For instance, BSR-seq assigned glossy 3 (gl3) gene of maize to ~ 2 Mb region and a single gene myb transcription factor was reported in this region. This gene was reported to control the genes involved in long-chain fatty acid synthesis in maize (Liu et al. 2012a, b). Similarly, BSR-Seq enabled fine mapping of grain protein content (GPC) region in wheat (GPC-B1) to 0.4 cM from previously reported 30 cM. This study pinpointed candidate genes (13–18 genes) for grain protein content in wheat (Trick et al. 2012). In another study, BSR-Seq approach identified marker associated with the Yr15 gene that imparts resistance to yellow rust in wheat. It facilitated fine mapping of this region to a shortest distance of 0.77 cM. These markers were also utilized to analyze germplasms and can be used in marker-assisted selection (Ramirez-Gonzalez et al. 2015). A more recent study on BSR-Seq allowed cloning of mutant genes in maize that are involved in plant growth via delineation of mapping interval and candidate SNPs from whole-genome sequencing of pooled F2 individuals (Klein et al. 2018). Thus, BSR-Seq approach is being widely adopted for rapid discovery of genes and markers linked with the target genes.
QTL-Seq for gene identification QTL-Seq integrates traditional bulk segregant analysis (BSA) with sequencing methods. This approach has been established as highly efficient for rapid discovery of candidate genes for the trait of interest. By allowing placement of QTL within a smaller genomic segment, it facilitates both detection of QTL and its fine mapping at a stretch. The procedure involves creation of extreme bulks by selecting lines with extreme low and high values of trait from a segregating population. The pools along with one of the parental genotypes are subsequently sequenced using NGS. A reference genome assembly for the parental genotype is developed based on sequencing data. Subsequently, SNP index for each bulk is computed based on the number of reads for a SNP that differs from the SNP in the parent reference assembly. (If 10 reads are having unique nucleotide as in the reference assembly, then SNP index is 0, i.e., 0/10.) Similarly, if all reads have a different nucleotide compared to the reference parent nucleotide, then the SNP index is 1 (10/10). Based on these analyses, Δ-SNP index value of high and low bulks is calculated and the candidate genomic region for the trait of interest is identified. This approach was successfully demonstrated for faster identification of QTLs for blast resistance in rice (Oryza sativa) (Takagi et al. 2013a). Similarly, rapid discovery of a major QTL for early flowering was undertaken in cucumber (Cucumis sativus) (Lu et al. 2014). In the case of chickpea (Cicer arietinum), Singh et al. (2016a) refined a 7 Mb QTL region on linkage group (LG) CaLG04 (previously identified using non-NGS-based markers) to a ~ 1 Mb region for root and seed weight traits in chickpea. Another QTL-seq study in chickpea delineated a 35 kb genomic region on CaLG01 controlling 100-seed weight (Das et al. 2015). In the case of pigeonpea (Cajanus cajan), sequencing of resistant and susceptible bulks in combination with WGRS data of four additional genotypes elucidated candidate genes associated with Fusarium wilt and sterility mosaic disease resistance (Singh et al. 2016b). Similarly, QTL-Seq approach was applied to fine-map bacterial wilt resistance genes and develop diagnostic markers for use in breeding in the case of groundnut (Luo et al. 2019a). Adoption of QTL-Seq is increasingly reported for delineating candidate QTLs for both qualitative and quantitative traits (Yang et al. 2017; Li et al. 2018; Zhang et al. 2018; Clevenger et al. 2018; Zhang et al. 2019; Luo et al. 2019b).
Mutation mapping techniques for gene identification
MutMap The MutMap technique was proposed by Abe et al. (2012) in rice to identify genomic regions governing important agronomic traits. In brief, this technique involves generation of a mutant population using chemical mutagen followed by the selection of line with desirable phenotype in M2 or in the subsequent generations. Such selected mutant is crossed with wild-type parent, and the F1 is selfed to generate an F2 population segregating for the mutant and wild-type phenotypes. DNA samples of F2 lines showing mutant phenotype are bulked and subjected to WGRS. The causative SNP for the specific trait is determined based on the SNP index. If all the short reads covering a particular genomic position share a SNP that differs from the reference, the SNP index is defined as 1. The identified genomic regions with the SNP index of 1 are the causal locus underlying the mutant phenotype.
MutMap+ This technique involves direct sequencing of M3 population instead of backcrossing with wild-type plant; thus, the mutants which are not amenable for crossing in MutMap technique can be utilized for the study. Similar to QTL-seq, mutant and wild-type bulks are prepared using M3 lines and subjected to WGRS. Genomic loci responsible for trait of interest are then inferred with the help of SNP index (Feik et al. 2013).
MutMap-Gap MutMap-Gap facilitates identification of the causal SNPs in the genomic regions that are missing from the reference genome sequence. The method involves: (i) identification of the sequences unmapped with the reference genome, (ii) delineation of the candidate genomic segment with MutMap approach, (iii) creation of a de novo assembly of the target region through combining short reads pinpointed in steps i and ii, (iv) alignment of the bulk-sequenced reads (of mutant individuals) to the ‘P + scaffolds’ reference (reference sequence combined with scaffolds obtained in step iv, and finally (v) identification of the causal mutation within the gap region (Takagi et al. 2013b).
MutChromSeq Another complexity reduction sequencing approach is mutant chromosome sequencing (MutChromSeq), which involves mutagenesis and screening for mutants followed by chromosome sorting of the selected mutant and sequencing the specific chromosome to identify the causative mutation (Steurnagel et al. 2017). Sánchez-Martín et al. (2016) compared sequence information of multiple independently derived mutant flow-sorted chromosomes which would allow the identification of induced, causal mutations without the need for positional fine mapping in barley and wheat. MutChromSeq can overcome the limitations of RNA Seq (tissue specific, time of sampling and sequencing depth) and exome sequencing (captures known genes).
Exome sequencing
Whole-exome sequencing allows us to find out the variations in the protein coding regions and thus enhances the identification of disease-causing mutations in the target gene. The cost-effectiveness of this approach stems from the fact that it involves sequencing of known targeted region instead of the whole genome. It is also extended to capture all the functional region of a genome including noncoding genes and regulatory elements such as promoters and enhancers (Warr et al. 2015). Several targeted genome sequencing techniques are available for faster mapping and gene identification, such as (i) Resistance gene enrichment sequencing (RenSeq) technology for rapid cloning: RenSeq is a NBS-LRR gene-specific R gene enrichment method, which allows rapid identification of disease resistance genes by targeted resequencing. This technique includes bait design using known NBS-LRR gene families followed by sequencing of the enriched samples NBS-LRR and genome annotation. As a proof-of-concept study, Jupe et al. (2013) demonstrated its utility in potato (Solanum tuberosum) and tomato. In this study, target enrichment library was prepared using 523 NB-LRR-like sequences from potato genome, 57 tomato NB-NRC domains, 9 characterized NB-LRR types from tomato, tobacco (Nicotiana tabacum) and pepper (Capsicum annum). The enriched samples against genomic DNA of the sequenced S. tuberosum Group Phureja clone were sequenced, and annotation was carried out. This resulted in successful enrichment of NB-LRRs from 438 to 755. (ii) MutRenSeq: Subsequently, Steuernagel et al. (2016) proposed mutational R gene enrichment sequencing (MutRenSeq) that combines chemical mutagenesis with exome capture and sequencing for rapid cloning of resistance genes such as stem rust resistance genes Sr22 and Sr45 in hexaploid wheat. MutRenSeq significantly reduces the duration of gene cloning process from 5–10 years to 2 years. (iii) SMART-RenSeq: Witek et al. (2016) employed SMART-RenSeq (single-molecule real-time RenSeq) to clone a gene responsible for resistance to Phytophthora infestans (Rpi-amr3i) causing late blight disease in potato. (iv) AgRenSeq: In order to develop broad range of disease resistance in domesticated crops, R genes from wild relatives can be utilized. To achieve this, association analysis was combined with RenSeq approach to develop AgRenSeq method (Arora et al. 2019). This technique involves screening of wild plants for variety of diseases and sequencing of the wild plants to look for resistance genes. It was successfully applied in wheat for the rapid identification of resistance genes for stem rust disease. Any crop with diverse germplasm can be subjected to AgRenSeq approach for R gene cloning. Recent examples have shown utility of RenSeq for improving disease resistance in plants, and similar technique for abiotic stress-tolerant gene identification will benefit crops affected with abiotic stress. (v) Targeted chromosome-based cloning (TACCA) via long-range assembly follows an approach similar to MutChromSeq where prior information about the mapped gene (flanking markers of a QTL) and its chromosomal location is used for chromosome sorting and sequencing. Thind et al. (2017) cloned leaf rust resistance gene Lr22a in wheat using this technique. Two SSR markers flanking Lr22a covering 0.48 cM interval on chromosome 2D were mapped previously, the chromosome 2D was sorted followed by sequencing, and the causative genes were identified within four months. These targeted sequencing approaches reduce time and cost in achieving faster gene discovery compared to WGRS if prior knowledge about the gene or loci of interest is available.
Gene editing for candidate gene characterization
Gene editing is a versatile tool identified in this decade for gene characterization and creation of novel alleles. Taking advantage of the sequencing technologies, the candidate genes are identified and validated genome wide, and almost any gene underlying any trait of interest can be dissected. For instance, Lou et al. (2017) reported the role of OsSAPK2 through creating loss of function mutants by CRISPR/Cas9 approach in rice. This study targeted the third exon of OsSAPK2 (SNF 1-RELATED PROTEIN KINASE 2) for sgRNA designing. The mutant lines (sapk2) were more sensitive to drought and reactive oxygen species (ROS) than wild-type plants. This study suggested that the OsSAPK2 will be a candidate gene for drought tolerance in rice. Recent reviews highlight increasing contributions of CRISPR/cas9 toward novel gene identification and characterization in various crops (Arora and Narula 2017; Jaganathan et al. 2018; Chen 2019; Varshney et al. 2019b).
