
Abstract
The synthesis of α-linolenic acid (ALA) requires the activity of ω-3 fatty acid desaturases (ω-3 FADs). The quality of peanut oil would be much improved if the content of ALA could be increased. A scan of the peanut genome revealed that it harbored 36 FAD genes, mapping to 16 of the species’ 20 chromosomes. A phylogenetic analysis concluded that these genes belonged to six sub-families, namely stearoyl-acyl-acyl carrier protein desaturases (SAD), FAD2, FAD3, FAD4/5, FAD6 and FAD7/8. Of these, FAD3 and FAD7/8 encoded ω-3 FADs, while genes belonging to the other four sub-families encoded ω-6 FADs. Based on RNA-Seq data, each of the 36 FAD genes was shown to be transcribed in non-stressed plants, but there was variation between them with respect to which organs they were transcribed in. Four ω-3 AhFAD3 genes were functionally characterized; when expressed in Arabidopsis thaliana protoplasts, each was localized mainly in the endoplasmic reticulum, while within peanut, the genes were more strongly transcribed in the developing seed than in either the root or the leaf. When constitutively expressed in Arabidopsis thaliana, both the total fatty acid content of the seed and the relative contribution of ALA were increased. The transgenic seedlings also exhibited an improved level of survival when challenged by salinity stress.
Similar content being viewed by others


Introduction
The seeds of peanut (Arachis hypogaea L.) represent an important source of culinary oil, the fatty acid content of which is dominated by oleic acid (C18:1, about 50% of the total oil content) and linoleic acid (C18:2, about 30% of the total oil content); its content of linolenic acid (C18:3) is very low (about 0.1%) (Li et al. 2012). α-linolenic acid (ALA) is an essential fatty acid in the human diet as it cannot be synthesized in the body, since humans lack any ω-3 fatty acid desaturases (ω-3 FADs) (Baker et al. 2016). Increasing the ALA content of peanut oil therefore represents a potential means of improving its quality.
In plant cells, ALA represents not only a component of storage and membrane lipids, but also is a precursor of the phytohormone jasmonic acid, which is central to the plant response to abiotic and biotic stress (Weber 2002). Several FAD3 and FAD7/8 genes have been identified in plants (Arondel et al. 1992; Chi et al. 2017; Hernández et al. 2016; Radovanovic et al. 2014; Román et al. 2012; Teixeira et al. 2010; Venegas-Calerón et al. 2010; Vrinten et al. 2005; Xue et al. 2018; Yang et al. 2012; Yurchenko et al. 2014). A number of examples have been presented which confirm that the activity of ω-3 FADs affects the plant stress response: these include the up-regulation by low temperature of a FAD gene in Arabidopsis thaliana plants (Chen et al. 2018; Gibson et al. 1994) and a demonstration that the over-expression of an ω-3 FAD improves chilling tolerance in tomato (Yu et al. 2009). Many FAD genes are known to experience alternative splicing (AS), a post-transcriptional phenomenon which is important for plant growth, development, signal transduction, flowering and the response to various environmental cues (Blencowe 2006; Reddy et al. 2013; Remy et al. 2014; Stamm et al. 2005; Tang et al. 2016; Yang et al. 2014; Zhang et al. 2016). For example, in soybean, the abundance of specific splicing variants of both GmFAD3A and GmFAD7 respond to low temperature stress (Román et al. 2012).
The genome sequences of the diploid progenitors of cultivated peanut (A. duranensis and A. ipaensis) (Bertioli et al. 2016; Chen et al. 2019), along with that of peanut itself, have recently been acquired (Bertioli et al. 2019; Zhuang et al. 2019), which now enables a systematic analysis of the complement of peanut FAD genes (AhFADs). In addition, four ER-located ω-3 AhFAD3 genes have been selected for an analysis of their gene structure and function.
Materials and Methods
Plant Materials
The peanut cultivar (cv.) ‘Fenghua1’ was used for gene cloning and the A. thaliana ecotype Col-0 for transformation.
Sequence Analysis of FADs
The complement of FAD genes in the genome of cultivated peanut was identified by using “fatty acid desaturase” as a search string in a search of www.peanutbase.org/ (Bertioli et al. 2019). Gene structures were derived using GSDS software (http://www.gsds.cbi.pku.edu.cn/), while TMHMM software (http://www.cbs.dtu.dk/services/TMHMM/) was used to locate transmembrane domains and ProtComp 9.0 (http://www.softberry.com/berry.phtml?topic=protcomppl&group=programs&subgroup=proloc) to predict the sub-cellular localization of gene products.
Phylogenetic Analysis
The Phytozome database (http://phytozome.jgi.doe.gov/pz/portal.html) was scanned for plant FAD protein sequences using “FAD” as a search string. Following the removal of incomplete sequences, an alignment was performed using the ClustalW program (http://www.genome.jp/tools-bin/clustalw). Mega6 software was used to construct a plant FAD phylogeny, based on the neighbor-joining method; statistical confidence in the clade branching points was obtained by running 1000 bootstrap replicates (Hall 2013).
