
Abstract
In this study, the equilibrium distributions of selected trace elements between molten iron-saturated copper alloy and selected iron silicate slags were measured, and the effects of silica fluxing on them. In addition to the copper and iron main components of the system, trace elements like antimony, gallium, germanium, gold, indium, and silver were added in experiments that spanned the temperature range of 1473–1573 K (1200–1300 °C). Experimental charges were quenched and prepared in polished mounts. In situ analyses of the resulting phases were made directly on the mounts without the need of phase separation prior to analysis. Electron probe X-ray microanalysis was used for concentrations at or above approximately 100 ppmw, and laser ablation-inductively coupled plasma-mass spectrometry for the lower concentrations in the slags. The very low slag concentrations of germanium, antimony, and indium obtained indicate that these elements can be removed from the slag by reduction, whereas gallium concentrations in the slag were high. Consequently, gallium removal from iron residues, such as zinc smelting jarosite, is difficult without volatilization. Based on the present observations, the industrial reduction processes for the treatment of smelting and refining slags as well as for the processing of iron residues, and extracting the reducible metal oxides and their metal values can be optimized. The target in fluxing should be to maintain the slag compositions with a silica concentration higher than about 28 wt%.
Similar content being viewed by others
Introduction
Slags produced from the smelting and refining process contain significant quantities of metals that need to be recovered in a separate slag cleaning step [1]. In copper smelting, slag cleaning is performed by either of two major technologies: slag milling and flotation [2], or reduction followed by settling in an electric furnace [3]. In contrast, slags produced in the nickel [4], lead [5], and tin [6] industries typically are cleaned in an electric furnace process using coke reduction. The increasing awareness to properly handle harmful industrial wastes [7] has also increased our attention to various hydrometallurgical iron residues produced in extraction technologies, particularly jarosite produced in zinc smelting [8,9,10,11,12,13,14]. The metals contained in these residues (e.g., Zn, Pb, and Cu) are recovered either from the flue dust or in metal or speiss forming during the reductive treatment of the iron slag.
Fundamental trace element distribution and solubility data concerning the reduction process of iron silicate slags, particularly at iron saturation, are scarce. The limiting phase boundary in the reduction process is the iron saturation surface where the only variable in isothermal conditions is the composition of the slag. In the simplest case of the Fe–O–SiO2 slags, this means its Fe:SiO2 ratio, and in case the slag is modified to enhance metal recoveries, then typically also its CaO:SiO2 and MgO:SiO2 ratios will be of interest. Previous relevant research includes Kim et al. [15] who studied modifiers and minor element behaviors between copper and iron silicate slags. Zhai et al. [16] as well as Banda et al. [17] studied the effects of CaO modification on cobalt recovery from slag. Surapunt et al. [18] measured zinc distributions between copper- and iron-saturated silicate slags in open and closed systems. Zajaczkowski et al. [19] studied the distribution of lead and copper in alumina-saturated flash smelting slag. Limited data are available also for MgO-saturated nickel smelting slags [20,21,22].
The aim of this study was to measure the equilibrium distributions of selected trace components over the temperature range 1473–1573 K (1200–1300 °C) between molten iron-saturated copper alloy and selected iron silicate slags, and to demonstrate the effects on these distributions at different silica flux concentrations (effectively by varying the Fe:SiO2 ratio). This is a fundamental study of reduction smelting of jarosite and other iron residues.
Experimental
The base slag–metal system used in this study, Cu–Fe–O2–SiO2, has four components (denoted as C) in its simplest form, which is increased by one when a trace element is added. Therefore, in the experimental setups, where three phases (denoted as f) are in equilibrium under isothermal and isobaric conditions, the degree of freedom or the number of free variable F is as follows:.
