
Abstract
The importance of adaptive mutations in molecular evolution is extensively debated. Recent developments in population genomics allow inferring rates of adaptive mutations by fitting a distribution of fitness effects to the observed patterns of polymorphism and divergence at sites under selection and sites assumed to evolve neutrally. Here, we summarize the current state-of-the-art of these methods and review the factors that affect the molecular rate of adaptation. Several studies have reported extensive cross-species variation in the proportion of adaptive amino-acid substitutions (α) and predicted that species with larger effective population sizes undergo less genetic drift and higher rates of adaptation. Disentangling the rates of positive and negative selection, however, revealed that mutations with deleterious effects are the main driver of this population size effect and that adaptive substitution rates vary comparatively little across species. Conversely, rates of adaptive substitution have been documented to vary substantially within genomes. On a genome-wide scale, gene density, recombination and mutation rate were observed to play a role in shaping molecular rates of adaptation, as predicted under models of linked selection. At the gene level, it has been reported that the gene functional category and the macromolecular structure substantially impact the rate of adaptive mutations. Here, we deliver a comprehensive review of methods used to infer the molecular adaptive rate, the potential drivers of adaptive evolution and how positive selection shapes molecular evolution within genes, across genes within species and between species.
Similar content being viewed by others


Introduction
After Darwin proposed that natural selection acts as a main driver of evolution, a major goal of evolutionary biologists has been to understand how beneficial mutations shape species adaptation to their environment. Over the years, the number of approaches used to detect positive selection has increased substantially, making use of the increasing amount of genome data available. In particular, methods have been developed to pinpoint genes, or positions within these genes, that exhibit a pattern of genetic variation statistically incompatible with a pure nearly-neutral scenario (Ohta 1992), where mutations are considered to be neutral, nearly neutral or deleterious (i.e. Nielsen et al. 2005; Ometto et al. 2005; Kosiol et al. 2008). The ecological relevance of such candidate genes can be further tested using functional annotations, when available, or experimentally, for instance, by using reverse genetics and ancestral allele reconstruction (i.e. Hilson et al. 2004; Nielsen et al. 2005; Voight et al. 2006; Roux et al. 2014). This allowed to detect instances of adaptive evolution in many functional categories, such as immune genes in ants (Roux et al. 2014) and in hominids (Nielsen et al. 2005), virulence associated genes in pathogens (Stukenbrock et al. 2011; Dong et al. 2014), and coat-color related genes in hares (Jones et al. 2018) and mice (Hoekstra et al. 2006). While such methods allow a detailed understanding of case-studies, they do not enable one to assess the genome-wide distribution of the fitness effects of mutations.
By contrast, mutation accumulation (MA) experiments are specifically designed to estimate a genome-wide rate of mutation and distribution of effects of mutations on fitness (i.e. Shaw et al. 2002; Bataillon 2003; Rutter et al. 2012). With this approach, one can infer (1) the number of mutations that led to the divergence between MA lines, and (2) the fitness effects of these mutations on the (fitness-related) trait of interest (i.e. viability or lifetime reproductive success; see Glossary). Previous studies have inferred the presence of beneficial mutations in MA line experiments both in the field and in greenhouse studies of A. thaliana (Shaw et al. 2002; Rutter et al. 2012). Nonetheless, MA approaches can only give insight on recent adaptive events, and, therefore, provide little information regarding the proportion of adaptive genetic differences between species. Furthermore, MA experiments yield too few beneficial mutations to be able to test for the occurrence of genomic regions where adaptive mutations are more likely to occur. Conversely, population genomic approaches only offer indirect insights on mutation rates and fitness effects but can leverage patterns of sequence variation between and within species to infer rates of adaptive evolution, thus providing knowledge on the drivers of adaptation at deeper scales of evolution.
The role of positive (a.k.a. Darwinian) selection in molecular evolution is still widely debated (Hey 1999; Gillespie 2000; Kern and Hahn 2018; Jensen et al. 2019). The neutral theory of molecular evolution (Kimura 1968) states that the bulk of segregating polymorphisms is either neutral or deleterious and that the genetic differences between species are explained mainly by neutral substitutions (see Glossary), while beneficial mutations are considered to be too rare to contribute much to the observed polymorphism and divergence. With an increasing amount of data becoming available, however, the question of whether adaptive mutations play a role in molecular evolution can be investigated with a greater precision. “How much of the genetic variation can be explained by adaptive evolution? What is the frequency of adaptive mutations along the genome? Are there regions where adaptive mutations are more likely to occur?” are some of the questions that can now be addressed with population genomics data and statistical methods for the inference of selection.