Transcription Profiling Based on RNA-Seq Data
The abundance of AhFAD transcripts was estimated from an RNA-Seq based peanut transcriptome (NCBI Sequence Read Archive SRP093901). The four sources of cDNA used for this purpose were prepared from the root and leaf of 12 day old cv. ‘Fenghua 1’ seedlings, and from seed of the same cultivar harvested 30 days after flowering (seed1) and 50 days after flowering (seed2). After the removal of adapter sequence and low quality reads, the remaining sequence data were aligned with the genome sequence of cv. ‘Tifrunner’ (www.peanutbase.org/peanut_genome) (Bertioli et al. 2019) using TopHat2 software (Kim et al. 2013). The aligned reads were assembled using the Cufflinks v2.2.1 program, and estimates of transcript abundance were based on the FPKM (fragments per kilobase million) parameter (Trapnell et al. 2010).
Isolation of AhFAD3 Genes
Four ω-3 AhFAD3 genes harbored by peanut cv. ‘Tifrunner’ (Arahy.BC0JZ1, Arahy.40PHQK, Arahy.0JDQ22 and Arahy.ZDHF3I) (https://www.peanutbase.org/) were isolated from cv. ‘Fenghua1’ and functionally analyzed: they have been redesignated here as AhFAD3-1 through -4. The AhFAD coding sequences were amplified in RT-PCRs based on the primer pairs FAD3-1/4-F/-R and FAD3-2/3-F/-R) (Table S1). The template provided in these reactions was cDNA prepared from seed harvested 30 days after flowering. Each 25 μL reaction contained 1 μL template (100 ng/μL), 2 μL 2.5 mM dNTP, 2.5 μL 10×TransTaq@ HiFi Buffer (http://www.transgenbiotech.com), 1 μL TransTaq@ HiFi DNA polymerase, 1 μL of each primer (10 μM) and 16.5 μL ddH2O. The reactions were given an initial denaturation (94 °C/5 min), followed by 28-30 cycles of 94 °C/30 s, 60 °C/30 s, 72 °C/35 s, and a final elongation of 72 °C/10 min. The amplicons were electrophoretically separated through a 1% agarose gel and appropriate fragments purified and submitted for sequencing to Sangon Biotech (Shanghai, China).
Total RNA Isolation and Fluorescence-Based Semi-Quantitative PCR
The root, stem, leaf and flower of cv. ‘Fenghua 1’ plants were sampled at 15, 30, 45 and 60 days after flowering to prepare total RNA, using a DP441 RNAprep Pure Plant kit (Tiangen, Beijing), and the resulting RNA converted into cDNA using a Thermo Scientific RevertAid First Strand cDNA Synthesis kit (https://www.thermofisher.com/cn/zh/home.html). An RT-PCR assay supplied by Agilent Technologies (Agilent Technologies, California, USA) was used to profile AhFAD3 transcription; the relevant primer pairs were FAD3-1/4-F/−R to amplify a fragment of FAD3-1 and -4, and FAD3-2/3-F/−R a fragment of FAD3-2 and -3 (Table S1). The primer pair Actin11-F/-R (Table S1) was used to amplify the reference Actin sequence (GenBank number GO264911). Each 20 μL reaction contained 10 μL TaqMan Fast qPCR Master Mix, 0.4 μL of each non-labeled primer (10 μM each), 0.4 μL of fluorescently-labeled primer (10 μM), 2 μL cDNA (100 ng/μL) and 6.8 μL ddH2O. The reactions were given an initial denaturation (94 °C/3 min), followed by 45 cycles of 94 °C/5 s, 57 °C/15 s, 72 °C/30 s, and a final elongation of 72 °C/10 min. Relative transcript abundances were estimated using the 2-ΔΔCT method (Livak and Schmittgen 2001). Each reaction was run in triplicate.
Sub-Cellular Localization of AhFADs
The open reading frames (lacking the stop codons) of AhFAD3-1 and -4, and FAD3-2 and -3 were PCR-amplified using the primer pairs PBSK-1/4-F/-R and PBSK-2/3-F/-R) (Table S1), respectively, and inserted into the multiple cloning site (MCS) of the pBSK+-35S-EGFP plant transient expression vector (kindly provided by Weicai Yang, Institute of Genetics and Developmental Biology, Chinese Academy of Science). The constructs were then transformed into A. thaliana mesophyll protoplasts using the PEG-calcium transfection method (Yoo et al. 2007). Fluorescence generated by the expression of GFP was observed using confocal laser scanning microscopy (Leica TCS SP5).
A. thaliana Transformation
Primer pairs AN-1/4-F/-R and AN-2/3-F/-R were used to amplify the open reading frames of, respectively, FAD3-1 and -4, and FAD3-2 and -3. The amplicons were inserted into the plant binary vector PRI101-AN (kindly provided by Minqin Wang, Shandong University, China) and from thence into Agrobacterium tumefaciens strain LBA4404 (Poirier et al. 2000). A. thaliana plants were transformed using the inflorescence infection method (Clough and Bent 1998). Progeny of putative transformants were plated on half strength Murashige and Skoog (1962) (MS) medium (Murashige and Skoog 1962) containing 100 mg/L kanamycin to select for T1 plants carrying the transgene, and the same selection procedure was imposed on the T2 and T3 generations. The derivation of seed fatty acid content and composition was carried out using seed set by transgene homozygous T3 plants. All primer sequences are given in Table S1.