The free variable will be set by the Fe:SiO2 ratio of the slag which will be fixed in each P,T point. The base system without trace elements is thus self-equilibrating and reaches equilibrium without control of the internal atmosphere composition. Consequently, an inert gas was used as the furnace atmosphere. The addition of each trace element adds one degree of freedom to the equilibrium system. The equilibrium was measured experimentally in this study as trace element concentrations of iron-saturated copper alloy and the slag.
The experiments were conducted at temperatures of 1473 K (1200 °C), 1523 K (1250 °C), and 1573 K (1300 °C) using four different Fe:SiO2 ratios (Fe:SiO2 = 3.1, 2.3, 1.8, and 1.5 (w/w)). Each experiment was repeated at each temperature at 1 atm total pressure, giving a total of 29 experiments, including the time series of five annealings for 1 h, 2 h, 3 h, 4 h, and 5 h for determining the necessary equilibration time.
Prior to the experiments, Fe2O3 was reduced by heating 10 g of Fe2O3 in a quartz crucible in a vertical tube furnace at 800 °C for 14.5 h in controlled \( {\text{PO}}_{2}\), obtained using CO (purity ≥ 99.97 vol%; Aga-Linde, Finland) and CO2 (purity ≥ 99.9993 vol%; Aga-Linde, Finland) gases with a volumetric ratio of 50:50 (flow rates for both gases were 100 mL/min (STP) during the experiments). The resulting material was analyzed by XRD using XPertPro software (Panalytical, The Netherlands) showing only peaks for FeO. Visually, the reddish color of Fe2O3 had changed to black after the reduction. The reduction of Fe2O3 decreases the evolution of oxygen during the initial periods of the equilibration experiments and shortens the equilibration time.
An iron crucible (length ≈ 15 mm, outer diameter of 5.8 mm or less) was prepared from 6 mm pure Fe rod, see Table 1, by cutting and drilling into a crucible shape. The iron-saturated iron–silicate based slag mixture was prepared by mixing prereduced FeO and SiO2 powders to form a slag, which initially contained 20, 25, 30, and 35 wt% SiO2. In addition, the trace elements Ag, Au, Ga (as Ga2O3), Ge, In, and Sb were simultaneously added as powders into the copper–slag mixtures, each assaying 1 wt% of the mass of copper. The slag–alloy mixture containing the trace elements and slag weighed 0.2 g each, and thus in the iron crucible, their corresponding weight ratio was initially 1:1.
The semisealed ampoule was formed from a quartz tube (8 mm OD, 6 mm ID; Heraeus: grade HSQ 300) by a hydrogen–oxygen torch. A 1.9 mm diameter hole was drilled into the ampoule 40 mm above the bottom of the tube using a dentist’s drill. The iron crucible containing the sample was inserted into the tube. Subsequently, the quartz tube was cut to a length of 50–80 mm, and a hook for suspension was formed on the top-end using a hydrogen–oxygen flame. This operation left the drilled hole as the only opening into the ampoule to the surrounding furnace atmosphere.
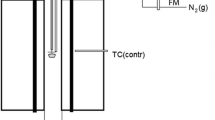
In this method, we assumed that the semiclosed ampoule forms a kinetic barrier for the volatile elements but allows equilibration with, e.g., the gaseous CO–CO2 mixtures if needed. Figure 1a shows a schematic of the semisealed ampoule arrangement containing the iron crucible with the sample in it. During equilibration, the gas phase above the liquid slag within the ampoule saturates with the volatile elements. The small hole decreases the removal rate of the saturated gas phase, by Knudsen-like gas diffusion through the hole, while the saturated gas phase reduces transfer of the volatile elements from the condensed phases into the gas flow. Thus, it was expected that the volatile elements equilibrate between slag and alloy without concentration gradients, and remain in the sample during annealing and after quenching in sufficient concentrations for in situ analysis with EPMA or LA-ICP-MS. In equilibrium, the distribution coefficients of the metals between the copper alloy, slag and metallic Fe, remains the same regardless of slow evaporation of the metals, provided the diffusional mass transfer in the slag and alloy are faster than the volatilization. Figure 1b illustrates basics of the new concept.