Evolving mapping resources for better resolution of genetic architectures
QTL mapping with standard mapping populations
An appropriate mapping population developed from genetically diverse and contrasting parents is a prerequisite of QTL identification (Collard et al. 2005). Various mapping populations such as F2, recombinant inbred line (RIL) and double haploid (DH) have been used for QTL identification (Varshney et al. 2009b). Although each population has its own advantages and disadvantages, a RIL population is the most commonly used for QTL mapping studies. Besides, the size of mapping population remains an important factor and depends on a variety of other factors such as type, genetic nature of target trait and access to genotyping and phenotyping facilities; a mapping population comprising 50–250 or more individuals is generally required for preliminary QTL mapping (Collard et al. 2005). A larger mapping population will result in high-resolution mapping of major and minor QTLs. However, the size of mapping population in the pre-NGS era for detecting the QTL was often limited to 200–300 individuals due to lack of high-throughput genotyping methods. Once the QTL for target trait is identified through coarse mapping, populations for fine mapping are developed with large number of progenies (~ 500 to < 10,000) to capture enough recombination to place QTL into a shorter genomic segment (Table 1). A growing body of research suggests NILs as the most preferred population for fine mapping studies as the genetic background is similar across the population except for the target genomic region. Such populations allow the effect of the QTL to be observed accurately, and resolving them into a shorter interval is possible (Fridman et al. 2000; Jander et al. 2002; Uga et al. 2013; Song et al. 2015).
Emergence of high-throughput genotyping platforms in the post-NGS era has dramatically transformed the methods employed previously for fine mapping of a candidate genomic segment. Genome-wide SNP markers are now available even for the crops that were earlier known as orphan crops (Varshney et al. 2013a; Bohra and Singh 2015; Varshney et al. 2019a). Therefore, use of genome-wide methodologies is widespread for RIL or F2 populations for fine genetic dissection of QTL regions, which was otherwise not possible during the pre-NGS era. Recently, several studies have reported fine mapping and identification of genes from RIL, F2 populations (Qi et al. 2014; Wang et al. 2018).
Innovative experimental designs for enhanced gene discovery
The major drawback of the biparental QTL analysis is that the QTLs are often placed to large chromosomal regions as the inference is derived from limited recombinational events. Further, limited genetic diversity and inadequate polymorphic markers hamper the level of precision with which QTL could be placed in the genome. Later, GWAS was widely used to overcome these issues as it permits consideration of broad genetic diversity for trait mapping. In recent times, experimental populations based on multiple founders have gained widespread attention to accelerate QTL mapping and gene discovery (Bohra 2013). These multi-parental populations offer balanced population structure over GWAS and allow profuse recombination as compared to traditional biparental populations (Bazakos et al. 2017; Wallace et al. 2018). These community genetic resources include multi-parent advanced generation inter-cross (MAGIC) and nested association mapping population (NAM).
MAGIC population is developed by crossing multiple founders through two-way, four-way and eight-way crossing, leading to attainment of a fully inbred recombinant population. The major advantages of MAGIC population include increased recombination, improved mapping resolution and greater allelic diversity (Cavanagh et al. 2008). MAGIC design was successfully implemented in plants for fine genetic mapping including Arabidopsis (Kover et al. 2009), wheat (Huang et al. 2012), rice (Bandillo et al. 2013), chickpea (Gaur et al. 2012), tomato (Pascual et al. 2015), cowpea (Huynh et al. 2018) and so forth. The current status of MAGIC populations in major crops along with the unique opportunities and challenges offered by such mapping resources has been thoroughly discussed elsewhere (Huang et al. 2015).
Another multi-parent mating design NAM involves crossing one common reference genotype with diverse founders to generate a series of “interconnected” segregating inbred families. Like MAGIC, NAM offers the advantages of both linkage analysis and association mapping approaches while overcoming the shortcomings of both approaches. The utility of NAM design for QTL mapping is well established in maize (Yu et al. 2008), and the design has been extended to other crops like wheat (Bajgain et al. 2016), sorghum (Bouchet et al. 2017), barley (Nice et al. 2017), oilseed rape (Hu et al. 2018) and soybean (Song et al. 2017; https://www.soybase.org/SoyNAM/).
Fine mapping and QTL cloning in the pre- and post-NGS era—notable examples
Recent advances in molecular biology, biotechnology and genomics have facilitated the cloning of QTLs in crop plants. Technological advances and the efforts toward QTL cloning (see Salvi and Tuberosa 2005; Price 2006) and beyond cloning (Anderson and Mitchell-Olds 2010) were critically appraised recently. We summarize fine mapping and cloning procedures followed during the pre- and post-NGS era in model crops like Arabidopsis and tomato followed by major cereals and legumes with few case studies.
Model plants/crops
Arabidopsis Arabidopsis (Arabidopsis thaliana) is the first plant genome sequenced, and the availability of the genome sequence of Arabidopsis has brought tremendous changes to the methods of fine mapping and cloning (Lukowitz et al. 2000). In the early 1990s, development of a genetic linkage map with 50 markers required great efforts (Bell and Ecker 1994). More than 4000 BC1F1 plants were assayed by Michaels and Amasino (1999) for positional cloning of flowering genes (FLC and FRI). Recombinants were identified using two flanking SSR markers. The marker numbers were increased by four yeast artificial chromosome (YAC) clones from this region. The cleaved amplified polymorphic sequence (CAPS) markers were generated, and further progeny testing delimited the FLC region to a 620 kb interval. Further, analysis of BAC clones in this region delineated a 10–20 kb region with three genes, among which MADS box transcription factor was found to play an important role in flowering in Arabidopsis. This research took almost 5 years from coarse mapping to positional cloning of the flowering gene. The availability of genome sequence of Arabidopsis provided ample marker resources for fine mapping and QTL cloning for the trait of interest (Jander et al. 2002). For instance, VTC2 gene responsible for ascorbic acid deficiency was fine-mapped using the DNA markers from Cereon Genomics within a time span of 2 years.
The key genomic regions associated with flowering time and rosette leaf number were identified using GBS-SNP-based examination of genome-wide crossover. This study identified recombination break points, and QTL analysis using a saturated linkage map determined the location of genes for flowering time and rosette leaf number within 9 and 26.9 kb, respectively. Genome-wide analysis with 215 K SNPs had uncovered candidate genes for time-dependent drought QTLs (Bac-Molenaar et al. 2016). As envisaged by Huang et al. (2009), enhanced precision in the identification of crossovers enabled by new genome-wide genotyping technologies will make current methods of QTL discovery and fine mapping faster, accurate and cost-efficient.
Tomato Tomato is one of the earliest targeted crop species for QTL studies, for which molecular markers were available in 1980s itself. The marker repertoire was enhanced from few RFLP markers to millions of SNP in the due course with the release of whole-genome sequence in 2012 (Tomato Genome Consortium 2012). Genomic resources including genome sequences, genome maps, QTLs and gene expression atlas are available in SGN (SOL Genomics Network) database; this provides a complete information of tomato and other crops that belong to Solanaceae (Mueller et al. 2005). In 1993, pto gene conferring resistance to P. syringae pv. tomato was cloned using a high-density map with RFLPs and a tomato YAC library (Martin et al. 1993). The Pto gene was genetically mapped using 251 F2 progenies; later, the cloned segment/gene was confirmed by analyzing a total of 1300 F2 plants, F3 families and 50 cultivars using markers spanning the identified YAC segment. This is the first report on map-based cloning of disease-resistant gene in plants. Cloning of fruit weight QTLs in tomato using map-based cloning was successfully performed, and progeny testing involved RFLP assay on a total of 3472 F2 plants with markers derived from a YAC contig (Alpert and Tanksley 1996). Later, Frary et al. (2000) screened this YAC contig with a cDNA library followed by cosmid library of L. pennellii (small fruited genotype), which identified a candidate gene ORFX, and the underlying mechanism was elucidated with complementation test of this gene. In another study, 7000 F2 lines were used for fine mapping of sugar content in tomato (Fridman et al. 2000). Using RFLP markers derived from BAC sequence, a shortest interval of 484 bp of an invertase gene was identified by progeny testing. However, the scenario has improved due to the availability of tomato genome sequence (Tomato genome consortium, 2012) and new sequencing techniques which allowed to precisely identify few candidate genes from a large set of genes. For instance, Sun et al. (2015) reported 12 candidate genes controlling tomato fruit shape and other morphological characters from a set of 122 annotated genes in 3.03 Mb region through RNA seq technique (Table 1).
Cereals Among cereals, extensive studies on fine mapping and QTL cloning have been conducted in rice, wheat, maize, barley, etc. The reference genomes have become available for almost all major cereal crops such as rice (IRGSP 2005), wheat (Choulet et al. 2010), maize (Schnable et al. 2009), sorghum (Paterson et al. 2009), barley (IBGSC 2012), pearl millet (Varshney et al. 2017). Availability of these reference genomes has greatly facilitated fine mapping and QTL cloning studies on various agronomic traits including response to biotic and abiotic stresses. Few of these genetic studies are listed in Table 1.