Here, we present the current state-of-the-art methods used to model the distribution of fitness effects (DFE) and infer the frequency of adaptive mutations. We then review evidence for variation in the rate of adaptive evolution within genes, within genomes and between species.
Synthesis of methods
In the following section, we review the methods that can be used to estimate the rate of adaptive evolution from sequence data. We distinguish two main approaches: phylogenetic methods, based on the divergence between multiple species; and population genetics approaches, which contrast within-species polymorphism to the divergence with an outgroup species.
Glossary
-
Mutation accumulation (MA): experimental design where a single inbred line is used to create various sub-lines that are propagated under conditions minimizing the opportunity for selection. MA lines are allowed to diverge independently for several generations. The number of mutations that led to the divergence between MA lines and the fitness effects of these mutations on the trait of interest influence the empirical distribution of the mean phenotypic value of the trait. If the trait measured is fitness or a fitness component, this setting can be used to infer the genome-wide mutation rates and the underlying distribution of fitness effects (DFE, see below).
-
Synonymous mutation: a mutation, in a protein-coding region, that leaves the amino-acid residue unchanged.
-
Non-synonymous mutation: a mutation, in a protein-coding region, that leads to a change in the amino-acid residue.
-
Substitution: a fixed difference between species.
-
Polymorphism: a mutation segregating within a population (or a species).
-
Positive selection: selective process by which a beneficial mutation increases in frequency within a population.
-
Adaptive evolution: at the molecular level, it occurs in a certain genomic region through the successive fixation of advantageous mutations (Charlesworth and Charlesworth 2010).
-
Negative/Purifying selection: natural selection against a deleterious mutation.
-
Distribution of fitness effects (DFE) of mutations: represents the distribution of the relative frequencies of selection coefficients (s), extending from strongly and weakly deleterious, through neutral mutations to slightly and strongly advantageous.
-
dN: number of non-synonymous substitutions per site.
-
dS: number of synonymous substitutions per site.
-
Dn: number of non-synonymous substitutions per gene/region.
-
Ds: number of synonymous substitutions per gene/region.
-
Pn: number of non-synonymous polymorphisms per gene/region.
-
Ps: number of synonymous polymorphisms per gene/region.
-
α: proportion of amino-acid substitutions that are adaptive.
-
Genetic drift: random changes in allele frequencies produced by the sampling of the genetic variants that compose a population every new generation.
-
Genetic draft: a process that induces allele frequency changes through recurrent selective sweeps at linked positions.
-
Selective sweep: the process by which a beneficial substitution reduces genetic diversity at linked positions.
-
Background selection: the process by which negatively selected deleterious mutations reduce neutral genetic diversity at linked positions.
-
ωa: rate of adaptive amino-acid non-synonymous substitutions relative to the mutation rate.
-
Ka+: rate of adaptive amino-acid substitutions, denoted as: αKa, where Ka represents an alternative notation of dN.
Quantifying the proportion of adaptive substitutions
-
(1)
Phylogenetic methods
The strength and direction of selection on the branch of a phylogenetic tree can be measured by contrasting the nonsynonymous (dN) and synonymous divergence (dS) in a given gene (e.g. Miyata et al. 1979; Li et al. 1985; Yang and Nielsen 2002; Eyre-Walker 2006). The dN/dS ratio, noted as ω, provides an estimate of the rate of nonsynonymous substitutions relative to the rate of synonymous substitutions. Assuming that mutation rates at synonymous and non-synonymous sites are constant and equal, and that synonymous substitutions are selectively neutral, genes with ω > 1 are considered to be evolving under positive selection, while genes with ω < 1 are evolving under negative selection. Because ω is based on averages of substitution rates across multiple nucleotide sites that undergo both positive and negative selection, this statistic can only detect strong positive selection (e.g. Yang and Nielsen 2002; Eyre-Walker 2006). As most nonsynonymous mutations are expected to be either neutral or deleterious, dN will tend to be much lower than dS, hence ω will tend to be globally lower than one (i.e. Yang and Nielsen 2002; Eyre-Walker 2006).