a A schematic of the cross section of the semiclosed silica ampoule, iron crucible. and the slag–(CuFe) alloy sample. b Schematic of the semiclosed gas phase (p is partial pressure of a species) arrangement and the expected behavior of volatiles in the experimental technique used
When pMe,sl, pM,M < pMe,inside, mass transfer and volatilization of the metal/oxide to the surrounding furnace atmosphere will occur, but it is assumed to be slow compared to vaporization processes on the slag surface. When pMe,inside < pMe,outside, the metal/oxide-containing gas moves outside the ampoule through the hole. In this experimental arrangement, the molten alloy–solid alloy equilibrium with FeO in the slag forms the prevailing oxygen pressure in the ampoule.
The equilibration experiments were conducted in a vertical tube furnace under N2 (purity ≥ 99.999 vol%; Aga-Linde, Finland) atmosphere using a similar setup and method as described earlier [23]. The semisealed ampoule containing the sample was introduced into the furnace work tube from its lower end and suspended there with a 0.5 mm Pt-wire. The ampoule was held in the low end, while the furnace was sealed, and the gas atmosphere flushed with flowing nitrogen for 30 min, at a flow rate of 200 mL/min. After flushing, the ampoule was raised to the hot zone of the furnace, equilibrated and quenched into a 273.15 K (0 °C) ice–water mixture. During the experiments, temperature of the sample was monitored using a calibrated, S-type (Pt/Pt90 + Rh10; ± 2 °C) thermocouple (Johnson-Matthey, UK).
Time required for equilibration was determined experimentally as 2 h based on changes in the slag composition. After quenching, the sample was dried, mounted into epoxy, cut, and polished using conventional wet metallographic methods. The sample surface was made conductive with carbon coating by evaporation.
Microstructure observations and preliminary analysis of chemical compositions of the phases were obtained using a Tescan MIRA 3 Scanning Electron Microscope (Tescan, Czech Republic) equipped with an UltraDry silicon drift energy dispersive spectrometer and a NSS microanalysis software (Thermo Fischer Scientific, USA). The accurate chemical phase compositional analyses were carried out by EPMA using a Cameca SX100 (Cameca SAS, France). The EPMA was operated using 20 kV accelerating voltage, 40 nA beam current, and 50–100 µm defocused electron beam. The external standard materials and analyzed lines used in the EPMA were: O Kα (hematite), Si Kα (quartz), Fe Kα (hematite), Cu Kα (Cu), Ag Lα (Ag), Sb Lα (SbTe), Au Lα (Au), Ga Kα (GaAs), Ge Kα (Ge), and In Lα (InAs). The analytical results of the primary EPMA WDS data were corrected using a PAP-based online matrix correction software [24] by Cameca.
The concentrations of all trace elements, apart from gallium, in the slag were below the detection limits of EPMA. Therefore, the slag phase was also analyzed with LA-ICP-MS technique. A Photon Machines Analyte Excite 193 nm 4 ns ArF laser ablation device (Teledyne CETAC Technologies, USA), coupled with a Nu AttoM single-collector ICP-MS (Nu Instruments Ltd., UK), housed at Geological Survey of Finland, was utilized. The laser energy was set to 30% of 5.0 mJ, resulting in a fluence of 2.5 J/cm2 on the sample surface. A spot size of 85 µm and operating frequency of 10 Hz were selected. The estimated detection limits by the equipment are collected in Table 2.
The mass spectrometer was operated in FastScan mode in low resolution (ΔM/M = 300) for higher sensitivity. An analysis protocol of 5 preablation pulses for removal of the carbon coating and possible contaminants from the surface, 20-s pause, 25-s gas background analysis, and 400 ablation pulses was adopted. At least six points were analyzed from the slag phase of each sample, and the averages and their standard deviations were used in the following chapters.
NIST 612 SRM [25] was used as the external standard and 29Si as the internal standard for analyses. NIST 610 SRM, USGS BHVO-2G, and BCR-2G reference glasses were analyzed as unknowns for monitoring data quality. The data collected from the mass spectrometer were analyzed using GLITTER software [26]. The trace element concentrations obtained by LA-ICP-MS and presented in this article have been calculated using the isotopes: 72,73Ge, 107,109Ag, 115In, 121,123Sb, and 197Au.
Results and Discussion
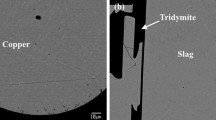
A typical microstructure of a quenched sample is shown in Fig. 2. The liquid Cu and slag are clearly visible and present in large areas. Microstructures in slow quenched areas of the samples show minor precipitation, more pronounced than in experiments where small, open crucibles were used [27]. Very small Fe3O4 precipitates were common in the slag, and the copper alloy in thick parts of the specimen indicated severe segregation. The well-quenched areas, showing microcrystalline or glassy structure in the SEM micrographs, were selected for the EPMA and LA-ICP-MS measurements.