Rice The first success story of gene cloning in rice appeared even before the availability of genome sequence information of rice. For instance, the gene Xa21 (identified from O. longistaminata (Khush et al. 1990)) was cloned using map-based cloning approach. The Hd1 locus responsible for photoperiod sensitivity was cloned using a map-based cloning approach on a large BC3F3 population (Yano et al. 2000). Similarly, analysis of 2807 BC3F4 plants led to mapping of heading QTL, Hd6 to a 26.4 kb region and complementation test confirmed its role in late heading in rice (Takahashi et al. 2001). In the post-NGS era, whole-genome resequencing of populations has been adopted in rice for quick identification of candidate genes. Several analytical frameworks were developed to tackle the deluge of the sequence information generated from such whole-genome sequencing projects. One such promising method was proposed by Huang et al. (2009), known as sliding window approach to identify recombination break points using low-coverage WGRS of 150 recombinant inbreds. The interval between two recombination break points is known as bin that served as markers for linkage map construction. This approach has accurately mapped the semi-dwarf gene “sd1” on chromosome 1 of rice genome. Another domestication-related gene required for red pericarp (Rc) was also cloned (Sweeney et al. 2006). cDNA cloning was also adopted to clone a novel bacterial blight resistance-related gene ME137 from O. meyeriana (He et al. 2013). Guo and colleagues cloned the qSD7-1 dormancy QTL underlying gene Os07g11020, which is annotated as a transcription factor and is the same as the red pericarp color gene Rc from wild rice (O. rufipogan) (https://portal.nifa.usda.gov/web/crisprojectpages/0214099-characterization-of-the-qsd7-1-seed-dormancy-gene-for-allelic-differentiation-and-regulatory-mechanism-in-isogenic-background-of-rice.html). Using map-based cloning approach, Dai et al. (2012) identified a major quantitative trait locus (QTL) LHD1 (late heading date 1), an allele of DTH8/Ghd8, which controls the late heading date of wild rice and encodes a putative HAP3/NF-YB/CBF-A subunit of the CCAAT-box-binding transcription factor. In another study, using map-based cloning approach, Wang et al. (2015) demonstrated map-based cloning of BPH29 gene, a B3 domain-containing recessive gene conferring brown planthopper resistance in rice. This study used an Indica rice introgression line RBPH54, derived from wild rice O. rufipogon with sustainable resistance to BPH. Cloning of An-2 gene that encodes the Lonely Guy-like protein 6 (OsLOGL6) into O. sativa ssp indica cv GuangLuAi4 (GLA4) demonstrated to have a large impact on reducing awn length and increasing tiller and grain numbers in domesticated rice (Gu et al. 2015).
With whole-genome sequence information and NGS protocols in place, several studies have reported characterization of important traits in rice including tillering traits (Yu et al. 2017), disease resistance (Kim et al. 2015; Prahalada et al. 2017), seed longevity (Sasaki et al. 2015), etc. McCouch et al. (2016) launched an open-access high-resolution platform which contains collection of diverse germplasm, high-density SNP marker data and bioinformatics tools for facilitating genome-wide association mapping in rice. For instance, GBS approach has been utilized for QTL identification using nearly 3000 SNPs on backcross inbred lines (BILs) for grain weight and grain length (Bhatia et al. 2018).
Wheat Fine genetic mapping of fusarium head blight (FHB) resistance QTL to short interval of 1.2 cM was reported in wheat (Liu et al. 2006). Recombinants were selected using SSR and STS markers from a large mapping population with 3156 lines derived from an F7 line (RI 63). This study also highlighted micro-collinearity among wheat, rice and barley with respect to the genomic region controlling FHB resistance. Similarly, positional cloning of VRN1 gene for vernalization in wheat employed a large population for progeny testing (3095 F2 plants) and comparative physical maps of rice and sorghum for collinearity analysis of VRN1 region (Yan et al. 2003). BAC contigs and bin-mapped markers from genetic map (ESTs, SSR and RFLP) were used followed by newly developed STS and CAPS markers to fine-map greenbug aphid resistance gene Gb3 using F2:3 population (Azhaguvel et al. 2012). This marker enrichment allowed mapping of Gb3 gene to a short interval of 1.1 cM of wheat chromosome arm 7DL. Using BSR-Seq, a set of 13–18 genes in syntenic cereal genomes for grain protein content (Trick et al. 2012). Similarly, powdery mildew resistance gene PmTm4 was fine-mapped into 0.66 cM interval using comparative genomics approaches on large F2 population (Xie et al. 2017). Furthermore, high-density linkage mapping based on NGS-derived markers has enabled fine mapping of major traits like fungal resistance (Cockram et al. 2015), powdery mildew resistance (Liang et al. 2016), awn shape and length (Yoshioka et al. 2017), flag leaf traits (Hussain et al. 2017) and stripe rust resistance (Ma et al. 2019).
Maize Plant architecture is an important phenomenon to study, especially in maize, understanding the genetic control of ear and tassel is important due to their role in grain yield. Doebley et al. (1997) cloned the teosinte branched 1 (tb 1) gene which explains the evolutionary changes during maize domestication. This gene was found to play an important role in repressing the growth of axillary organ and enable the formation of female inflorescences. Initially, QTL for tb1 was mapped on F2 population; further, the QTL was introgressed into different genetic backgrounds to validate the QTL and complementation test has confirmed the role of tb1 in maize architecture. A similar analysis of fruit case/ear structure on 3106 F2 plants delimited a single gene, teosinte glume architecture (tga1) into 1024 bp controlling ear structure from a wild teosinte to domesticated maize (Wang et al. 2005). Salvi et al. (2007) have cloned a major flowering-time quantitative trait locus, vegetative to generative transition 1 (Vgt1) in maize. The cloned region was confined to ~ 2 kb noncoding region positioned 70 kb upstream from Ap2-like transcription factor. A large F2 population comprising 10,000 lines derived from N28 × NIL C22-4 was used for the QTL cloning study. NIL C22-4 was obtained through the twenty cycles of straight backcrossing of N28 and Gaspé Flint (an early flowering variety). Tassel and ear architecture were dissected using an early-generation population genotyped with low-coverage GBS assay (Chen et al. 2014). This study reports candidate genes involved in tassel structure in addition to confirming several reported QTLs in the shortest physical interval with less time, cost and effort. Recently, BSR-Seq was applied in maize to understand herbicide resistance mechanism, and cytochrome P450 gene (CYP81A9) was identified to be the candidate gene of Nss associated with nicosulfuron sensitivity in maize (Liu et al. 2019). Applying combination of approaches like linkage mapping and genome-wide association studies has shown its efficacy in narrowing down of the target regions in a short span of time.
Barley In the pre-NGS era, the best approach for mapping traits in crops with limited genomic resources relied on exploiting the syntenic relationships with the model crops that have whole-genome sequence information. Hinze et al. (1991) mapped the resistance loci mlo on chromosome 4 (2.7 cM) for powdery mildew resistance using RFLP markers on backcross lines. High-resolution mapping of Rym4/Rym5 locus conferring resistance to the barley yellow mosaic virus complex (BaMMV, BaYMV and BaYMV-2) were reported by Pellio et al. (2005). Two high-resolution mapping populations of 1040 F2 and 3884 F2 lines were developed for mapping rym4 and rym5, respectively. Combinations of markers including RAPD, AFLP, SSR and CAPS were employed for marker saturation and screening; further, closely linked markers were converted to STS markers. The homozygous recombinants were characterized for disease resistance. This study delimited the Rym4/Rym5 locus into less than 2 cM on genetic map and paved a way for positional cloning. Boron tolerance (Bot1) gene was cloned using DH lines and a large mapping population comprising 6720 lines (Sutton et al. 2007). Bot1 was reported to be the responsible gene for boron tolerance by controlling the net entry of boron into the roots and the disposal of boron from leaves in African barley landrace Sahara 3771. Fine mapping of a semi-dwarfing gene sdw3 to 0.04 cM was achieved using synteny between barley and other cereal genomes such as rice, sorghum and brassica (Vu et al. 2010). Crops with complex genomes like barley and wheat have been greatly benefitted by the NGS-based protocols like GBS (Poland and Rife 2012). GBS analysis was used to map plant height QTL on RIL population (Liu et al. 2014). Using barley genome assembly, the genes located within the QTL region were identified by mapping the flanking markers on the genome. Similarly, Liller et al. (2017) have fine-mapped a QTL (AL7.1) for awn length to < 0.9 cM on NILs using SNP markers derived from barley consensus map. Barley genome has been sequenced very recently (Mascher et al. 2017) and being explored for fine trait mapping. BSA-Seq analysis suggested nine confident genes resulting from fine mapping of the locus Rha2 for cereal cyst nematode (CNN) in barley (Van Gansbeke et al. 2019). A seminal paper by Pourkheirandish et al. (2015) in barley reported fine mapping of btr1 and btr2 genes to genomic intervals of 1.2 kb and 4.9 kb, respectively, on chromosome 3H by analyzing more than 10,000 F2 individuals. The genetic complementation tests validating the identities of btr1 and btr2 as ORF 1 and ORF 3, respectively, elucidated key changes occurred during domestication in barley in terms of rachis brittleness and seed dispersal system.