Fatty Acid Composition
The fatty acid content of both non-transgenic and T3 transgenic A. thaliana was obtained using the gas chromatography protocol described by Zheng et al. (2017).
Salinity Tolerance of Transgenic A. thaliana
Seed of both non-transgenic and T3 transgenic A. thaliana were sterilized and plated on half strength MS medium for 2-3 weeks. Uniform seedings were then transferred to the same medium containing either 0, 100, 125, 150, 175, 200 or 225 mM NaCl. The seedling survival rate was estimated after a two week exposure. The experiment was run in triplicate.
Promoter Cloning and GUS Staining
Genomic DNA was prepared from peanut plants using a Plant Genome DNA Extraction kit (TIANGEN Biotechnology Co., Beijing, China). The primer pairs FAD3-1-F/-R, FAD3-2-F/-R, FAD3-3-F/-R and FAD3-4-F/-R (Table S1) were used to amplify the up-stream sequence of each of the four FAD genes. The fragments were inserted to the MCS site of the pCAMBIA1381Z plasmid which also harbors the GUS. The resulting constructs were transformed into A. tumefaciens strain LBA4404, and from thence into A. thaliana, as described above. Progeny of putative transformants were raised on half strength MS medium containing 100 mg/L kanamycin to select for T1 plants carrying the transgene, and the same selection procedure was repeated in the T2 and T3 generations. Whole plants, flowers, pods and seeds of transgene homozygous T3 plants were subjected to GUS staining (Jefferson et al. 1987). All primer sequences are given in Table S1.
Results
FAD Genes in the Cultivated Peanunt Genome
The search for AhFAD genes in the cultivated peanut genome identified 36 candidate genes distributed over 16 of the 20 chromosomes (Table S2); each of the two subgenomes harbored 18 genes. Four of these genes mapped to each of chromosomes Arahy.17 and Arahy.19, while Arahy. 04, Arahy.07, Arahy.10 and Arahy.14 each harbored only one gene. The predicted product length of the set of genes varied from 75 (Arahy.3BHF0I.1) to 456 (Arahy.722ASC.1) residues (Table S2). The Arahy.3BHF0I.1 product was an incomplete protein, while those of both Arahy.NPA3T0.1 and Arahy.ZRK7H3.1 were shorter than the other AhFAD2s. Each of the AhFAD sequences belonged to one of the six subfamilies, namely SAD, FAD2, FAD3, FAD4/5, FAD6 or FAD7/8 (Fig. 1). Both FAD3 and FAD7/8 genes encode ω-3 FADs, while those belonging to the other four subfamilies encode ω-6 FADs. The largest subfamily was the SADs (12 members, including three pairs of homeologs: Arahy.XVN2S5 / Arahy.9C266U, Arahy.E24IL6/Arahy.BR0SNA and Arahy.7G971V/Arahy.H654N0) and three sets of paralogs (Arahy.RN8HKV/Arahy.7J26PX, Arahy.JF7GVB/Arahy.1R1L75 and Arahy.KNYU1K/Arahy.072NVT). The FAD4/5 subfamily comprised only three members, including the homeologs Arahy.K3W3ZZ/Arahy.GU7MJ6. The FAD2 subfamily comprised seven members, including three pairs of homoelogs (Arahy.BY45PL/Arahy.8TPQ4A, Arahy.S1Y1PZ/Arahy.9P5B67 and Arahy.42CZAS/Arahy.5913QL). The FAD6 subfamily included six members, including one pair of homeologs (Arahy.V3YSAY/Arahy.FHD422). The FAD3 subfamily comprised the two pairs of homeologs Arahy.ZDHF3I/Arahy.BC0JZ1 and Arahy.40PHQK/Arahy.0JDQ22. Finally, the FAD7/8 subfamily had four members, represented by two pairs of homeologs (Arahy.WLZ7Z3/Arahy.722ASC and Arahy.9ET73H/Arahy.1R706V). Five of the FADs showed evidence of AS: each of Arahy.XVN2S5, Arahy.V3YSAY, Arahy.ZDHF3I, Arahy.BC0JZ1 and Arahy.WLZ7Z3 generated two isoforms (Table S2). None of the splicing variants differed from its non-spliced isoform in terms of the number of transmembrane domains encoded.

The gene structure of AhFADs. Clade I: SAD subfamily; clade II: FAD4/5 subfamily; clade III: FAD6 subfamily; clade IV: FAD2 subfamily; clade V: FAD3 subfamily; clade VI: FAD7/8 subfamily. The phylogenetic tree was generated using amino acid sequence of the proteins. Red lines marked the four genes studied in this paper
The number of introns present in the various genes varied (Fig. 1). In members of the SAD subfamily, the number ranged from one to three, with alternative splicing affecting the exon number contributing to the transcripts formed by Arahy.XVN2S5.1 and Arahy.XVN2S5.2. Among members of the FAD2 subfamily, the number was one or two, while among members of the FAD4/5 subfamily, it was three or four. The highest number of introns was associated with members of the FAD6 subfamily (six to ten) (except for Arahy.3BHF0I.1). Genes belonging to the FAD3 and FAD7/8 subfamilies were interrupted by six to eight introns, with AS affecting the exon number contributing to some of the transcripts.