Microstructures of slag–metal–iron crucible interfaces after equilibration and quenching from 1473 (1200) and 1573 K (1300 °C) (white-Cu alloy, light gray-Fe(s) and dark gray-slag)
Solubilities of the Base Metals
The final concentration of copper in reducing conditions of the slag cleaning at copper saturation is not at all well known in the literature. That endpoint is on the iron saturation boundary in the Fe–O–SiO2 system. Therefore, the copper concentration of the iron silicate slag was measured as a function of silica concentration of the slag at 1473 (1200) to 1573 K (1300 °C), in saturation with solid iron. The results are shown in Fig. 3 as a function of measured silica concentration of the slag, calculated from the EPMA data. A comparison of the present observations with Surapunt et al. [18] shows excellent agreement at 1200–1250 °C.

Copper solubility in Fe–O–SiO2 slags at iron and copper saturations at 1473–1573 K (1200–1300 °C); data by Surapunt et al. from [18]
The solubility of iron in liquid copper in these experiments was constantly controlled in the presence of the solid iron crucible that held the experimental charges. Therefore, the composition of the slag or its iron oxide activity does not affect the solubility of iron in the molten copper-based alloy. The present results with the individual standard uncertainty of each point (1 × σ) are shown in Fig. 4.

Solubility of iron in molten copper at iron saturation in the present experiments at 1473–1573 K (1200–1300 °C)
Solubility of iron in liquid copper was also measured in this slag–copper alloy–solid iron equilibrium system, which contained 2.6–2.9 wt% other alloying elements in the metallic copper phase. The average values obtained were 4.0, 5.5 and 7.4 wt% [Fe]Cu at temperatures 1473 K (1200 °C), 1523 K (1250 °C) and 1573 K (1300 °C), respectively. The values are in good agreement with the experimental data from the pure binary Cu–Fe system by Oelsen et al. [28], which has been evaluated as the most reliable experimental dataset by several critical assessments [29,30,31]. It is evident that in addition to silver and gold [32, 33], also other alloying elements of copper increase the activity coefficient of iron in copper alloys, which explains the approximately 0.5 wt% lower iron solubilities measured in the present case, compared to the data of Oelsen et al. [28] from pure binary copper–iron alloys.
Solubilities of the Trace Elements in the Slag
The equilibrium concentrations of trace elements in the slag at copper and iron saturation predict the lowest concentrations achievable in the reduction process of slag cleaning with copper alloy as the collector metal. Thus, they can be regarded as the limiting minimum values in the industrial operations as well, see, e.g., Hughes [34]. Additionally the selected slag-to-metal ratio determines the maximum recoveries in the collector metal, in the absence of volatilization [35].
The trace element solubilities in the slags from Cu + 1 wt% Me copper alloy obtained in this study indicate a general decreasing trend when silica concentration is increased. This means that the losses of valuable metals increase when the slag used in the smelting is more iron oxide rich than the orthosilicate composition of the Fe–O–SiO2 system or have a Fe:SiO2 > 1.86 (w/w) or 29.5 wt% SiO2.
The solubilities of silver and gold in the slag as a function of silica concentration at various temperatures are shown in Figs. 5 and 6. The experimental uncertainties of silver concentrations were high in some samples, but no obvious reason was found in their micrographs. The silver concentration obtained at iron saturation is relatively high compared to the other precious and platinum group metals [36], between 5 and 20 ppm. Gold dissolved in iron silicate slags much less, and its concentration was always below 0.3 ppm. Slight tendencies toward lower solubilities at low temperatures can be recognized in the present experimental observations. The obtained gold concentration is much lower than earlier data on metallurgical slags [37] if recalculated to the solubility values of pure gold.

Concentrations of silver in iron silicate slag at copper and iron saturations at 1473–1573 K (1200–1300 °C)

Concentration of gold in iron silicate slags under copper- and iron saturation conditions at 1473–1573 K (1200–1300 °C)
The concentrations of germanium and indium in the slag at iron saturation were in the ranges of 10–30 ppm and 60–70 ppm, respectively. The experimental data at 1200–1300 °C are shown in Figs. 7 and 8 and plotted as a function of the measured silica concentrations of the iron silicate slag. A sample at 1200 °C had higher uncertainties than the other experiments, but no indication of the source of high scatter could be identified from its microstructure. A solubility study at 1500 °C and carbon–CO(g) saturation in iron-free alumina–lime silicate melts [38] suggests much smaller indium concentrations, but its oxygen partial pressure was much lower, \( \log_{10} {\text{PO}}_{2} /{\text{atm}} = - 16\,{\text{to}}\, - 17,\) than that in the present study (Figs. 7, 8).

Concentration of germanium in iron silicate slag at copper and iron saturation

Concentration of indium in iron silicate slag at copper and iron saturation
The concentration of antimony in slag at iron saturation obtained in this study was around 1–4 ppm (Fig. 9). Copper was not effective collector for gallium, and its concentration in the slag remained at as high as around 2000 ppm or 0.2 wt% (Fig. 10). The obtained concentration of antimony is much lower than reported by Goto et al. [39] at 1200 and 1250 °C in silica-saturated iron silicate slags, close to iron saturation of about 6 wt% Fe3O4 in the slag.

Concentration of antimony in iron silicate slag at copper and iron saturation

Concentrations of gallium in iron silicate slags at copper and iron saturations; EPMA data
Distribution Coefficients for the Trace Elements
The distribution coefficient of an element Me in metal alloy–slag systems is defined in the current study as
where [] refers to the metal alloy (m) and () to the iron silicate slag (s) melt. Its relationships with the thermodynamic properties of element Me in the equilibrium phases have been discussed earlier elsewhere [40, 41].
Due to the relatively high solubility of silver in iron silicate slags, even at metallic iron saturation, its distribution coefficient is much less than that of gold, see Figs. 11 and 12. The experimental scatter in the distribution coefficient values is relatively large. It suggests evident post quenching mass transfer, due to insufficient quenching rate of the sample and as consequence of the large mass of the sample-ampoule arrangement. The distribution coefficient values obtained for silver, however, are clearly higher than obtained in earlier open crucible arrangements, e.g., Yazawa [40], Avarmaa et al. [27] and Sukhomlinov et al. [42]. This fact is also supported by the present high concentrations of silver measured in the slag and alloy compared with the fully open experimental assemblies where Ag volatilization was significant [27].