Sorghum Several studies have reported QTLs in sorghum for abiotic and biotic stress tolerance and other agronomically important traits; however, only a few of these could reach the level of fine mapping and QTL cloning. In recent years, researchers have increasingly adopted WGRS for building high-density genome maps that serve as foundation to locate QTLs with greater precision and accuracy in sorghum (Zou et al. 2012; Hilley et al. 2017). Fine structure of sorghum aluminum tolerance locus Altsb was elucidated following association mapping in a panel of 254 accessions. This study accentuates genetic manipulation of a precisely mapped 6 kb genomic region to confer aluminum tolerance in molecular breeding (Caniato et al. 2014). Fine mapping of shoot fly resistance and stay-green mechanism of terminal drought tolerance-related traits on chromosome SBI-10 was successfully achieved using GBS-SNP-based high-density linkage map on high-resolution fine mapping cross (Kiranmayee 2016). Genome sequencing of sorghum has paved the way for developing molecular markers linked to specific traits by extracting the DNA sequence for the region of interest. For instance, Han et al. (2015) delimited the qGW1 region to 101 kb region for grain weight in sorghum using SSR markers derived from whole-genome sequence. Similarly, by using SSRs from genome sequence, Li et al. (2016a) mapped qDor7 QTL for seed dormancy trait onto a genomic region spanning 96 kb with 16 candidate genes. Following linkage mapping and GWAS in two RILs and a diverse panel with GBS, Boyels et al. (2017) found genomic regions controlling grain quality traits. A more recent GWA study in sorghum using GBS-SNPs revealed resistance genes for grain mold fungal disease resistance (Nida et al. 2019).
Legumes
Legumes have lagged far behind those of cereals with respect to fine mapping and QTL cloning. However, fine mapping studies have gained momentum in recent years and the progress is likely to accelerate following the availability of whole-genome sequences of these legume species including soybean (Schmutz et al. 2010), pigeonpea (Varshney et al. 2012), chickpea (Varshney et al. 2013b), common bean (Schmutz et al. 2014) and groundnut (Bertioli et al. 2019; Zhuang et al. 2019).
Soybean is a well-studied crop compared to other legumes, and therefore several studies on fine mapping and QTL cloning have been conducted in soybean. For instance, three candidate genes for root knot nematode resistance were identified by using GBS on a RIL population (Xu et al. 2013). NILs were used for map-based cloning of flowering and maturity gene (Watanabe et al. 2011; Tardivel et al. 2014). Two genomic loci responsible for phytophthora resistance, namely RpsUN1 and RpsUN2, were fine-mapped into 151 and 36 kb regions, respectively (Li et al. 2016b). QTL-Seq approach identified a novel Phytophthora sojae resistance gene RpsHC18, revealing its precise location on chromosome 3 in soybean (Zhong et al. 2018). Positional cloning in soybean with informative recombinants at the Rhg4 locus enabled Liu et al. (2012a, b) to define an 8 kb region on chromosome 8 controlling resistance to soybean cyst nematode (SCN). Confirmatory evidence for the causative gene underlying Rhg4 locus, i.e., serine hydroxymethyltransferase (SHMT), was provided through mutant screens, gene expression and gene silencing experiments. More recent use of WGRS data of 106 soybean accessions by the same group has established major role of CNVs in rhg1 (GmSNAP18) and Rhg4 (GmSHMT08) loci in combination with epistasis and promoter variation for broad-based resistance against SCN (Patil et al. 2019).
In chickpea, skim sequencing has been done on one RIL population to refine the QTL region controlling drought component traits (Kale et al. 2015). This study resolved a broad 7.74 Mb QTL region into ~ 300 kb short segment containing 26 genes. Furthermore, QTL-Seq combined with WGRS identified candidate genes for 100-seed weight (100SDW) and root traits in chickpea (Singh et al. 2016a). These QTLs were co-mapped with the earlier identified QTLs for 100SDW and root traits. Another QTL-Seq study in chickpea identified QTLs for Ascochyta blight on five chromosomes (Ca1, Ca2, Ca4, Ca6 and Ca7) among which QTLs on Ca1, Ca4, Ca6 and Ca7 were overlapped with the earlier identified QTLs using conventional QTL mapping (Deokar et al. 2019). In pigeonpea, marker densities of the genetic maps have been improved incredibly with the recent adoption of NGS techniques (Saxena et al. 2017). Consequently, QTL analysis using high-density genetic linkage maps led authors to detect QTLs in shorter genomic region for disease response (fusarium wilt, sterility mosaic disease: Saxena et al. 2017) and flower, seed-related traits (Yadav et al. 2019). QTL-Seq approach has been used for fine mapping of various important traits in groundnut. Based on the non-synonymous SNPs found between the extreme bulks, allele-specific diagnostic markers were reported for three SNPs for rust and one SNP for LLS (Pandey et al. 2017b). More recently, QTL-Seq by Zhao et al. (2019) localizes AhTc1 gene in peanut controlling purple testa to a 4.7 Mb region and the underlying J3K16L gene was confirmed through bulked segregant RNA sequencing (BSR-seq) and gene overexpression analyses. A similar QTL-Seq approach in groundnut was associated with 2.4 Mb and 0.74 Mb genomic regions on the pseudomolecules B05 and A09, respectively, with fresh seed dormancy trait (Kumar et al. 2019).
Is map-based cloning still relevant in the post-NGS era?
In the past two decades, map-based cloning of QTLs/loci for agronomic traits was very popular and several laboratories around the world accomplished fine mapping and cloning of genes by investing > 10 years or so (Salvi and Tuberosa 2005, 2007). One of the main reasons for this included availability of limited markers and requirement of higher costs on sequencing technologies. Identification of millions of SNPs for genetic mapping experiments has now become a common phenomenon. Similarly, new methods of sequencing have brought the costs on sequencing dramatically low. These advances in our opinion have provided a radical change and great opportunity in the way of creating experimental designs and genetic mapping procedures.
For fine mapping in the past, the coarse mapping-based information was used for refining the underlying genomic region by bringing more and more markers. Subsequently, when the QTL region was used to be delimited to very small region on genetic maps, the markers from these regions were used to be deployed for screening large-insert libraries developed using YAC or BAC clones. After identification of positive BAC clones, Sanger sequencing of those clones used provides sequencing of those regions, then predicts the genes and finally shortlists and validates those genes responsible for QTL. However, new ways were introduced in the post-NGS era to avoid these cumbersome procedures. Massive discovery of genome-wide genetic markers like SNPs facilitates quick development of high-density genome maps. QTL mapping using these high-density maps can refine the preliminary QTL regions into candidate gene identification in a faster manner. For instance, if we need to place a QTL into as finer as 10 kb, in case of Arabidopsis we may need ~ 12,500 well-placed markers and in case of papaya ~ 37,200, for rice ~ 43,000 markers, for grape ~ 50,500, for chickpea ~ 73,000, and in the case of large genomes like maize, we may need ~ 2,30,000 and for soybean ~ 1,11,500. Discovery and mapping of such a huge number of genetic markers is now possible by WGRS of entire population (Peters et al. 2003).
The WGRS-based strategy permits placement of a QTL in a genomic region as fine as 10 kb or even lesser. Comparison of those genomic regions with the genome assemblies (as for majority of crops, reference genomes have become available now) can easily identify the well-annotated genes in those regions. In fact, rapid trait mapping approaches such as QTL-Seq can identify the well-annotated genes directly on genome assembly. Such methodological leaps in our opinion indicate non-requirement of traditional cloning methods that seek initial coarse mapping of the genomic region followed by fine mapping to reach candidate loci. The recent post-NGS technologies definitely can help to reduce the time of QTL cloning, and more importantly, one can even bypass the standard fine mapping processes as the primary mapping itself could pinpoint the genes in the QTL region with great precision.
Conclusion
Fine mapping and QTL cloning were instrumental in understanding the functional mechanism of important plant phenotypes in the past decade. However, sequencing technologies have revolutionized genomics and breeding research in the last decade than in the last 150 years (https://www.lifetechnologies.com/in/en/home/life-science/agricultural-biotechnology/discovery-of-high-density-molecular-markers.html). The resources and time invested to attain candidate genes have been reduced tremendously with these NGS technologies. In the recent past, fine mapping of QTL has become less complicated and rapid due to a variety of reasons: (i) availability of reference genomes for majority of crop species, (ii) availability of high-resolution mapping populations such as MAGIC and NAM, (ii) possibility of construction of genetic maps and undertaking QTL analysis with high marker densities, (iii) possibility of landing directly to genes in QTL regions using rapid trait mapping approaches such as QTL-seq and RenSeq, (iv) availability of gene annotation information for majority of crops to easily shortlist possible genes from the candidate gene lists and (v) availability of mutant populations in several crops and possibility of using fast gene editing approaches for validating gene function. The projects that had taken considerable time (10–15 years) to fine-map, clone QTL and identify a candidate gene now can be completed within a time of 10–20 months.