In order to consider variation in selective constraints in space and time, models have been developed to account for variable selective pressure among sites (Nielsen and Yang 1998; Yang et al. 2000, 2005), branches (Yang and Nielsen 1998), or both (so-called branch-site models; Yang and Nielsen 2002; Zhang et al. 2005; Kosakovsky Pond et al. 2011). In site-based models, the ω ratio varies across sites and positive selection is inferred at a specific site if the average dN is higher than dS over all lineages. In branch-based models, the ω ratio varies among lineages and positive selection is detected if the average dN is higher than dS across all sites in a certain branch or a series of branches defining a lineage in a phylogenetic tree. In turn, branch-site models allow the ω ratio to vary both across sites and lineages. Using this framework, distinct models can be compared to test for the occurrence of positive selection at particular sites or branches (e.g. Yang and Nielsen 2002; Zhang et al. 2005). Although these methods detect adaptation at the site level, it has been shown that they are conservative in measuring selection over a certain region and/or lineage (Rodrigue and Lartillot 2017). This higher conservatism could be due to adaptive processes not being concentrated on a small number of sites but rather scattered across a large number of positions in a certain genomic region (Rodrigue and Lartillot 2017). Moreover, branch-site models assume that evolution on the majority of branches is neutral and that adaptive processes are rare and usually isolated. Hence, events of frequent adaptation over long evolutionary periods would not be captured, leading to underestimates of the rate of adaptive evolution in the tested proteins (Nielsen and Yang 1998; Yang et al. 2000, 2005; Rodrigue and Lartillot 2017). Besides, as these approaches are based on multiple-species alignments, the analysis is focused on genes that are shared by all species, which are more ancient and typically more conserved. Rapidly evolving genes are typically discarded from such analysis since their alignment becomes less reliable as the divergence between species increases.
-
(2)
Population genetics methods
-
a.
The McDonald and Kreitman (MK) test
-
a.
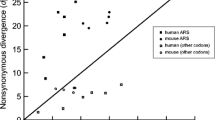
Population genetic methods pioneered by Hudson, Kreitman, and Aguadé (1987) test a neutral evolution scenario by comparing the number of polymorphic sites within a population with the number of substitutions with a distinct species (HKA test). Under a neutral scenario, the relative amount of polymorphism and divergence is constant between loci. The HKA test compares these values between at least two genomic regions to test this prediction (Hudson et al. 1987). McDonald and Kreitman (1991) first extended this approach to detect adaptive protein evolution (Fig. 1). The so-called MK test requires data from as little as two closely-related species, typically including several individuals in the study species and one individual from an outgroup species. It compares the number of polymorphisms to the number of substitutions for a locus in two classes of sites: synonymous, which are assumed to evolve neutrally, and non-synonymous, which are potentially under selection (McDonald and Kreitman 1991). The number of nonsynonymous substitutions is denoted as Dn, the number of synonymous substitutions as Ds, the number of nonsynonymous polymorphisms as Pn and the number of synonymous polymorphisms as Ps (see Glossary), leading to the so-called MK-table:

Timeline presenting the state-of-the-art population genetic methods to infer the rate of adaptive evolution (top) and the major findings on the factors impacting the variation of the molecular adaptive rate across species, along the genome, between and within genes (bottom). References for DFE methods can be found in Table 1. Light orange boxes correspond to the variation of the molecular adaptive rate between species; dark orange boxes represent the variation along the genome; blue boxes represent variation between protein-coding genes; and the green box correspond to the factors impacting the molecular adaptive rate at the intra-genic level. References for these studies can be found in the corresponding section in the main text. α: proportion of adaptive amino-acid substitutions; Ne: effective population size; s: selection coefficient; ωa: rate of adaptive non-synonymous substitutions; RSA: relative solvent accessibility. References: (1) Hudson et al. 1987; (2) McDonald and Kreitman 1991; (3) Sawyer and Hartl 1992, Charlesworth 1994, Smith and Eyre-Walker 2002, Fay et al. 2001, Bustamante et al. 2002, Sawyer et al. 2003; (4) Bustamante et al. 2002; (5) Charlesworth and Eyre-Walker 2006; (6) Williamson 2003, Nielsen and Yang 2003; (7) i.e. Hvilsom et al. 2012, Eyre-Walker and Keightley 2009; (8) Halligan et al. 2010; (9) Gossmann et al. 2010, Strasburg et al. 2011; (10) Carneiro et al. 2012; (11) Enard et al. 2014; (12) Galtier 2016. Species figures were taken from PhyloPic (http://www.phylopic.org)
Polymorphisms | Substitutions | |
|---|---|---|
Synonymous | Ps | Ds |
Non-synonymous | Pn | Dn |
Under a scenario where all mutations are either strongly deleterious or neutral, Dn/Ds is expected to be equal to Pn/Ps. Conversely, Dn/Ds higher than Pn/Ps is taken as a signature of positive selection, and Dn/Ds lower than Pn/Ps can be observed in case of balancing selection. As a beneficial mutation reaches fixation at a faster rate than a neutral mutation, it contributes comparatively more to divergence than to polymorphism levels (McDonald and Kreitman 1991; Eyre-Walker 2006).