Distribution coefficients of silver between copper and slag at iron saturations

Distribution coefficients of gold between copper and slag at iron saturations
A small temperature dependence is also found in the present results for the distribution coefficients of silver and gold so that low temperatures favor trace element deportment in the metal alloy, except at low silica concentrations.
The distribution coefficients of gallium and indium are shown in Figs. 13 and 14. A clear dependence of the Fe:SiO2 ratio can be observed in the case of gallium, but it is very small for indium. Also the temperature dependencies follow a similar pattern. A high processing temperature seems to favor the deportment of gallium in copper alloy, but in the case of indium, temperature has only a small impact on the distribution equilibria. According to the solubility measurements by Ko et al. [38], silica increases the solubility in iron-free silicate slags which is an opposite trend to that found in the present study.

Distribution coefficients of gallium between copper and slag at iron saturations; EPMA data

Distribution coefficients of indium between copper and slag at iron saturations
The copper–slag distribution coefficients for germanium and antimony are presented in Figs. 15 and 16. Both elements distribute predominantly in the copper alloy with very high distribution coefficients of a few hundreds and thousands, respectively. Also the influence of the slag Fe:SiO2 ratio is significant in both cases. The obtained distribution coefficients for antimony are on an order of magnitude larger than that reported by Goto et al. [39] at 1200–1250 °C and silica saturation, but well in line with a recent publication from this group [43]. Shuva et al. [44] compared the previous germanium distribution experiments in copper and lead smelting slags but misinterpreted the data by Yan and Swinbourne [45]. The present observations indicate a stronger deportment to the metal alloy compared to the earlier data at MgO saturation and 5–19 wt% CaO. The evident reasons for this are the slow cooling post equilibration and concentration gradients in a system where most germanium was lost during the equilibration. The present observations are, however, in good agreement with geochemical data [46].

Distribution coefficients of germanium between copper and slag at iron saturations at 1473–1573 K (1200–1300 °C)

Distribution coefficient of antimony between copper and slag at iron saturations at 1473–1573 K (1200–1300 °C)
A part of the FeO–Fe2O3–SiO2 isothermal section at 1573 K (1300 °C) in ferric oxide lean systems is shown in Fig. 17, where the corners of the triangle have been defined as 90 wt% Fe + 10 wt% SiO2, 40 wt% FeO + 60 wt% SiO2, and 40 wt% FeO + 50 wt% Fe2O3 + 10 wt% SiO2. In addition to phase boundaries, based on Mtox database of MTDATA software [47], calculated isoactivity contours of FeO and SiO2 have been superimposed on the graph.