All but one SAD subfamily member (the exception was Arahy.BR0SNA.1) encoded no transmembrane domains. All but one of the FAD2 genes encoded five or six transmembrane domains; the exception was Arahy.7E0HBM.1 (three transmembrane domains). FAD3 genes encoded four or five transmembrane domains, while FAD4/5 genes encoded between two and four. Of the six FAD6 genes, two encoded four transmembrane domains, but the other four encoded fewer than this, probably because these latter sequences were incomplete. The FAD7/8 genes all encoded four transmembrane domains (Table S2).
With respect to the predicted location of expression, the FAD2 and FAD3 gene products likely were deposited in the endoplasmic reticulum (ER), whereas those belonging to either the SAD, FAD4/5, FAD6 or FAD7/8 subfamilies were expressed in the chloroplast (Table S2).
Phylogenetic Analysis of the FADs
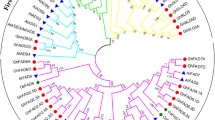
The scan of the Phytozome database resulted in a set of 227 potential FAD sequences (Fig. 2), harbored by nine dicotyledonous species, namely A. thaliana, Gossypium raimondii, Cucumis sativus, Solanum lycopersicum, Helianthus annuus, Glycine max and the three Arachis species hypogaea, duranensis and ipaensis. The analysis revealed two large clades, one of which clustered the genes encoding the soluble, transmembrane domain-lacking SADs, and the other the insoluble FADs, which included at least one transmembrane domain. The 12 Arachis spp. SADs were clustered into two secondary clades, of which one contained exclusively Arachis spp. sequences. Five secondary FAD clades were recognized: the FAD4/5s occupied a phylogenetic position closest to the root, indicating this group’s more ancient origin, while the FAD3s and FAD7/8s appeared to be the most recently evolved groups. Although the FAD3s and FAD7/8s shared both a similar gene structure and nucleotide sequence, most of the FAD3s encoded a product deposited in the ER whereas the FAD7/8 products were expressed in the chloroplast. The number of FAD2 and FAD5 sequences present in the sunflower genome is notably greater than in any of the other species surveyed.

The phylogeny tree of plant FAD genes. Ath: A. thaliana, Gra: Gossypium raimondii, Csa: Cucumis sativus, Sly: Solanum lycopersicum, Han: Helianthus annuus, Gma: Glycine max, Ahy: A. hypogaea, Adu: A. duranensis, Aip: A. ipaensis
Transcriptional Behavior of the Peanut FADs
The transcriptional behavior of each of the 36 AhFADs, as deduced from RNA-Seq data, is illustrated in Fig. 3. No transcript was detected for eight of the genes. Both Arahy.7G971V and Arahy.H654NO were strongly transcribed in each of the organs surveyed, and especially so in the seed2 sample, suggesting that their products make a substantial contribution to seed development and the accumulation of oil. Arahy.E24IL6 and Arahy.BR0SNA were also strongly transcribed in both the seed1 and seed2 samples, implying they too may encode products important for lipid synthesis. Transcript of all seven FAD2 members was detected, in particular that of Arahy.8TPQ4A, which was strongly transcribed in all four organs. Of the three FAD4/5s detected, Arahy.ND3SKJ and Arahy.GU7MJ6 were notable in that they were well represented in the leaf sample. Transcript of only two of the six FAD6s was detected, and the abundance of these transcripts was highest in the leaf sample. Transcript of all four FAD3s was detected, most notably that of Arahy.0JDQ22. Transcript of the four FAD7/8s was represented in all four organs: Arahy.WLZ7Z3 and Arahy.722ASC transcript was particularly abundant in the leaf.

Transcription profiling of 28 AhFAD genes in various organs of the plant. Seed1, 30 days after flowering; Seed2, 50 days after flowering; Root and Leaf, 12 day old of cv. ‘Fenghua 1’ seedlings
Isolation of AhFAD3 Sequences and their Transcriptional Behavior
The four AhFAD3 genes Arahy.BC0JZ1, Arahy.40PHQK, Arahy.0JDQ22 and Arahy.ZDHF3I (equivalent to AhFAD3-1 through -4) were targeted for PCR-based cloning from cv. ‘Fenghua 1’ plants. Their sequences have been submitted to GenBank under accession numbers MH085086-88, MK757485. AhFAD3-1 and -4 map to the homoelogs Arahy.17 and Arahy.07, respectively (Table S2); their sequences shared a nucleotide identity of 99.6% and their predicted translation products differed by just two residues (Fig. 4a). Both gene products included the same conserved domains and featured a single transmembrane domain, but differed with respect to both their pI and their molecular weight (Table 1). AhFAD3-2 and -3 similarly map to a pair of homoelogs (chromosomes Arahy.11 and Arahy.01); their level of nucleotide identity was 96.7%, as was the level of peptide similarity between their predicted products (Fig. 4b). A stretch of five residues lying close to the protein’s C terminus was present in AhFAD3-3, but not in -2. The two polypeptides harbored the same conserved domains, but differed with respect to the number of transmembrane domains present, their pI and their molecular weights (Table 1). Given the high sequence similarity between each pair of genes, an analysis of their transcription in the vegetative tissue and in developing seed was based on aggregating the abundance of AhFAD3-1 and -4 transcript, and similarly that of AhFAD3-2 and -3 (Fig. 5). Both pairs of homeologs were most strongly transcribed early during seed development (15–30 days after flowering). The highest abundance of AhFAD3-1/4 transcript was present in the root sample, whereas that of AhFAD3-2/3 was in the flower. Neither pair was strongly transcribed in the stem.