References
Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H et al (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol 30:174–178
Alpert KB, Tanksley SD (1996) High-resolution mapping and isolation of a yeast artificial chromosome contig containing fw2.2: a major fruit weight quantitative trait locus in tomato. Proc Natl Acad Sci USA 93:15503–15507
Anderson JT, Mitchell-Olds T (2010) Beyond QTL cloning. PLoS Genet 6:11
Arora L, Narula A (2017) Gene editing and crop improvement using CRISPR-Cas9 system. Front Plant Sci 8:1932
Arora S, Steuernagel B, Gaurav K, Chandramohan S et al (2019) Resistance gene cloning from a wild crop relative by sequence capture and association genetics. Nat Biotechnol 37:139–143
Ashikari M, Sakakibara H, Lin S et al (2005) Cytokinin oxidase regulates rice grain production. Science 309:741–745
Azhaguvel P, Rud JC, Ma Y, Luo MC, Weng Y (2012) Fine genetic mapping of greenbug aphid-resistance gene Gb3 in Aegilops tauschii. Theor Appl Genet 124:555–564
Bac-Molenaar JA, Granier C, Keurentjes JJ, Vreugdenhil D (2016) Genome-wide association mapping of time-dependent growth responses to moderate drought stress in Arabidopsis. Plant Cell Environ 39:88–102
Bajgain P, Rouse MN, Tsilo TJ, Macharia GK, Bhavani S, Jin Y, Anderson JA (2016) Nested association mapping of stem rust resistance in wheat using genotyping by sequencing. PLoS ONE 11:e0155760
Bandillo N, Raghavan C, Muyco PA et al (2013) Multi-parent advanced generation inter-cross (MAGIC) populations in rice: progress and potential for genetics research and breeding. Rice 6:11
Barrero JM, Cavanagh C, Verbyla KL et al (2015) Transcriptomic analysis of wheat near-isogenic lines identifies PM19-A1 and A2 as candidates for a major dormancy QTL. Genome Biol 16:93
Bazakos C, Hanemian M, Trontin C, Jiménez-Gómez JM, Loudet O (2017) New strategies and tools in quantitative genetics: how to go from the phenotype to the genotype. Annu Rev Plant Biol 68:435–455
Bell CJ, Ecker JR (1994) Assignment of 30 microsatellite markers to the linkage map of Arabidopsis. Genomics 19:137–144
Bernatzky R, Tanksley SD (1986) Toward a saturated linkage map in tomato based on isozymes and random cDNA sequences. Genetics 112:887–898
Bertioli DJ, Jenkins J, Clevenger J, Dudchenko O, Gao D, Seijo G, Samoluk SS (2019) The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat Genet 51:877
Bhatia D, Wing RA, Yu Y et al (2018) Genotyping by sequencing of rice interspecific backcross inbred lines identifies QTLs for grain weight and grain length. Euphytica 214:41
Bohra A (2013) Emerging paradigms in genomics-based crop improvement. Sci World J 585467:17
Bohra A, Singh NP (2015) Whole genome sequences in pulse crops: a global community resource to expedite translational genomics and knowledge-based crop improvement. Biotechnol Lett 37:1529–1539
Bouchet S, Olatoye MO, Marla SR et al (2017) Increased power to dissect adaptive traits in global sorghum diversity using a nested association mapping population. Genetics 206:573–585
Boyles RE, Pfeiffer BK, Cooper EA, Rauh BL, Zielinski KJ, Myers MT, Brenton Z, Rooney WL, Kresovich S (2017) Genetic dissection of sorghum grain quality traits using diverse and segregating populations. Theor Appl Genet 130:697–716
Cai C, Zhu G, Zhang T, Guo W (2017) High-density 80 K SNP array is a powerful tool for genotyping G. hirsutum accessions and genome analysis. BMC Genomics 18:654
Caniato FF, Hamblin MT, Guimaraes CT et al (2014) Association mapping provides insights into the origin and the fine structure of the sorghum aluminum tolerance locus AltSB. PLoS ONE 9:e87438
Cavanagh C, Morell M, Mackay I, Powell W (2008) From mutations to MAGIC: resources for gene discovery, validation and delivery in crop plants. Curr Opin Plant Biol 11:215–221
Chen SJ (2019) Minimizing off-target effects in CRISPR-Cas9 genome editing. Cell Biol Toxicol 35:399–401. https://doi.org/10.1007/s10565-019-09486-4
Chen Z, Wang B, Dong XL et al (2014) An ultra-high density bin-map for rapid QTL mapping for tassel and ear architecture in a large F2 maize population. BMC Genom 15:433
Choulet F, Wicker T, Rustenholz C et al (2010) Megabase level sequencing reveals contrasted organization and evolution patterns of the wheat gene and transposable element spaces. Plant Cell 22:1686–1701
Clevenger J, Chu Y, Chavarro C, Botton S, Culbreath A, Isleib TG, Holbrook CC, Ozias-Akins P (2018) Mapping late leaf spot resistance in peanut (Arachis hypogaea) using QTL-seq reveals markers for marker-assisted selection. Front Plant Sci 9:83
Cobb JN, DeClerck G, Greenberg A, Clark R, McCouch S (2013) Next-generation phenotyping: requirements and strategies for enhancing our understanding of genotype–phenotype relationships and its relevance to crop improvement. Theor Appl Genet 126:867–887
Cockram J, Scuderi A, Barber T et al (2015) Fine-Mapping the wheat snn1 locus conferring sensitivity to the parastagonospora nodorum necrotrophic effector sntox1 using an eight founder multiparent advanced generation inter-cross population. G3-Genes Genom Genet 5:2257–2266
Collard BCY, Jahufer MZZ, Brouwer JB, Pang ECK (2005) An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: the basic concepts. Euphytica 142:169–196
Dai X, Ding Y, Tan L, Fu Y, Liu F, Zhu Z, Sun X, Sun X, Gu P, Cai H, Sun C (2012) LHD1, an allele of DTH8/Ghd8, controls late heading date in common wild rice (Oryza rufipogon). J Integr Plant Biol 54:790–799
Das S, Upadhyaya HD, Bajaj D et al (2015) Deploying QTL-seq for rapid delineation of a potential candidate gene underlying major trait-associated QTL in chickpea. DNA Res 22:193–203
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML (2011) Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet 12:499–510
Deokar A, Sagi M, Daba K, Tar’an B (2019) QTL sequencing strategy to map genomic regions associated with resistance to ascochyta blight in chickpea. Plant Biotechnol J 17:275–288
Dinka SJ, Campbell MA, Demers T, Raizada MN (2007) Predicting the size of the progeny mapping population required to positionally clone a gene. Genetics 176:2035–2054
Doebley J, Stec A, Hubbard L (1997) The evolution of apical dominance in maize. Nature 386:485–488
Doi K, Izawa T, Fuse T et al (2004) Ehd1, a B-type response regulator in rice, confers short-day promotion of flowering and controls FT-like gene expression independently of Hd1. Genes Dev 18:926–936
Eshed Y, Zamir D (1995) An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated QTL. Genetics 141:1147–1162
Fekih R, Takagi H, Tamiru M, Abe A, Natsume S et al (2013) MutMap+: genetic mapping and mutant identification without crossing in rice. PLoS ONE 10:e68529
Francia E, Barabaschi D, Tondelli A et al (2007) Fine mapping of a HvCBF gene cluster at the frost resistance locus Fr-H2 in barley. Theor Appl Genet 115:1083–1091
Frary A, Nesbitt TC, Frary A et al (2000) fw22: a quantitative trait locus key to the evolution of tomato fruit size. Science 289:85–88
Fridman E, Pleban T, Zamir D (2000) A recombination hotspot delimits a wild-quantitative trait locus for tomato sugar to 484 bp within an invertase gene. Proc Natl Acad Sci USA 97:4718–4723
Garg R, Jain M (2013) RNA-Seq for transcriptome analysis in non-model plants. In: Legume Genomics, Humana Press Totowa, pp 43–58
Gaur PM, Jukanti AK, Varshney RK (2012) Impact of genomic technologies on chickpea breeding strategies. Agronomy 2:199–221
Goodwin S, McPherson JD, McCombie WR (2016) Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 17:333
Gu B, Zhou T, Luo J, Liu H, Wang Y, Shangguan Y, Zhu J, Li Y, Sang T, Wang Z, Han B (2015) An-2 encodes a cytokinin synthesis enzyme that regulates awn length and grain production in rice. Mol Plant 8:1635–1650
Guo L, Ma L, Jiang H, Zeng D, Hu J, Wu L, Gao Z, Zhang G, Qian Q (2009) Genetic analysis and fine mapping of two genes for grain shape and weight in rice. J Integr Plant Biol 51:45–51
Gupta PK, Varshney RK (2000) The development and use of microsatellite markers for genetics and plant breeding with emphasis on bread wheat. Euphytica 113:163–185
Han L, Chen J, Mace ES et al (2015) Fine mapping of qGW1, a major QTL for grain weight in sorghum. Theor Appl Genet 128:1813
He B, Huang X, Li D, Jiang C, Yu T, Yin F, Li W, Cheng Z (2013) The cDNA cloning of a novel bacterial blight-resistance gene ME137. Acta Biochm Biophy Sin 45:422–424
Hilley JL, Weers BD, Truong SK et al (2017) Sorghum dw2 encodes a protein kinase regulator of stem internode length. Sci Rep 7:4616
Hinze K, Thomson RD, Ritter E, Salamini F, Schulze-Lefert P (1991) RFLP-mediated targeting of the mlo resistance locus in barley (Hordeum vulgare). Proc Natl Acad Sci USA 88:3691–3695
Hu J, Guo C, Wang B et al (2018) Genetic properties of a nested association mapping population constructed with semi-winter and spring oilseed rapes. Front Plant Sci 9:1740
Huang X, Feng Q, Qian Q et al (2009) High-throughput genotyping by whole-genome resequencing. Genome Res 19:1068–1076