Calculated isoactivity contours of silica and ferrous oxide in the system FeO-Fe2O3-SiO2 at 1573 K (1300 °C) and in 1 atm total pressure (dotted lines) SiO2 and (continuous line) FeO (standard states: SiO2(s, tridymite) and FeO(l); Mtox database [47])
As Fig. 17 indicates, the properties of the slag ‘matrix’ change significantly when its composition is moved from halite (FeO) saturation of low silica toward the silica-saturated assays, particularly at iron saturation where its silica activity is increased from about a(SiO2) = 0.15 to 1 (with solid tridymite as standard state). At the same time, the activity of ferrous oxide decreases from about a(FeO) = 0.8 to 0.35 (referred to pure, liquid FeO). In Fig. 17, the isoactivity contours of ferrous oxide (from 0.3 to 0.8) are reproduced with labels of two decimals and those of silica (from 0.150 to 0.950) with three decimals.
Based on those fundamental features, it is easy to understand the obtained trends of the trace element concentrations in the iron silicate slags at iron saturation. Due to the experimental setup, the reduction process of the oxide of metal Me and solubility is controlled by the equilibrium:
On the raw material side, the only variable is the activity of the metal oxide, and thus its final concentration in the slag, whereas on the product side, low activity coefficient of Me in copper and low activity of FeOx in the slag promote the depletion of the slag from the trace element. It allows for generation of a stable slag without excessive leaching of its harmful metal content [48, 49]. An excessive addition of silica must be avoided, due to, e.g., the increasing viscosity of the slag [50] and the decreasing reaction rates.
Conclusions
Molten silicate solutions are nonideal solution phases, due to the strong polymerization or associates formed by SiO2 with basic oxides, such as CaO and MgO, but also with weaker silicate formers, e.g., CoO, FeO, and NiO [51]. This implies that no free basic oxides, or their cations, are present in the silicate slag compositions rich in SiO2, and thus, the thermodynamic activity coefficients of the basic components and, in particular, strong silicate formers under these conditions are very much below unity.
The residual concentrations of precious metals, germanium, and antimony as well as indium obtained at iron saturation, i.e., in highly reducing conditions, indicate that they can be recovered from the slag almost completely, but gallium concentrations in the slag at iron saturation were high. Its removal from iron residues, such as jarosite from zinc smelting, with a reasonable yield is thus difficult without volatilization during processing.
The results indicate the key importance of the Fe:SiO2 ratio of the iron silicate slag to the residual concentration of a trace element in the endpoint of slag cleaning by chemical reduction. With strong silicate formers as trace elements, more basic fluxing components are needed to release the trace element from the slag. A typical example for this is lead smelting where lime is used for increasing lead recoveries from the slag. The solubilities of the minor components grow significantly when the slag assay is more basic than the orthosilicate composition (e.g., Fe2SiO4), all ‘bridging oxygen’ has been consumed in the silicate network by the basic oxide for the formation of SiO44− tetrahedra, and ‘free’ iron oxides are present in the molten silicate structure [52].
The industrial reduction technologies used for processing of iron residues and extracting reducible metal oxides and their metal values require a proper fluxing. Based on the present observations on the iron-saturated iron silicate slags, the target should be to maintain the slag compositions higher than about 28 wt% SiO2 by fluxing with silica. Also the residual concentration of lead from the iron residue is low in the silicate slag under those conditions [23].
It is obvious that several trace metals tend to vaporize during the smelting process [53] and are transferred to the off-gas stream where they form flue dust at low temperatures, in the heat-recovery boiler and the subsequent electrostatic precipitator.
References
Shen H, Forssberg E (2003) An overview of recovery of metals from slags. Waste Manag 23(10):933–949
Subramanian K, Themelis N (1972) Copper recovery by flotation. JOM 24(4):33–38
Demetrio S, Ahumada J, Durán M, Mast E, Rojas U, Sanhueza J, Reyes P, Morales E (2000) Slag cleaning: the Chilean copper smelter experience. JOM 52(8):20–25
Warner AE, Diaz C, Dalvi A, Mackey P, Tarasov A, Jones R (2007) JOM world nonferrous smelter survey Part IV: nickel: sulfide. JOM 59(4):58–72
Willis GM (1980) The physical chemistry of lead smelting. In: Cigan JM, Mackey P, O’Keefe TJ (eds) Proceedings of lead-zinc-tin ’80. The Metallurgical Society of AIME, Warrendale, pp 457–476