-
b.
Extensions of the MK-test: Estimation of the proportion of amino-acid substitutions (α)
By applying a derivative of the MK-table, Charlesworth (1994) estimated the proportion of amino-acid substitutions that are driven by positive selection, a measure referred to as α (Fig. 1; see Glossary) (Charlesworth 1994; Smith and Eyre-Walker 2002): α = 1 − (DsPn)/(DnPs). However, as the levels of nucleotide diversity and amino-acid divergence are generally low, the numbers of polymorphic sites and nonsynonymous substitutions are very small for most genes taken individually. Hence, estimates of α for single genes have inherently large sampling variances, leading to the need for pooling data across many genes (Stoletzki and Eyre-Walker 2011). Such pooling is often done by summing counts of polymorphisms and divergence in each category (Fay et al. 2001) or by taking the average across genes (Smith and Eyre-Walker 2002). By using a different parametrization of the MK test, Sawyer and Hartl (1992) used a Poisson random field (PRF) model to derive expectations for the counts of Dn, Ds, Pn and Ps by considering the processes of mutation, selection, and genetic drift (see Glossary) acting independently and simultaneously at multiple sites (Sawyer and Hartl 1992). From the PRF model, one can relate the scaled selection coefficient (γ = Ne S, where Ne represents the effective population size and s the selection coefficient) and counts of polymorphism and divergence. Based on this approach, Bayesian models were developed where the posterior distribution of scaled selection coefficients for a given locus is inferred either by assuming a fixed-effects model, where γ is constant across sites (Bustamante et al. 2002); or a random-effects model, where γ of each new mutation is drawn from a single underlying normal distribution (Sawyer et al. 2003).
However, a limitation of these approaches is that they do not account for the segregation of slightly deleterious mutations, which can bias estimates of α in a demography-dependent manner (Eyre-Walker and Keightley 2009). On the one hand, α can be underestimated if the population size has been relatively constant or decreased since the divergence from the outgroup species, because slightly deleterious mutations may be observed as polymorphisms while having a much lower chance of fixation when compared to neutral mutations. This, however, can be controlled by removing polymorphisms segregating at low frequencies (Charlesworth 1994; Smith and Eyre-Walker 2002). On the other hand, α can be overestimated if the tested population experienced a demographic expansion: as the level of polymorphism is much lower, it leads to an apparent excess of substitutions (Eyre-Walker 2002). Modelling of the full range of the fitness effects of mutations and proper accounting of the underlying demography of the sample is, therefore, needed to achieve more accurate estimates of α.
Inferring α and the distribution of fitness effects (DFE) from the site frequency spectrum (SFS)
In the following, we briefly present methods that are specifically designed to infer the distribution of fitness effects from the frequency of the derived alleles across the genome in order to estimate the rate of adaptive evolution.
-
a.
The folded/unfolded Site Frequency Spectrum (SFS)
The site frequency spectrum (SFS) is used to summarize the levels of polymorphisms in a sample of individuals. It represents the empirical distribution of the allelic frequencies for a given set of loci in the population. If the information on the ancestral allele at each variable position is available, the unfolded SFS can be computed, where the set of counts of the derived allele will be given. Conversely, if the ancestral allele cannot be inferred, the folded SFS may be calculated instead, representing the distribution of the minor allele frequencies. In these approaches, the SFS of potentially selected sites is compared to a neutral SFS. Most methods do so by comparing a non-synonymous to a synonymous SFS, however, this can also be done by contrasting genic with intergenic regions (Racimo and Schraiber 2014) or protein-binding with non-binding sites (Jenkins et al. 1995). The shape of both SFS provides crucial information on the underlying population genetic processes, such as demography and selection (Schraiber and Akey 2015; Barroso et al. 2019). For instance, slightly deleterious mutations segregate more often at low frequencies relative to neutral ones, while positively selected mutations are typically segregating at a higher frequency. But demography can also impact the SFS. For example, an expanding population has an excess of rare variants relative to what is expected in a stable population (Tajima 1989; Schraiber and Akey 2015; Barroso et al. 2019). The challenge is, therefore, to distinguish between the effect of selection and demography. This is done by assuming a neutral reference, for instance, the synonymous SFS, to which a demographic model is fitted. Selection is then inferred from the non-synonymous SFS. This assumption, together with the assumption of site independence is central to all methods inferring the distribution of fitness effects from the SFS.