Amino acids alignment of the four cloned AhFAD3 products. a AhFAD3-1 vs AhFAD3-4, two amino acids differences. b AhFAD3-2 vs AhFAD3-3, eight single amino acids differencs and one deletion/insertion fragment

Transcription profiling of the four AhFAD3 genes in various organs of the plant. It was difficult to discern the abundance of AhFAD3-1 and -4 transcript, and similarly that of AhFAD3-2 and -3, so the blue column represented the summary of AhFAD3-1 and -4, and the red one represented that of AhFAD3-2 and -3
Functional Analysis of the AhFAD3s
When each of the AhFAD3-GFP fusion transgenes was expressed in A. thaliana protoplasts, most of the GFP signal was concentrated in the ER, but was also detected in the cell membranes and the cytoplasm (Fig. 6). Subsequently, each of the genes was constitutively expressed in A. thaliana to determine their contribution to the seeds’ FA content and composition. In both non-transgenic and transgenic seeds, nine fatty acids were detected, namely C16:0 (palmitic acid), C18:0 (stearic acid), C18:1 (oleic acid), C18:2 (linoleic acid), C18:3(linolenic acid), C20:0 (arachidic acid), C20:1 (eicosenic acid), C20:2 (eicosadienoic acid) and C22:1 (erucic acid) (Fig. 7). The most abundant fatty acids were C18:2 (30.5%) and C18:3 (19.1%). The effect of the transgenes was to raise the total FA content of the seed by between 24.8% and 33.3%, reflecting a rise in the content of each of the individual FAs, but especially that of C18:3 (Fig. 7a). The relative contribution of the various fatty acids was also altered, with that of C18:3 increasing markedly and that of C18:2 decreasing (Fig. 7b). An analysis of the AhFAD3 promoter sequences was carried out by linking each to GUS and expressing the resulting transgenes in A. thaliana. The whole plant, the flowers, the siliques and the seeds all tested positive for GUS activity (Fig. 8), consistent with the promoters all being capable of driving constitutive expression.

The sub-cellular localization of AhFAD3-GFP fusion protein in transgenic A. thaliana protoplasts. “GFP”: green fluorescent protein signal, “ER”: red fluorescence of endoplasmic reticulum, “Bright-field”: bright field image, “Merged”: merged set of images

The FA constitution and content in the oil of seeds set by A. thaliana plants harboring an AhFAD3 transgene. a FA content (mg/g dry weight), C18:3 with the greatest increase. b Proportion of individual FAs (%), the percentage of C18:3 increased significantly whereas C18:2 decreased compared with the control

AhFAD3 promoter activity assay. A. thaliana plants harboring a pAhFAD3-GUS fusion transgene stained for GUS activity (blue color). Control: non-transgenic A. thaliana seedlings
Over-Expressing AhFAD3s Increased the Salinity Tolerance of the Transgenic A. thaliana
In transgenic A. thaliana seedlings raised in the presence of less than 150 mM NaCl, there was no significant difference in the survival rate of non-transgenic and transgenic plants, both of which mostly (>99%) survived unscathed (Fig. 9). At higher concentrations of NaCl, however, the survival rate of the transgenic seedlings was higher than that of the non-transgenic ones: in the presence of 175 mM NaCl, the survival rate of the transgenic seedlings was about 10.2–20.5% greater than that of the non-transgenic ones; at 200 mM NaCl, the survival rate of the transgenic seedlings, though much reduced, remained 5.6–9.0% higher than that of the non-transgenic ones.

The survival rate of A. thaliana seedlings harboring an FAD3 transgene challenged by a range of NaCl concentrations. Columns indicate means±SE (n = 3). *: mean differs significantly (P < 0.05) from that of the non-transgenic seedlings
Discussion
Cultivated peanut is an allotetraploid formed following a natural hybridization between the two diploid species Ar. duranensis and Ar. ipaensis (Bertioli et al. 2016). The completion of the genome sequences of both progenitor species and of the cultivated peanut itself offers the opportunity to characterize the membership of gene families such as the FADs (Bertioli et al. 2016; Chen et al. 2019). A previous report has documented that A. duranensis harbors 15 FAD genes, while A. ipaensis harbors 16 (Ruan et al. 2018). Here, a similar analysis of the cultivated peanut genome has revealed 36 FADs, with 18 represented in each of the two subgenomes (Table S2). The additional five genes comprised one FAD2 (Arahy.7E0HBM) and four FAD6s (Arahy.NPA3T0, Arahy.ZRK7H3, Arahy.3BHF0I and Arahy.93DW2D). The structure of the latter genes differed sufficiently from that of the other two FAD6s, to form a recognizably novel sub-clade (Fig. 1); their absence in both of the two progenitor genomes implies their evolution post the formation of the cultivated peanut alloploid. Curiously, there was no evidence of any transcript from any of them, at least in the four organs surveyed (Fig. 3), raising the possibility that they are pseudogenes. FAD genes are known to be readily duplicated: for instance, in cucumber, 12 of the 14 FAD5s present appear to cluster closely together, suggestive of a frequent occurrence of gene duplication events following speciation (Dong et al. 2016). Similarly, the cotton ω-3 FAD gene family appears to have undergone rapid expansion (Yurchenko et al. 2014).