Huang BE, George AW, Forrest KL, Kilian A, Hayden MJ, Morell MK, Cavanagh CR (2012) A multiparent advanced generation inter-cross population for genetic analysis in wheat. Plant Biotechnol J 10:826–839
Huang BE, Verbyla KL, Verbyla AP, Raghavan C, Singh VK, Gaur P, Leung H, Varshney RK, Cavanagh CR (2015) MAGIC populations in crops: current status and future prospects. Theor Appl Genet 128:999–1017
Hussain W, Baenzigar S, Belamkar V et al (2017) Genotyping-by-sequencing derived high-density linkage map and its application to QTL mapping of flag leaf traits in bread wheat. Sci Rep 7:16394
Huynh BL, Ehlers JD, Huang BE et al (2018) A multi-parent advanced generation inter-cross (MAGIC) population for genetic analysis and improvement of cowpea (Vigna unguiculata L Walp). Plant J 93:1129–1142
IBGSC (2012) A physical, genetic and functional sequence assembly of the barley genome. Nature 491:711–716
International Rice Genome Sequencing Project (2005) The map-based sequence of the rice genome. Nature 436:793–800
Jaganathan D, Ramasamy K, Sellamuthu G, Jayabalan S, Venkataraman G (2018) CRISPR for crop improvement: an update review. Front Plant Sci 9:985
Jander G, Norris SR, Rounsley SD et al (2002) Arabidopsis map-based cloning in the post-genome era. Plant Physiol 129:440–450
Jeong N, Suh SJ, Kim M et al (2012) Ln is a key regulator of leaflet shape and number of seeds per pod in soybean. Plant Cell 24:4807–4818
Jupe F, Witek K, Verweij W et al (2013) Resistance gene enrichment sequencing (RenSeq) enables re-annotation of the NB-LRR gene family from sequenced plant genomes and rapid mapping of resistance loci in segregating populations. Plant J 76:530–544
Kale S, Jaganathan D, Ruperao P et al (2015) Prioritization of candidate genes in “QTL-hotspot” region for drought tolerance in chickpea (Cicer arietinum L.). Sci Rep 5:15296
Kamolsukyunyong W, Ruanjaichon V, Siangliw M, Kawasaki S, Sasaki T, Vanavichit A, Tragoonrung S (2001) Mapping of quantitative trait locus related to submergence tolerance in rice with aid of chromosome walking. DNA Res 8:163–171
Keller B, Manzanares C, Jara C, Lobaton JD, Studer B, Raatz B (2015) Fine-mapping of a major QTL controlling angular leaf spot resistance in common bean (Phaseolus vulgaris L.). Theor Appl Genet 128:813–826
Khush GS, Bacalangco E, Ogawa T (1990) A new gene for resistance to bacterial blight from it O. longistaminata. Rice Genet Newsletters 7:121–122
Kim SM, Suh JP, Qin Y et al (2015) Identification and fine-mapping of a new resistance gene, Xa40, conferring resistance to bacterial blight races in rice (Oryza sativa L.). Theor Appl Genet 128:1933
Kiranmayee KNS (2016) Fine mapping of shoot fly resistance and stay-green QTLS on Sorghum Chromosome SBI-10. Dissertation, Osmania University
Klein H, Xiao Y, Conklin PA et al (2018) Bulked-segregant analysis coupled to whole genome sequencing (BSA-Seq) for rapid gene cloning in maize. G3-Genes Genom Genet 8:3583–3592
Kover PX, Valdar W, Trakalo J, Scarcelli N, Ehrenreich IM, Purugganan MD, Durrant C, Mott R (2009) A multiparent advanced generation inter-cross to fine-map quantitative traits in Arabidopsis thaliana. PLoS Genet 5:e1000551
Kulwal PL, Thudi M, Varshney RK (2012) Crop Breeding for Sustainable Agriculture, Genomics Interventions in. In: Meyers RA (ed) Encyclopedia of sustainability science and technology. Springer, New York, pp 6–29
Kumar R, Janila P, Vishwakarma MK, Khan AW, Manohar SS, Gangurde SS, Variath MT, Shasidhar Y, Pandey MK, Varshney RK (2019) Whole genome re-sequencing-based QTL-seq identified candidate genes and molecular markers for fresh seed dormancy in groundnut. Plant Biotechnol J. https://doi.org/10.1111/pbi.13266
Li L, Lin F, Wang W, Ping J, Fitzgerald JC, Zhao M, Li S, Sun L, Cai C, Ma J (2016a) Fine mapping and candidate gene analysis of two loci conferring resistance to Phytophthora sojae in soybean. Theor Appl Genet 129:2379–2386
Li P, Mace ES, Guo Y, Han L, Wang M, He Y, Chen J, Yuyama N, Jordan DR, Cai H (2016b) Fine Mapping of qDor7, a major qtl affecting seed dormancy in sorghum (Sorghum bicolor (L.) Moench). Trop Plant Biol 9:109–116
Li P, Du C, Zhang Y, Yin S, Zhang E, Fang H, Lin D, Xu C, Yang Z (2018) Combined bulked segregant sequencing and traditional linkage analysis for identification of candidate gene for purple leaf sheath in maize. PLoS ONE 13:e0190670
Liang J, Fu B, Tang W et al (2016) Fine mapping of two wheat powdery mildew resistance genes located at the Pm1 cluster. Plant Genome 9:2
Liller CB, Walla A, Boer MP et al (2017) Fine mapping of a major QTL for awn length in barley using a multiparent mapping population. Theor Appl Genet 130:269–281
Liu S, Zhang X, Pumphrey MO et al (2006) Complex microcolinearity among wheat, rice, and barley revealed by fine mapping of the genomic region harboring a major QTL for resistance to Fusarium head blight in wheat. Funct Integr Genom 6:83–89
Liu S, Kandoth PK, Warren SD, Yeckel G, Heinz R, Alden J, Yang C, Jamai A, El-Mellouki T, Juvale PS (2012a) A soybean cyst nematode resistance gene points to a new mechanism of plant resistance to pathogens. Nature 492:256
Liu S, Yeh CT, Tang HM, Nettleton D, Schnable PS (2012b) Gene mapping via bulked segregant RNA-Seq (BSR-Seq). PLoS ONE 7:e36406
Liu H, Bayer M, Druka A et al (2014) An evaluation of genotyping by sequencing (GBS) to map the Breviaristatum-e (ari-e) locus in cultivated barley. BMC Genom 15:104
Liu X, Bi B, Xu X et al (2019) Rapid identification of a candidate nicosulfuron sensitivity gene (Nss) in maize (Zea mays L.) via combining bulked segregant analysis and RNA-seq. Theor Appl Genet 132:1351–1361
Lou D, Wang H, Liang G, Yu D (2017) OsSAPK2 confers abscisic acid sensitivity and tolerance to drought stress in rice. Front Plant Sci 8:993
Lu H, Lin T, Klein J et al (2014) QTL-seq identifies an early flowering QTL located near Flowering Locus T in cucumber. Theor Appl Genet 127:1491–1499
Lukowitz W, Gillmor CS, Scheible WG (2000) Positional cloning in arabidopsis why it feels good to have a genome initiative working for you. Plant Physiol 123:795–805
Luo H, Pandey MK, Khan AW (2019a) Next-generation sequencing identified genomic region and diagnostic markers for resistance to bacterial wilt on chromosome B02 in peanut (Arachis hypogaea L.). Plant Biotechnol J 17(12):2356–2369. https://doi.org/10.1111/pbi.13153
Luo H, Pandey MK, Khan AW et al (2019b) Discovery of genomic regions and candidate genes for stable QTLs controlling shelling percentage using QTL-seq approach in cultivated peanut (Arachis hypogaea L.). Plant Biotechnol J 17(7):1248–1260
Ma J, Qin N, Cai B et al (2019) Identification and validation of a novel major QTL for all-stage stripe rust resistance on 1BL in the winter wheat line. Theor Appl Genet 132:1363–1373
Martin GB, Brommonschenkel SH, Chunwongse J, Frary A, Ganal MW, Spivey R, Wu T, Earle ED, Tanksley SD (1993) Map-based cloning of a protein kinase gene conferring disease resistance in tomato. Science 262:1432–1436
Mascher M, Gundlach H, Himmelbach A et al (2017) A chromosome conformation capture ordered sequence of the barley genome. Nature 544:427–433
McCouch S, Wright MH, Tung CW et al (2016) Open access resources for genome wide association mapping in rice. Nat Commun 7:10532
Michael TP, Jackson S (2013) The first 50 plant genomes. Plant Genome 6:1–7
Michaels SD, Amasino RM (1999) FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 11:949–956
Mueller LA, Solow TH, Taylor N et al (2005) The SOL Genomics Network: a comparative resource for Solanaceae biology and beyond. Plant Physiol 138:1310–1317
Munos S, Ronc N, Botton E et al (2011) Increase in tomato locule number is controlled by two single-Nucleotide nucleotide polymorphisms located near Wuschel. Plant Physiol 156:2244–2254
Nair SK, Babu R, Magorokosho C et al (2015) Fine mapping of Msv1, a major QTL for resistance to Maize Streak Virus leads to development of production markers for breeding pipelines. Theor Appl Genet 128:1839–1854
Nice LM, Steffenson BJ, Blake TK, Horsley RD, Smith KP, Muehlbauer GJ (2017) Mapping agronomic traits in a wild barley advanced backcross-nested association mapping population. Crop Sci 57:1199–2210
Nida H, Girma G, Mekonen M et al (2019) Identification of sorghum grain mold resistance loci through genome wide association mapping. J Cereal Sci 85:295–304
Pandey MK, Agarwal G, Kale SM et al (2017a) Development and evaluation of a high-density genotyping ‘Axiom_Arachis’ array with 58 K SNPs for accelerating genetics and breeding in groundnut. Sci Rep 7:40577
Pandey MK, Khan AW, Singh VK, Vishwakarma MK, Shasidhar Y, Kumar V, Garg V, Bhat RS, Chitikineni A, Janila P (2017b) QTL-seq approach identified genomic regions and diagnostic markers for rust and late leaf spot resistance in groundnut (Arachis hypogaea L.). Plant Biotechnol J 15:927–941
Park ST, Kim J (2016) Trends in next-generation sequencing and a new era for whole genome sequencing. Int Neurourol J 20(Suppl 2):S76–S83