-
b.
The use of divergence data
The number of substitutions is usually computed at the codon level, distinguishing nonsynonymous from synonymous substitutions, or an equivalent if non-coding DNA is used, by comparing the study species with at least one outgroup species. The outgroup sequences have to be selected with care. First, a closely-related outgroup species can potentially bias estimates of the rate of adaptive substitutions due to potentially shared polymorphisms. Second, a distantly-related outgroup species may lead to an underestimation of the divergence, and consequently of the rate of adaptive evolution, due to the possible presence of multiple “invisible” substitutions between the two species. One can potentially overcome this limitation by using multiple outgroup species, in order to span several levels of divergence and get more accurate estimates of the local substitution rate (Keightley and Jackson 2018). Moreover, if the divergence between the outgroup and the ingroup species is too high, we may suffer from the same bias as phylogenetic methods towards the more conserved genes, as fast evolving genes will not yield reliable sequence alignments. This would potentially underestimate the rate of adaptive substitutions by losing information on lineage-specific genes.
-
c.
First likelihood models of DFE accounting for slightly deleterious mutations
The first likelihood model used to estimate the molecular rate of adaptive evolution was developed by Bierne and Eyre-Walker (2004) (Fig. 1). The authors developed an extension of the MK test allowing nonsynonymous mutations to be potentially strongly advantageous. This model assumes that, for a given gene, estimates of Dn, Ds, Pn and Ps are Poisson distributed and infers the number of adaptive amino-acid substitutions (η) and α by assuming that the selection parameters are either constant across all loci or that they follow a certain DFE, in this case, a Gamma or a Beta distribution (see Box 1). Welch (2006) extended the method developed by Bierne and Eyre-Walker (2004) by including models with a continuous distribution of selection coefficients and a two weighted spikes probability distribution of α, where α takes the value α0 or α1 with probabilities q and 1 − q (Eqs. 4 and 8, respectively; Welch 2006). This likelihood framework has the advantage of enabling the comparison between nested models (Mangel and Hilborn 1996; Barton 2000): to test the occurrence of positive selection, we compare a model that potentially includes adaptive substitutions (η or α > 0) with a neutral model (η or α = 0) (Bierne and Eyre-Walker 2004; Welch 2006).
Further extensions of these methods model a deleterious DFE in the form of a Gamma distribution (Eyre-Walker et al. 2006; Keightley and Eyre-Walker 2007; Eyre-Walker and Keightley 2009). Each mutation arising at a site is ascribed a scaled selection coefficient, 4Nes, where the effective population size (Ne) is constant among loci, and s is drawn from an underlying DFE to be estimated from the data. Moreover, the SFS jointly estimates demographic parameters that allow for temporal changes in the effective population size (Eyre-Walker et al. 2006; Keightley and Eyre-Walker 2007; Eyre-Walker and Keightley 2009). These models come together in two of the most widely used inference methods: DoFE and dfe-alpha (Fig. 1, Table 1).
-
d.