Although the peanut genome harbors more than 30 FADs, but under non-stress conditions the transcripts of every one of them could not be detected, with a few of the genes being transcribed very strongly in one or more organs of the plant (Fig. 3), as also revealed by an earlier study (Yurchenko et al. 2014). In the olive fruit, FAD3A appears to be the key gene responsible for accumulation of C18:3 in the seed, while FAD7 contributes most materially in the mesocarp (Hernández et al. 2016). C18:1 and C18:2 represent the major components of peanut seed oil, accounting for about 80% of its total fatty acid content. AhSAD and AhFAD2 are largely responsible for the synthesis of these components, so it was not unexpected to find that the two AhSAD genes Arahy.7G971V and Arahy.H654N0 and the AhFAD2 gene Arahy.H654N0 were all strongly transcribed in the developing seed. So far, four separate AhFAD2 genes have been identified as present in the peanut genome, and the inactivation of one of them is known to change the FA constitution of the seed oil (Chi et al. 2011; Wang et al. 2015). The synthesis of ALA, a very minor component of the oil (accounting for <0.1%) but an essential fatty acid in the human daily diet, is catalyzed by ω-3 FADs (FAD3/4 s and 7/8 s), genes which are transcribed in the seed at a very low level; the implication is that an attempt to boost the content of C18:3 through the manipulation of FAD genes will require increasing the intensity with which one or more of the ω-3 FADs are transcribed. The four AhFAD3 genes which have been functionally characterized here were shown to be active largely in the ER (Fig. 6), but their constitutive expression A. thaliana did succeed in increasing both the representation of C18:3 in, and the total fatty acid content of the seed oil (Fig. 7).
The importance of ALA for the plant’s metabolism is that represents a precursor of the phytohormone jasmonic acid, which contributes in many ways to both development and the response to abiotic stress (Weber 2002). The desaturation of FAs in the membrane is used by some plants as a strategy for surviving low temperature stress. In A. thaliana, the ads2 mutant produces a reduced level of unsaturated fatty acids and a higher one of saturated fatty acids; the mutant is dwarfed and sterile when exposed to a sub-optimal temperature (Chen and Thelen 2013). FAD2 has been shown to be required by A. thaliana to combat salinity stress, because it helps to maintain the level of desaturation of vacuolar and plasma membrane fatty acids necessary for the functioning of Na+/H+ exchangers (Zhang et al. 2012). In cucumber, some FAD genes can be induced by low temperature and repressed by high temperature, which has been taken to imply that FADs form part of the species’ machinery to cope with temperature stress (Dong et al. 2016). In sunflower, in contrast, a FAD2 gene has been reported as being repressible by both low temperature and salinity (Zhou et al. 2017). Meanwhile, in cotton, one FAD7/8 responds positively to low temperature, whereas a second one scarcely responds to either of these stresses (Yurchenko et al. 2014). It has earlier been demonstrated that in peanut, some – but not all – AhFADs respond to a variety of stress agents (Chi et al. 2017). Here, it has been shown that the heterologous expression in A. thaliana of each of four AhFAD3s had a positive impact on the survival of NaCl-challenged seedlings (Fig. 9), implying that their products may in some way contribute to the ability of peanut plants to combat salinity stress.
In conclusion, a scan of the recently completed peanut genome has revealed the presence of 36 likely AhFAD genes, most of which were represented by transcript in some, or all parts of the plant. Four members of AhFAD3 subfamily were functionally characterized in some detail: their expression was concentrated largely in the ER, and their transcript abundance was typically higher in seed than in either root or leaf tissue. When heterologously expressed in A. thaliana, each had the effect of increasing both the representation of ALA and the total FA content of the seed oil. They also acted to enhance the survival of seedlings grown in a medium containing >150 mM NaCl.
Data Availability
The sequences of the four ω-3 AhFAD3 genes have been submitted to GenBank under accession numbers MH085086 through MH085088, MK757485. All the supporting data are included as additional files.