Floyd J (1980) The physical chemistry of tin smelting. In: Cigan JM, Mackey P, O’Keefe TJ (eds) Proceedings of lead-zinc-tin ’80. The Metallurgical Society of AIME, Warrendale, pp 508–531
Navarro A, Cardellach E, Mendoza J, Corbella M, Domènech L (2008) Metal mobilization from base-metal smelting slag dumps in Sierra Almagrera (Almeriá, Spain). Appl Geochem 23(4):895–913
Choi CY (1996) Evolution of iron separation processes in zinc smelting at Korea Zinc. In: Dutrizac J, Harris G (eds) Iron control and disposal. CIM, Montreal, pp 199–210
Esna-Ashari M (1966) Thermische Behandlung von zinkhaltigen Jarosit-Rückständen im CONTOP-Schmeltzzyklon. Erzmetall 49(5):314–321
Peacey J, Hancock P (1996) Review of pyrometallurgical processes for treating iron residues from electrolytic zinc plants. In: Dutrizac J, Harris G (eds) Iron control and disposal. CIM, Montreal, pp 17–35
Choi CY, Lee YH (1999) Treatment of zinc residues by Ausmelt Technology at Onsan zinc refinery. In: Gaballah I, Hager J, Solozbal R (eds) Proceedings of REWAS ’99. TMS, Warrendale, pp 1613–1622
Kim B, Jeong S, Lee J, Shin D, Moon N (2012) Behaviors of lead and zinc in top submerged lance (TSL) plant at Sukpo zinc refinery. Mater Trans 53(5):985–990
Creedy S, Glinin A, Matusewicz R, Hughes S, Reuter M (2013) Outotec Ausmelt technology for treating zinc residues. Erzmetall 66(4):230–235
Wang X, Xie K, Ma W, Yang M, Zeng P, Cao Y (2013) Recovery of zinc and other valuable metals from zinc leaching residue by top blowing fuming method. Miner Process Extr Metall 122(3):174–178
Kim G, Sohn H (1998) Effects of CaO, Al2O3, and MgO additions on the copper solubility, ferric/ferrous ratio, and minor-element behavior of iron-silicate slags. Metall Mater Trans B 29B(3):583–590
Zhai X, Li N, Zhang X, Fu Y, Jiang L (2011) Recovery of cobalt from converter slag of chambishi copper smelter using reduction smelting process. Trans Nonferrous Met Soc China 21(10):2117–2121
Banda W, Morgan N, Eksteen J (2002) The role of slag modifiers on the selective recovery of cobalt and copper from waste smelter slag. Miner Eng 15(11):899–907
Surapunt S, Takeda Y, Itagaki K (1995) Dissolution of zinc in CaO-SiO2-FeOx slag coexisting with liquid Cu-Zn-Fe (iron-saturation) alloy. Shigen-to-Sozai 111(8):553–558
Zajaczkowski A, Botor J, Czernecki J (2006) Thermodynamics of copper and lead in alumina saturated flash smelting slag. Can Metall Q 43(3):417–430
Henao H, Hino M, Itagaki K (2001) Distribution of Ni, Cr, Mn, Co and Cu between Fe-Ni alloy and FeOx-MgO-SiO2 base slags. Mater Trans 42(9):1959–1966
Li G, Tsukihashi F (2001) Distribution equilibria of Fe Co, and Ni between MgO-saturated FeOx-MgO-SiO2 slag and Ni alloy. ISIJ Int 41(11):1303–1308
Kitamura S, Kuriyama H, Maruoka N, Yamaguchi K, Hasaegawa A (2008) Distribution of cobalt between MgO-saturated FeOx-MgO-CaO-SiO2 slag and Fe-Cu-Co molten alloy. Mater Trans 49(11):2636–2641
Hellstén N, Taskinen P, Johto H, Jokilaakso A (2018) Trace metal distributions in nickel slag cleaning. In: Davis B, Moats M, Wang S (eds) Extraction 2018: proceedings of the first global conference on extractive metallurgy. TMS, Pittsburgh, pp 379–389
Pouchou JL, Pichoir F (1986) Basic expression of “PAP” computation for quantitative EPMA. In: Brown JD, Packwood RH (eds) Proceedings of the 11th international congress on X-ray optics and microanalysis (ICXOM). University of Western Ontario, Ontario, pp 249–256
Jochum KP, Weis U, Stoll B, Kuzmin D, Yang Q, Raczek I, Jacob DE, Stracke A, Birbaum K, Frick DA, Günther D, Enzweiler J (2011) Determination of reference values for NIST SRM 610-617 glasses following ISO guidelines. Geostand Geoanal Res 35(4):397–429
Van Achterberg E, Ryan C, Jackson S, Griffin W (2001) Data reduction software for LA-ICP-MS. Laser ablation ICP-MS in the earth science. Mineralogical Association of Canada, St John, pp 239–243
Avarmaa K, O’Brien H, Taskinen P (2016) Equilibria of gold and silver between molten copper and FeOx-SiO2-Al2O3 slag in WEEE smelting at 1300 °C. In: Reddy R, Chaubal P, Pistorius PC, Pal U (eds) Advances in molten slags, fluxes, and salts: proceedings of the 10th international conference on molten slags, fluxes, and salts. TMS, Pittsburgh, pp 193–202
Oelsen W, Schürmann E, Florin C (1961) Zur Kalorimetrie und Thermodynamik der Eisen-Kupfer-Legierungen. Arch Eisenhüttenwes 32(10):719–728
Chen Q, Jin Z (1995) The Fe-Cu system: a thermodynamic evaluation. Metall Mater Trans A 26A(2):417–426
Turchanin M, Agraval P, Nikolaenko I (2003) Thermodynamics of alloys and phase equilibria in the copper-iron system. J Phase Equilib 24(4):307–319
Shubhank K, Kang Y (2014) Critical evaluation and thermodynamic optimization of Fe-Cu, Cu-C, and Fe-C binary systems and Fe-Cu-C ternary system. CALPHAD 45:127–137