Extensions accounting for beneficial mutations
The fitness effect of new mutations is unlikely to be uniform within a given gene, but is rather expected to vary according to the sequence context and the nature of the functional changes that are incurred. It is, therefore, also important to consider the contribution of beneficial mutations to the SFS in addition to deleterious mutations. Some model-based inference methods account for mutations with positive effects in the DFE. Some of these distributions are theoretically motivated by explicit fitness landscape models (see Bataillon and Bailey (2014) for a review of theoretically plausible distributions) while others are motivated by statistical convenience (to fit the data with a flexible distribution). An extension of the dfe-alpha method described above (Schneider et al. 2011) uses the unfolded SFS together with divergence data to model a Gamma DFE that also accounts for positively selected mutations (Table 1, Fig. 1). The Grapes method (Galtier 2016) can be used with both unfolded and folded SFS combined with divergence data (which is optional when the unfolded SFS is used) to model five different DFE, including the traditional Gamma distribution of deleterious mutations and four other models that account for mutations with beneficial effects (Table 1, Fig. 1). Galtier (2016) analyzed the performance of these models over 44 different datasets and observed that the GammaExponential model, which combines a Gamma distribution of deleterious mutations with an exponential distribution of beneficial mutations, and the ScaledBeta model, which uses a Beta-shaped distribution of slightly deleterious and advantageous mutations, were the ones with the best AIC scores, thus highlighting the important role of beneficial mutations in shaping the SFS. Using a similar framework, polyDFE (Tataru et al. 2017) infers the DFE from an unfolded SFS but does not require divergence data, thus allowing the estimation of the molecular adaptive rate on the branch of the study species. PolyDFE can model different DFE, including a model comprising a combination of gamma and exponential distributions to model mutations with negative and positive effects, respectively (Table 1, Fig. 1). At the level of non-coding DNA, INSIGHT (Gronau et al. 2013) contrasts the unfolded SFS and divergence in the non-coding elements of interest with those in flanking neutral sites. This method applies a generative probabilistic model by pooling data across non-coding elements considering the within-genome variation in mutation rates and coalescent times. INSIGHT models a categorical DFE, where each site is assumed to evolve under one of four different selective processes: neutral drift, strong negative selection, weak negative selection or positive selection (Table 1).
Despite their similarity, the methods above make slightly different assumptions when modeling polymorphism (SFS counts) and divergence (divergent sites relative to an outgroup). All methods assume a Poisson random field model and that the polymorphism data can be summarized by counts of the unfolded or folded SFS. Grapes, dfe-alpha and DoFE assume that the SFS is known without error, while polyDFE can model an independent rate of misorientation in the data, and INSIGHT uses a low dimensional projection of the SFS, by treating the ancestral allele as a hidden random variable in the model. Demography is either modeled via a set of nuisance parameters (Grapes, polyDFE) or assuming a fixed demographic model featuring a specific change of population size back in time that is also estimated (DFE-alpha, DoFE). Last but not least, most methods model a single SFS (synonymous versus non-synonymous) across genes, but a recent extension of polyDFE allows for fitting jointly several SFS datasets simultaneously (Tataru and Bataillon 2019). This can be used to determine whether distinct genomic regions and/or species share a common DFE, or provide evidence for differences in DFE among genomic regions/species.
-
e.
aMK and ABC-MK models
The previously described methods assume that sites evolve independently. However, there has been growing evidence that selection at linked sites might be shaping genome-wide patterns of polymorphism (Barton 1995; Andolfatto 2007; Macpherson et al. 2007). Theoretical and empirical studies showed that, besides genetic drift and purifying selection, the frequency of a given allele can also be affected by recurrent selective sweeps at closely linked positions, a process known as genetic draft (see Glossary) (Gillespie 2000). Moreover, background selection (see Glossary) can also affect polymorphism levels at neutral sites if slightly deleterious mutations are segregating, creating interference at linked sites (Charlesworth et al. 1993; Bustamante et al. 2005; Keightley and Eyre-Walker 2007; Charlesworth 2012). Messer and Petrov (2012) developed an extension of the MK test that accounts for the effects of background selection and genetic draft on the levels of polymorphisms. They define α(x) as a function of the frequency of the derived mutation: α(x) = 1 − (d0 · p(x))/d · p0(x), where p(x) and p0(x) represent the polymorphism levels at nonsynonymous and synonymous sites, for a specific derived allele frequency x. Here, any bias affecting the synonymous and nonsynonymous SFS, either demography or selection at linked sites, will be excluded, as α(x) only depends on the ratio p(x)/p0(x). The asymptotic value of α(x) is then estimated in the limit x → 1, where it should converge to the true value of α under the MK assumptions: in practice, this is done by fitting an exponential function to the data, given by: α(x) ≈ α + bexp(− cx). This function, however, assumes that all deleterious mutations have the same selection coefficient and that levels of nonsynonymous mutations decrease roughly exponentially with increasing frequency of neutral polymorphisms. Uricchio et al. (2019) extended this method by exploring the impact of background selection on the rate of adaptation using an approximate Bayesian computation (ABC) method, which the authors call ABC-MK (Table 1, Fig. 1). As in the αMK approach, this model is less sensitive to the demography of the population. Besides, it separately infers α for both weakly and strongly beneficial alleles, thus accounting for the strength of selection. To do so, ABC-MK assumes that deleterious mutations are gamma-distributed and allows α to follow a continuous distribution, from weakly to strongly beneficial mutations. As these models are less sensitive to the uncertainty associated with the demography of the population, they have the power to deliver more robust estimates of the molecular rate of adaptation on non-model organisms.