References
Arondel V, Lemieux B, Hwang I, Gibson S, Goodman HM, Somerville CR (1992) Map-based cloning of a gene controlling omega-3 fatty acid desaturation in Arabidopsis. Science 258:1353–1355. https://doi.org/10.1126/science.1455229
Baker EJ, Miles EA, Burdge GC, Yaqoob P, Calder PC (2016) Metabolism and functional effects of plant-derived omega-3 fatty acids in humans. Prog Lipid Res 64:30–56. https://doi.org/10.1016/j.plipres
Bertioli DJ et al (2016) The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat Genet 48:438–446. https://doi.org/10.1038/ng.3517
Bertioli DJ et al (2019) The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat Genet 51:877–884. https://doi.org/10.1038/s41588-019-0405-z
Blencowe BJ (2006) Alternative splicing: new insights from global analyses. Cell 126:37–47. https://doi.org/10.1016/j.cell.2006.06.023
Chen M, Thelen JJ (2013) ACYL-LIPID DESATURASE2 is required for chilling and freezing tolerance in Arabidopsis. Plant Cell 25:1430–1444. https://doi.org/10.1105/tpc.113.111179
Chen L et al (2018) Identification and characterization of a plastidial ω-3 fatty acid desaturase EgFAD8 from oil palm (Elaeis guineensis Jacq.) and its promoter response to light and low temperature. PLoS One 13:e0196693
Chen X et al (2019) Sequencing of cultivated peanut, Arachis hypogaea, yields insights into genome evolution and oil improvement. Mol Plant 12:920–934. https://doi.org/10.1016/j.molp.2019.03.005
Chi XY, Yang QL, Pan LJ, Chen MN, He YN, Yang Z, Yu SL (2011) Isolation and characterization of fatty acid desaturase genes from peanut (Arachis hypogaea L.). Plant Cell Rep 30:1393–1404
Chi X et al (2017) Isolation and functional analysis of fatty acid desaturase genes from peanut (Arachis hypogaea L.). PLoS One 12:e0189759. https://doi.org/10.1371/journal.pone.0189759
Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16:735–742
Dong CJ, Cao N, Zhang ZG, Shang QM (2016) Characterization of the fatty acid desaturase genes in cucumber: structure, phylogeny, and expression patterns. PLoS One 11:e0149917. https://doi.org/10.1371/journal.pone.0149917
Gibson S, Arondel V, Iba K, Somerville C (1994) Cloning of a temperature-regulated gene encoding a chloroplast omega-3 desaturase from Arabidopsis thaliana. Plant Physiol 106:1615–1621
Hall BG (2013) Building phylogenetic trees from molecular data with MEGA. Mol Biol Evol 30:1229–1235
Hernández ML, Sicardo MD, Martinez-Rivas JM (2016) Differential contribution of endoplasmic reticulum and chloroplast omega-3 fatty acid desaturase genes to the linolenic acid content of olive (Olea europaea). Fruit Plant Cell Physiol 57:138–151. https://doi.org/10.1093/pcp/pcv159
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6:3901–3907
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36
Li L, Peng ZY, Chen B, Wang YY, Zhang B, Bi YP (2012) Accumulation pattern of fatty acids during the seed development of peanut. Acta Agriculturae Boreali-Sinica 27:173–177 (in Chinese)
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25:402–408
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassay with tobacco tissue culture. Physiol Plant 15:473–497
Poirier Y, Ventre G, Nawrath C (2000) High-frequency linkage of co-expressing T-DNA transgenic Arabidopsis thaliana transformed by vaccum-infiltration of Agrobacterium tumefaciens. Theor Appl Genet 100:487–493
Radovanovic N, Thambugala D, Duguid S, Loewen E, Cloutier S (2014) Functional characterization of flax fatty acid desaturase FAD2 and FAD3 isoforms expressed in yeast reveals a broad diversity in activity. Mol Biotechnol 56:609–620
Reddy ASN, Marquez Y, Kalyna M, Barta A (2013) Complexity of the alternative splicing landscape in plants. Plant Cell 25:3657–3683. https://doi.org/10.1105/tpc.113
Remy E et al (2014) Intron retention in the 5′UTR of the novel ZIF2 transporter enhances translation to promote zinc tolerance in arabidopsis. PLoS Genet 10:e1004375
Román Á, Andreu V, Hernández ML, Lagunas B, Picorel R, Martínez-Rivas JM, Alfonso M (2012) Contribution of the different omega-3 fatty acid desaturase genes to the cold response in soybean. J Exp Bot 63:4973–4982
Ruan J, Shan L, Li XG, Guo F, Meng JJ, Wan SB, Peng ZY (2018) Genome-wide identification and expression pattern analysis of peanut FAD gene family. Shandong Agric Sci 50:1–9 (in Chinese)
Stamm S et al (2005) Function of alternative splicing. Gene 344:1–20. https://doi.org/10.1016/j.gene.2004.10.022
Tang W et al (2016) Comprehensive transcriptome profiling reveals long noncoding RNA expression and alternative splicing regulation during fruit development and ripening in kiwifruit (Actinidia chinensis). Front Plant Sci 7:335. https://doi.org/10.3389/fpls.2016.00335
Teixeira MC, Carvalho IS, Brodelius M (2010) Omega-3 fatty acid desaturase genes isolated from purslane (Portulaca oleracea L.): expression in different tissues and response to cold and wound stress. J Agric Food Chem 58:1870–1877
Trapnell C et al (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515