Arita M, Tanaka M, Goto K, Someno M (1981) Activity and diffusivity measurements of copper in γ and δ Fe by equilibration between solid Fe and liquid Ag. Metall Trans A 12A(3):497–504
Capobianco C, Drake M, de Aro J (1999) Siderophile geochemistry of Ga, Ge and Si: cationic oxidation states in silicate melts and the effect of composition in iron-nickel alloys. Geochim Cosmochim Acta 43(17):2667–2672
Hughes S (2000) Applying Ausmelt technology to recover Cu, Ni, and Co from slags. JOM 52(8):30–33
Wegscheider S, Steinlechner S, Leuchtenmüller M (2016) Innovative concept for the recovery of silver and indium by a combined treatment of jarosite and electric arc dust. JOM 69(2):388–394
Borisov A, Palme H (2000) Solubilities of noble metals in Fe-containing silicate melts as derived from experiments in Fe-free systems. Am Miner 85(11–12):1665–1673
Swinbourne DR, Yan S, Salim S (2005) The solubility of gold in metallurgical slags. Miner Process Extr Metall 114(1):23–29
Ko KY, Park JH, Hong SH (2011) Dissolution mechanism of indium in molten CaO-SiO2-Al2O3 slag. In: Kongoli F (ed) Fray international symposium, vol 1. Flogen, Mont-Royal, pp 287–298
Goto S, Ogawa O, Inoue Y, Ohara H (1979) On the equilibria between Cu-Sb alloys and silica-saturated iron silicate slags. Shigen-to-Sozai 95(4):205–211
Yazawa A (1980) Distribution of various elements between copper, matte and slag. Erzmetall 33(7–8):377–382
Piskunen P, Avarmaa K, O’Brien H, Klemettinen L, Johto H, Taskinen P (2017) Precious metals distributions in direct nickel matte smelting with low-copper mattes. Metall Mater Trans B 49B(1):98–112
Sukhomlinov D, Taskinen P (2017) Distribution of Ni, Co, Ag, Au, Pt and Pd between copper metal and silica saturated iron silicate slag. In: Proceedings of the EMC 2017, June 25–28, Leipzig, vol 3. GDMB, Clausthal-Zellerfeld, pp 1029–1038
Klemettinen L, Avarmaa K, O’Brien H, Taskinen P, Jokilaakso A (2019) Behavior of tin and antimony in secondary copper smelting process. Minerals 9(1):39
Shuva MAH, Rhamdhani MA, Brooks GA, Masood S, Reuter MA (2016) Thermodynamics behavior of germanium during equilibrium reactions between FeOx-CaO-SiO2-MgO slag and molten copper. Metall Mater Trans B 47B(5):2889–2903
Yan S, Swinbourne DR (2003) Distributions of germanium under lead smelting conditions. Miner Process Extr Metall 112(2):75–80
Holzapfel C, Courtial P, Dingwell DB, Chakraborty S, Palme H (2001) Experimental determination of partial molar volumes of Ga2O3 and GeO2 in silicate melts: implications for the pressure dependence of metal-silicate partition coefficients. Chem Geol 174(1):33–49
Gisby J, Taskinen P, Pihlasalo J, Li Z, Tyrer M, Pearce J, Avarmaa K, Björklund P, Davies H, Korpi M, Martin S, Pesonen L, Robinson J (2017) MTDATA and the prediction of phase equilibria in oxide systems: 30 years of industrial collaboration. Metall Mater Trans B 48B(1):91–98
Pisciella P, Crisucci S, Karamanov A, Pelino M (2001) Chemical durability of glasses obtained by vitrification of industrial wastes. Waste Manag 21(1):1–9
Mombelli D, Mapelli C, Cecca C, Barella S, Gruttadauria A, Ragona M, Pisu M, Viola A (2018) Characterization of cast iron and slag produced by jarosite sludges reduction via arc transferred plasma (ATP) reactor. J Environ Chem Eng 6(1):773–783
Mills K (2011) The estimation of slag properties. In: Jones RT, Hoed P (eds) Southern African pyrometallurgy 2011. The SA Institution of Mining and Metallurgy, Johannesburg, pp S1–S52
Gaskell D (1981) J Tien, J Elliott (eds) The thermodynamic properties and structures of slags. Metallurgical treatises. The Metallurgical Society of AIME, Warrendale, pp 59–77
Masson C (1984) The thermodynamic properties and structures of slags. In: Fine H, Gaskell D (eds) Proceedings of the 2nd international symposium metallurgical slags and fluxes. TMS, Warrendale, pp 3–44
Avarmaa K, Yliaho S, Taskinen P (2018) Recoveries of rare elements Ga, Ge, In and Sn from waste electric and electronic equipment through secondary copper smelting. Waste Manage 71(1):400–410
Acknowledgements
Open access funding provided by Aalto University. The authors are indebted to the Business Finland (former Tekes) and Boliden Kokkola for financial support within the framework of the program CM Eco (Grant # 7405/we31/2016). The EPMA analyses were carried out by Mr Lassi Pakkanen of GTK (Geological Survey of Finland, Espoo).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
The contributing editor for this article was Markus Reuter.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hellstén, N., Klemettinen, L., Sukhomlinov, D. et al. Slag Cleaning Equilibria in Iron Silicate Slag–Copper Systems. J. Sustain. Metall. 5, 463–473 (2019). https://doi.org/10.1007/s40831-019-00237-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40831-019-00237-7