-
f.
Statistics used to infer the rate of adaptive substitutions
From the above-described methods, three major statistics are often used to qualify the rate of adaptive non-synonymous substitutions: ωa, α and Ka+. The rate of adaptive non-synonymous substitutions relative to the mutation rate, denoted as ωa, is given by ω − ωna, where ωna represents the fraction of the ω ratio contributed by neutral and deleterious mutations. The proportion of positively selected amino-acid substitutions, α, is then estimated as ωa/ω. Finally, Ka+ represents the rate of adaptive amino-acid substitutions and is given by αKa, where Ka is an alternative symbol of dN, which is the number of non-synonymous substitutions per site. Each of these statistics has its limitations. For instance, α depends both on ωa and ωna, thus differences in α may be due to variations in any of the two rates or both, making it unsuitable for distinguishing the impact of negative and positive selection. On the other hand, ωa is normalized by the mutation rate and, therefore, cannot be used to assess the impact of the mutation rate itself, which is an important varying factor along the genome. In this case, Ka+ is more appropriate (Castellano et al. 2016).
Between-species variation in the molecular adaptive rate
Several studies investigated the prevalence of positive selection in the evolution of distinct species. Here, we provide a summary of their main conclusions.
-
a.
Drosophila
Building on a long history of genetic studies, the Drosophila species complex was used in some of the pioneering research on adaptive evolution (Haudry et al. 2019). Brookfield and Sharp (1994) were the first to use the MK test to scan for signs of positive selection in Drosophila. They reported that three out of the seven genes analyzed had an excess of non-synonymous substitutions, thus suggesting that adaptive evolution was pervasive. By studying 35 genes, Smith and Eyre-Walker (2002) confirmed this hypothesis by reporting that ~ 45% of the amino-acid substitutions between D. simulans and D. yakuba were driven by positive selection. In the same year, Fay et al. (2002) estimated that ~ 70% of the amino-acid substitutions between D. simulans and D. melanogaster were adaptive. Further genome-wide studies also reported similar levels of adaptive evolution in the Drosophila genome (reviewed in Sella et al. 2009): 25 ± 20% (Bierne and Eyre-Walker 2004; Shapiro et al. 2007); 40 ± 10% (Welch 2006b); ~ 50% (Andolfatto 2007). Looking at the divergence between D. pseudoobscura and D. affinis, Haddrill et al. (2010) estimated even higher values of α, suggesting that 70–90% of the amino-acid substitutions differentiating the two species were driven by positive selection. By applying a Bayesian approach (Sawyer and Hartl 1992; Bustamante et al. 2001), Sawyer et al. (2003) estimated that ~ 94% of the substitutions were adaptive, although weakly selected (Nes ≈ 5, where s is the selection coefficient). It has been suggested, however, that these values of α could be overestimated if the current Ne is larger than the ancestral species (Eyre-Walker 2006; Rousselle et al. 2018). Nonetheless, analyses across the Drosophila genus led to similar estimates of α and, at least for D. melanogaster, the population size was inferred to have decreased (Akashi 1996; Haudry et al. 2019). Moreover, a recent study considering the past demography of the ancestral species found similar values of α to those previously reported in D. melanogaster (~ 49%, Zhen et al. 2018). These studies, therefore, provide evidence that positive selection may indeed be a prevalent mode of evolution in Drosophila genus.
-
b.
Hominids
Alongside Drosophila, humans and apes have been focal species for studies of adaptive evolution. Fay et al. (2001) reported that ~ 35% of the fixed amino-acid differences between humans and old-world monkeys were positively selected. This study, however, had the shortcoming of using a very conserved set of polymorphisms, which can overestimate the rate of non-synonymous substitutions, and consequently α (Eyre-Walker 2006). Conversely, several studies proposed that the rate of adaptive evolution is almost zero in chimpanzees (Mikkelsen et al. 2005; Hvilsom et al. 2012; Castellano et al. 2019) and within hominids (Zhang and Li 2005; Boyko et al. 2008; Eyre-Walker and Keightley 2009), suggesting that only ~ 10% of the fixed differences between humans and chimpanzees are adaptive (Bustamante et al. 2005; Boyko et al. 2008). In turn, Enard et al. (2014) found genome-wide signals of positive selection in the human genome after correcting for the effects of background selection and suggested that adaptation in humans is mainly driven by regulatory rather than by coding differences. A recent study using an improved modeling of segregating weakly deleterious mutations and accounting for the demographic history of the ancestral species reported an α value around 20%, which is consistent when using the chimpanzee or the macaque as the outgroup species (Zhen et al. 2018). The authors argued that considering the same population size for the outgroup and ancestral species could bias estimations of α, especially in humans, where the human ancestral population is known to be much smaller than that of, for example, chimpanzees or macaques. We discuss in more detail these differences across studies in the last section of this topic (f).