Venegas-Calerón M, Beaudoin F, Garcés R, Napier JA, Martínez-Force E (2010) The sunflower plastidial omega3-fatty acid desaturase (HaFAD7) contains the signalling determinants required for targeting to, and retention in, the endoplasmic reticulum membrane in yeast but requires co-expressed ferredoxin for activity. Phytochemistry 71:1050–1058
Vrinten P, Hu Z, Munchinsky M-A, Rowland G, Qiu X (2005) Two FAD3 desaturase genes control the level of linolenic acid in flax seed. Plant Physiol 139:79–87. https://doi.org/10.1104/pp.105.064451
Wang Y, Zhang XG, Zhao YL, Prakash CS, He GH, Yin DM (2015) Insights into the novel members of the FAD2 gene family involved in high-oleate fluxes in peanut. Genome 58:375–383
Weber H (2002) Fatty acid-derived signals in plants. Trends Plant Sci 7:217–224
Xue Y et al (2018) Omega-3 fatty acid desaturase gene family from two omega-3 sources, Salvia hispanica and Perilla frutescens: cloning, characterization and expression. PLoS One 13:e0191432. https://doi.org/10.1371/journal.pone.0191432
Yang QL et al (2012) Identification of FAD2 and FAD3 genes in Brassica napus genome and development of allele-specific markers for high oleic and low linolenic acid contents. Theor Appl Genet 125:715–729
Yang S, Tang F, Zhu H (2014) Alternative splicing in plant immunity. Int J Mol Sci 15:10424–10445. https://doi.org/10.3390/ijms150610424
Yoo SD, Cho YH, Sheen J (2007) Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat Protoc 2:1565–1572
Yu C, Wang HS, Yang S, Tang XF, Duan M, Meng QW (2009) Overexpression of endoplasmic reticulum omega-3 fatty acid desaturase gene improves chilling tolerance in tomato. Plant Physiol Biochem 47:1102–1112
Yurchenko OP et al (2014) Genome-wide analysis of the omega-3 fatty acid desaturase gene family in Gossypium. BMC Plant Biol 14:312. https://doi.org/10.1186/s12870-014-0312-5
Zhang J, Liu H, Sun J, Li B, Zhu Q, Chen SL, Zhang HX (2012) Arabidopsis fatty acid desaturase FAD2 is required for salt tolerance during seed germination and early seedling growth. PLoS One 7:e30355. https://doi.org/10.1371/journal.pone.0030355.g001
Zhang Q, Zhang X, Wang S, Tan C, Zhou G, Li C (2016) Involvement of alternative splicing in barley seed germination. PLoS One 11:e0152824. https://doi.org/10.1371/journal.pone.0152824
Zheng L et al (2017) Discovery of a new mechanism for regulation of plant triacylglycerol metabolism: the peanut diacylglycerol acyltransferase-1 gene family transcriptome is highly enriched in alternative splicing variants. J Plant Physiol 219:62–70. https://doi.org/10.1016/j.jplph.2017.09.009
Zhou XP, Wang MY, Feng L, Zhu JB, Sun L (2017) Cloning and expression of HaFAD2-2 gene from Helianthus annuus L. Acta Bot Boreal-Occident Sin 37:0845–0850 (in Chinese)
Zhuang W et al (2019) The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. Nat Genet 51:865–876. https://doi.org/10.1038/s41588-019-0402-2
Funding
This work was supported by the National Key R&D Program of China (2018YFD1000900), Shandong Province Germplasm Innovation (2017LZN035), the earmarked fund for Modern Agroindustry Technology Research System (CARS-13), and Agricultural Science and Technology Innovation Project of Shandong Academy of Agricultural Sciences (CXGC2018B05, CXGC2018E13). The founders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. PCR amplification, sequencing and RT-PCR were performed by Jian Ruan and Haiying Tian. Vector construction and sub-cellular localization were performed by Lei Shan and Jingjing Meng. A. thaliana transformation, screening of positive strains and salinity resistance test were performed by Feng Guo. Fatty acid composition analysis was performed by Zhimeng Zhang. Promoter cloning and GUS-staining were performed by Hong Ding. Design and drafting of the manuscript, revised the manuscript and gave final approval of the version to be published were performed by Zhenying Peng, Shubo Wan and Xinguo Li. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics Approval and Consent to Participate
Peanut cultivar ‘Fenghua1’ was kindly provided by Prof. Yongshan Wan, Shandong Agricultural University. ‘Fenghua1’ is a good peanut cultivar and widely planted in North China, and the seeds can be bought and sold at will.
Consent for Publication
Not applicable.
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key Message
Cultivated peanut genome harbors 36 AhFAD genes which belonged to six sub-families. Synthesis of α-linolenic acid requires the activity of ω-3 fatty acid desaturases (ω-3 FADs). Four ω-3 AhFAD3 genes were functionally characterized and localized mainly in the endoplasmic reticulum. Over-expression in Arabidopsis increased α-linolenic acid content, improved salt-resistance.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peng, Z., Ruan, J., Tian, H. et al. The Family of Peanut Fatty Acid Desaturase Genes and a Functional Analysis of Four ω-3 AhFAD3 Members. Plant Mol Biol Rep 38, 209–221 (2020). https://doi.org/10.1007/s11105-019-01191-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-019-01191-0