Pascual L, Desplat N, Huang BE et al (2015) Potential of a tomato MAGIC population to decipher the genetic control of quantitative traits and detect causal variants in the resequencing era. Plant Biotechnol J 13:565–577
Paterson AH, Landers ES, Hewitt JD, Paterson S, Lincoln SE, Tanksley SD (1988) Resolution of quantitative traits into Mendelian factors by using a complete linkage map of restriction fragment length polymorphisms. Nature 335:721–726
Paterson AH, Bowers JE, Bruggmann R et al (2009) The Sorghum bicolor genome and the diversification of grasses. Nature 457:551–556
Patil GB, Lakhssassi N, Wan J, Song L, Zhou Z, Klepadlo M et al (2019) Whole genome re-sequencing reveals the impact of the interaction of copy number variants of the rhg1 and Rhg4 genes on broad-based resistance to soybean cyst nematode. Plant Biotechnol J 17:1595–1611
Pellio B, Streng S, Bauer E, Stein N, Perovic D, Schiemann A, Friedt W, Ordon F, Graner A (2005) High-resolution mapping of the Rym4/Rym5 locus conferring resistance to the barley yellow mosaic virus complex (BaMMV, BaYMV, BaYMV-2) in barley (Hordeum vulgare ssp. vulgare L.). Theor Appl Genet 110:283–293
Peters JL, Cnudde C, Gerats T (2003) Forward genetics and map-based cloning approaches. Trends Plant Sci 8:484–491
Poland JA, Rife T (2012) Genotyping-by-sequencing for plant breeding and genetics. Plant Genome 5:92–102
Pourkheirandish M, Hensel G, Kilian B et al (2015) Evolution of the grain dispersal system in barley. Cell 162:527–539
Prahalada GD, Shivakumar N, Lohithaswa HC et al (2017) Identification and fine mapping of a new gene, BPH31 conferring resistance to brown planthopper biotype 4 of India to improve rice Oryza sativa L. Rice 10:41
Price AH (2006) Believe it or not, QTLs are accurate! Trends Plant Sci 11:213–216
Qi X, Li M, Xie M et al (2014) Identification of a novel salt tolerance gene in wild soybean by whole-genome sequencing. Nat Commun 5:4340
Ramirez-Gonzalez RH, Segovia V, Bird N et al (2015) RNA-Seq bulked segregant analysis enables the identification of high-resolution genetic markers for breeding in hexaploid wheat. Plant Biotechnol J 13:613–624
Rasheed A, Hao Y, Xia X, Khan A, Xu Y, Varshney RK, He Z (2017) Crop breeding chips and genotyping platforms: progress, challenges, and perspectives. Mol Plant 10:1047–1064
Rimbert H, Darrier B, Navarro J, Kitt J, Choulet F, Leveugle M, Duarte J, Riviere N, Eversole K, Consortium IWGS (2018) High throughput SNP discovery and genotyping in hexaploid wheat. PloS PLoS One 13:e0186329
Roder MS, Huang XQ, Borner A (2008) Fine mapping of the region on wheat chromosome 7D controlling grain weight. Funct Integr Genom 8:79–86. https://doi.org/10.1007/s10142-007-0053-8
Roorkiwal M, Jain A, Kale SM, Doddamani D, Chitikineni A, Thudi M, Varshney RK (2018) Development and evaluation of high-density Axiom® CicerSNP array for high-resolution genetic mapping and breeding applications in chickpea. Plant Biotechnol J 16:890–901
Rowan BA, Patel V, Weigel D et al (2015) Rapid and inexpensive whole-genome genotyping-by-sequencing for crossover localization and fine-scale genetic mapping. G3-Genes Genom Genet 13:385–398
Salvi S, Tuberosa R (2005) To clone or not to clone plant QTLs: present and future challenges. Trends Plant Sci 10:293–301
Salvi S, Tuberosa R (2007) Cloning QTLs in Plants. In: Tuberosa R (ed) Genomics-Assisted Crop Improvement (Varshney RK. Springer, Dordrecht, pp 207–226
Salvi S, Sponza G, Morgante M, Tomes D, Niu X, Fengler KA, Meeley R, Ananiev EV, Svitashev S, Bruggemann E (2007) Conserved noncoding genomic sequences associated with a flowering-time quantitative trait locus in maize. Proc Natl Acad Sci USA 104:11376–11381
Sánchez-Martín J, Steuernagel B, Ghosh S et al (2016) Rapid gene isolation in barley and wheat by mutant chromosome sequencing. Genome Biol 17:221
Sasaki K, Takeuchi Y, Miura K et al (2015) Fine mapping of a major quantitative trait locus, qLG-9, that controls seed longevity in rice (Oryza sativa L.). Theor Appl Genet 128:769–778
Saxena RK, Kale SM, Kumar V et al (2017) Genotyping-by-sequencing of three mapping populations for identification of candidate genomic regions for resistance to sterility mosaic disease in pigeonpea. Sci Rep 7:1813
Saxena RK, Rathore A, Bohra A et al (2018) Development and application of high-density axiom Cajanus SNP array with 56 K SNPs to understand the genome architecture of released cultivars and founder genotypes. Plant Genome. https://doi.org/10.3835/plantgenome2018.01.0005
Schmalenbach I, March TJ, Bringezu T et al (2011) High-resolution genotyping of wild barley introgression lines and fine-mapping of the threshability locus thresh-1 using the Illumina GoldenGate assay. G3-Genes Genom Genet 1:187–196
Schmutz J, Cannon SB, Schlueter J et al (2010) Genome sequence of the palaeopolyploid soybean. Nature 463:178–183
Schmutz J, McClean PE, Mamidi S et al (2014) A reference genome for common bean and genome-wide analysis of dual domestications. Nat Genet 46:707–713
Schnable PS, Ware D, Fulton RS et al (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326:1112–1115
Senthilvel S, Ghosh A, Shaik M, Shaw RK, Bagali PG (2019) Development and validation of an SNP genotyping array and construction of a high-density linkage map in castor. Sci Rep 9:3003
Shirasawa K, Hirakawa H (2013) DNA marker applications to molecular genetics and genomics in tomato. Breed Sci 63:21–30
Singh VK, Khan AW, Jaganathan D et al (2016a) QTL-seq for rapid identification of candidate genes for 100-seed weight and root/total plant dry weight ratio under rainfed conditions in chickpea. Plant Biotechnol J 14:2110–2119
Singh VK, Khan AW, Saxena RK et al (2016b) Next-generation sequencing for identification of candidate genes for Fusarium wilt and sterility mosaic disease in pigeonpea (Cajanus cajan). Plant Biotechnol J 14:1183–1194
Song XJ, Huang W, Shi M, Zhu MZ, Lin HX (2007) A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet 39:623–630
Song XJ, Kuroha T, Ayano M et al (2015) Rare allele of a previously unidentified histone H4 acetyltransferase enhances grain weight, yield, and plant biomass in rice. Proc Natl Acad Sci USA 112:76–81
Song Q, Yan L, Quigley C et al (2017) Genetic characterization of the soybean nested association mapping population. Plant Genome 10:2
Spindel J, Wright M, Chen C et al (2013) Bridging the genotyping gap: using genotyping by sequencing (GBS) to add high-density SNP markers and new value to traditional bi-parental mapping and breeding populations. Theor Appl Genet 126:2699–2716
Steuernagel B, Periyannan SK, Hernández-Pinzón I, Witek K et al (2016) Rapid cloning of disease-resistance genes in plants using mutagenesis and sequence capture. Nat Biotechnol 34:652–655
Steuernagel B, Vrána J, Karafiátová M, Wulff BBH, Dolezel J (2017) Rapid gene isolation using MutChromSeq. Methods Mol Biol 1659:231–243
Sun L, Rodriguez GR, Clevenger JP, Illa-Berenguer E, Lin J, Blakeslee JJ, Liu W, Fei Z, Wijeratne A, Meulia T, van der Knaap E (2015) Candidate gene selection and detailed morphological evaluations of fs8.1, a quantitative trait locus controlling tomato fruit shape. J Exp Bot 66:6471–6482
Sutton T, Baumann U, Hayes J et al (2007) Boron-toxicity tolerance in barley arising from efflux transporter amplification. Science 318:1446–1449
Sweeney MT, Thomson MJ, Pfeil BE, McCouch S (2006) Caught red-handed: Rc encodes a basic helix-loop-helix protein conditioning red pericarp in rice. Plant Cell 18:283–294
Takagi H, Abe A, Yoshida K et al (2013a) QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J 74:174–183
Takagi H, Uemura A, Yaegashi H, Tamiru M, Abe A et al (2013b) MutMap-Gap: whole-genome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast resistance gene Pii. New Phytol 200:276–283
Takahashi Y, Shomura A, Sasaki T, Yano M (2001) Hd6, a rice quantitative trait locus involved in photoperiod sensitivity, encodes the alpha subunit of protein kinase CK2. Proc Natl Acad Sci USA 98:7922–7927
Tao Y, Liu Q, Wang H et al (2013) Identification and fine-mapping of a QTL, qMrdd1, that confers recessive resistance to maize rough dwarf disease. BMC Plant Biol 13:145
Taramino G, Sauer M, Stauffer JL et al (2007) The maize (Zea mays L) RTCS gene encodes a LOB domain protein that is a key regulator of embryonic seminal and post-embryonic shoot-borne root initiation. Plant J 50:649–659
Tardivel A, Sonah H, Belzile F, O’Donoughue LS (2014) Rapid identification of alleles at the soybean maturity gene e3 using genotyping by sequencing and a haplotype-based approach. Plant Genome 7:2
Thind AK, Wicker T, Simkova H, Fossati D, Moullet O, Brabant C, Vrana J, Dolezel J, Krattinger SG (2017) Rapid cloning of genes in hexaploid wheat using cultivar-specific long-range chromosome assembly. Nat Biotechnol 35(8):793–796. https://doi.org/10.1038/nbt.3877
Tomato Genome Consortium (2012) The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485:635–641
Trick M, Adamski NM, Mugford SG et al (2012) Combining SNP discovery from next-generation sequencing data with bulked segregant analysis (BSA) to fine-map genes in polyploid wheat. BMC Plant Biol 12:14
Uga Y, Okuno K, Yano M (2011) Dro1, a major QTL involved in deep rooting of rice under upland field conditions. J Exp Bot 62:2485–2494
Uga Y, Sugimoto K, Ogawa S et al (2013) Control of root system architecture by DEEPER ROOTING 1 increases rice yield under drought conditions. Nat Genet 45:1097–1102
Van Gansbeke B, Khoo KHP, Lewis JG, Chalmers KJ, Mather DE (2019) Fine mapping of Rha2 in barley reveals candidate genes for resistance against cereal cyst nematode. Theor Appl Genet 132:1309–1320
Varshney RK (2016) Exciting journey of 10 years from genomes to fields and markets: some success stories of genomics-assisted breeding in chickpea, pigeonpea and groundnut. Plant Sci 242:98–107
Varshney RK, Hoisington DA, Nayak SN, Graner A (2009a) Molecular plant breeding: methodology and achievements. In: Somers D, Langridge P, Gustafson PJ (eds) Methods in molecular biology: plant genomics. Humana, Totowa, pp 283–304
Varshney RK, Nayak SN, May GD, Jackson SA (2009b) Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol 27:522–530
Varshney RK, Chen W, Li Y et al (2012) Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat Biotechnol 30:83–89
Varshney RK, MuraliMohan S, Gaur PM et al (2013a) Achievements and prospects of genomics-assisted breeding in three legume crops of the semi-arid tropics. Biotechnol Adv 31(8):1120–1134
Varshney RK, Song C, Saxena RK et al (2013b) Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 31:240–246
Varshney RK, Thudi M, Nayak SN et al (2014) Genetic dissection of drought tolerance in chickpea (Cicer arietinum L.). Theor Appl Genet 127:445–462
Varshney RK, Shi C, Thudi M et al (2017) Pearl millet genome sequence provides a resource to improve agronomic traits in arid environments. Nat Biotechnol 35:969–97609
Varshney RK, Godwin ID, Mohapatra T, Jones JDG, McCouch SR (2019a) A SWEET solution to rice blight. Nat Biotechnol 37:1280–1282
Varshney RK, Pandey MK, Bohra A et al (2019b) Towards sequence-based breeding in legumes in post-genome sequencing era. Theor Appl Genet 132:797–816
Vu GT, Wicker T, Buchmann JP, Chandler PM, Matsumoto T, Graner A, Stein N (2010) Fine mapping and syntenic integration of the semi-dwarfing gene sdw3 of barley. Funct Int Genom 10:509–521
Wallace JG, Rodgers-Melnick E, Buckler ES (2018) On the road to Breeding 40: unraveling the good, the bad, and the boring of crop quantitative genomics. Annu Rev Genet 52:421–444
Wang H, Nussbaum-Wagler T, Li B et al (2005) The origin of the naked grains of maize. Nature 436:714–719
Wang C, Wen G, Lin X, Liu X, Zhang D (2009) Identification and fine mapping of the new bacterial blight resistance gene, Xa31(t), in rice. Eur J Plant Pathol 123:235–240
Wang Y, Cao L, Zhang Y, Cao C, Liu F, Huang F, Qiu Y, Li R, Lou X (2015) Map-based cloning and characterization of BPH29, a B3 domain-containing recessive gene conferring brown planthopper resistance in rice. J Exp Bot 66:6035–6045
Wang J, Zhang X, Lin Z (2018) QTL mapping in a maize F2 population using Genotyping-by-Sequencing and a modified fine-mapping strategy. Plant Sci 276:171–180
Warr A et al (2015) Exome sequencing: current and future perspectives. G3-Genes Genom Genet 5:1543–1550
Watanabe S, Xia Z, Hideshima R et al (2011) A map-based cloning strategy employing a residual heterozygous line reveals that the GIGANTEA gene is involved in soybean maturity and flowering. Genet 188:395–407
Witek K, Jupe F, Witek KI et al (2016) Accelerated cloning of a potato late blight–resistance gene using RenSeq and SMRT sequencing. Nat Biotechnol 34:656–660
Xie JZ, Wang LL, Wang Y et al (2017) Fine mapping of powdery mildew resistance gene PmTm4 in wheat using comparative genomics. J Integr Agr 16:540–550
Xu K, Xu X, Fukao T et al (2006) Sub1A is an ethylene-response-factor-like gene that confers submergence tolerance to rice. Nature 442:705–708. https://doi.org/10.1038/nature04920
Xu X, Zeng L, Tao Y et al (2013) Pinpointing genes underlying the quantitative trait loci for root-knot nematode resistance in palaeopolyploid soybean by whole genome resequencing. Proc Natl Acad Sci 110:13469–13474
Xue W, Xing Y, Weng X et al (2008) Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat Genet 40:761–767
Yadav P, Saxena KB, Hingane A, Kumar C, Kandalkar VS, Varshney RK, Saxena RK (2019) An “Axiom Cajanus SNP Array” based high density genetic map and QTL mapping for high-selfing flower and seed quality traits in pigeonpea. BMC Genom 20:235
Yan L, Loukoianov A, Tranquilli G, Helguera M, Fahima T, Dubcovsky J (2003) Positional cloning of the wheat vernalization gene VRN1. Proc Natl Acad Sci USA 100:6263–6268
Yang X, Caro M, Hutton SF et al (2014) Fine mapping of the tomato yellow leaf curl virus resistance gene—on chromosome 11 of tomato. Mol Breed 34:749–760
Yang H, Li C, Lam H et al (2015) Sequencing consolidates molecular markers with plant breeding practice. Theor Appl Genet 128:779–795
Yang X, Xia X, Zhang Z et al (2017) QTL Mapping by whole genome re-sequencing and analysis of candidate genes for nitrogen use efficiency in rice. Front Plant Sci 8:1634
Yano M, Katayose Y, Ashikari M et al (2000) Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene Constans. Plant Cell 12:2473–2483
Yoshioka M, Iehisa JCM, Ohno R, Kimura T, Enoki H, Nishimura S, Nasuda S, Takumi S (2017) Three dominant awnless genes in common wheat: fine mapping, interaction and contribution to diversity in awn shape and length. PLoS ONE 12:e0176148
Yu J, Holland JB, McMullen MD, Buckler ES (2008) Genetic design and statistical power of nested association mapping in maize. Genetics 178:539–551
Yu H, Qiu Z, Xu Q et al (2017) Fine mapping of Low Tiller 1, a gene controlling tillering and panicle branching in rice. Plant Growth Regul 83:93–104
Yuan Y, Scheben A, Batley J, Edwards D (2019) Using genomics to adapt crops to climate change. In: Sarkar A, Sensarma S, vanLoon G (eds) Sustainable solutions for food security. Springer, Cham, pp 91–109
Zhang J, Ku L, Han Z et al (2014) The ZmCLA4 gene in the qLA4-1 QTL controls leaf angle in maize (Zea mays L.). J Exp Bot 65:5063–5076
Zhang X, Liu G, Zhang L, Xia C, Zhao T, Jia J, Liu X, Kong X (2018) Fine Mapping of a Novel Heading Date Gene, TaHdm605, in Hexaploid Wheat. Front Plant Sci 9:1059
Zhang H, Wang X, Pan Q, Li P, Liu Y, Lu X, Wang P (2019) QTG-Seq accelerates QTL fine mapping through QTL partitioning and whole-genome sequencing of bulked segregant samples. Mol Plant 12:426–437
Zhao Y, Ma J, Li M et al (2019) Whole-genome resequencing-based QTL-seq identified AhTc1 gene encoding a R2R3-MYB transcription factor controlling peanut purple testa colour. Plant Biotechnol J. https://doi.org/10.1111/pbi.13175
Zhong C, Sun S, Li P et al (2018) Next-generation sequencing to identify candidate genes and develop diagnostic markers for a novel Phytophthora resistance gene, RpsHC18, in soybean. Theor Appl Genet 131:525–538
Zhu BF, Si L, Wang Z et al (2011) Genetic control of a transition from black to straw-white seed hull in rice domestication. Plant Physiol 155:1301–1311
Zhuang W, Chen H, Yang M, Wang J, Pandey MK, Zhang C, Chang WC et al (2019) The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. Nat Genet 51:865–876
Zou G, Zhai G, Feng Q et al (2012) Identification of QTLs for eight agronomically important traits using an ultra-high-density map based on SNPs generated from high-throughput sequencing in sorghum under contrasting photoperiods. J Exp Bot 63:5451–5462
Acknowledgements
The authors are thankful to Australia–India Strategic Research Fund (AISRF) project funded by the Department of Science and Technology (DST) and Tropical Legumes I (TLI) project funded by Bill and Melinda Gates Foundation (BMGF) through CGIAR-Generation Challenge Program (GCP; http://www.generationcp.org/). The Department of Biotechnology (DBT) is greatly acknowledged for providing the grant and fellowship to the first author. This work has been carried out as part of the CGIAR Research Program on Grain Legumes and Dryland Cereals. ICRISAT is a member of the CGIAR Consortium.
Author information
Authors and Affiliations
Contributions
DJ and AB involved in writing; RKV, MT and DJ conceived the concept. All authors read and approved the MS.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical statement
This article does not contain any human or animal studies.
Additional information
Communicated by Albrecht E. Melchinger.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jaganathan, D., Bohra, A., Thudi, M. et al. Fine mapping and gene cloning in the post-NGS era: advances and prospects. Theor Appl Genet 133, 1791–1810 (2020). https://doi.org/10.1007/s00122-020-03560-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-020-03560-w