-
c.
Non-primate mammals
Halligan et al. (2010) reported that 57% of the amino-acid substitutions were adaptively driven in Mus musculus castaneus, a species of murid rodents. In two subspecies of the European rabbit, Oryctolagus cuniculus algirus and O. c. cuniculus, more than 60% of the amino-acid substitutions were found to be adaptive (Carneiro et al. 2012). Furthermore, a study performed on 44 non-model organisms, reported a mean value of α of around 50% in twelve mammal species (Galtier 2016).
-
d.
Plants
Studies of plants led to a huge variation in the inferred rate of molecular adaptation across species. High rates of adaptive evolution have been measured for the grand sheperd’s-purse (Slotte et al. 2010), the European aspen (Ingvarsson 2010) and species of sunflowers (Gossmann et al. 2010; Strasburg et al. 2011), where more than 30% of the amino-acid substitutions were estimated to be driven by positive selection. For the majority of plant species studied, though, α was observed to be close to zero (Gossmann et al. 2010). For example, in Arabidopsis thaliana, amino-acid substitutions are predominantly deleterious (Bustamante et al. 2002) with an average adaptive substitution rate very close to zero (Slotte et al. 2011). Authors proposed that this could be due to the Arabidopsis mating system, which by having a high frequency of inbreeding makes it harder to remove deleterious mutations (Bustamante et al. 2002). There are studies, however, reporting signs of adaptive evolution in the Arabidopsis genome. Barrier et al. (2003) found signs of positive selection in ~ 5% of the genes and Moutinho et al. (2019) showed that rates of adaptive evolution of sites at the surface of proteins are higher than the average across the genome, thus suggesting that some regions of the Arabidopsis genome are undergoing positive selection.
Slightly deleterious mutations were also observed to be prevalent in the genomes of A. lyrata (Barnaud et al. 2008; Foxe et al. 2008), Sorghum bicolor (Hamblin et al. 2006), and Zea species, (Bijlsma et al. 1986; Ross-Ibarra et al. 2009), thus suggesting very low rates of adaptive evolution also for these organisms. The reason behind such low rates of adaptive evolution in plant species is still unclear and further studies are needed to link plant adaptation at the ecological and molecular levels.
-
e.
Other species
The rate of adaptive evolution was also studied in a wide range of other organisms. For yeast (Liti et al. 2009) and the giant Galapagos tortoise (Loire et al. 2013), α was observed to be close to zero. Conversely, studies on the sea squirt (Tsagkogeorga et al. 2012) and enterobacteria (Charlesworth and Eyre-Walker 2006) reported that ~ 50% of the amino-acid substitutions are adaptive. For viruses, a high rate of adaptive substitutions is also observed: Williamson (2003) suggested that ~ 50% of the substitutions in the env gene of HIV-1 were positively selected. By accounting for the distribution of dN/dS across codons, Nielsen and Yang (2003) inferred slightly higher rates of adaptive evolution (75%). Moreover, they reported an α of about 85% in the hemagglutinin gene of the human influenza virus.
-
f.
What causes the across species variation of the rate of molecular adaptive evolution?
In the previous sections, we gave an overview of the wide range of data obtained across taxa, highlighting the great variation in the inferred rate of adaptive evolution across species (Fig. 2a). The factors determining this variability, however, remain unclear. Several studies have proposed that cross-species variation is explained by differences in effective population size (Eyre-Walker 2006; Eyre-Walker and Keightley 2009; Gossmann et al. 2012). According to this hypothesis, species with smaller Ne accumulate more weakly deleterious mutations simply by chance, thus increasing ωna and consequently reducing estimates of α. Conversely, large-Ne species are under more efficient purifying selection, hence removing mutations with negative effects from the allele pool at a faster rate. By performing a study on 44 different species, Galtier (2016) confirmed this hypothesis by showing that Ne was positively correlated with α and ωna, but not ωa.

Variation of the rate of adaptive non-synonymous substitutions (ωa; in black) and the rate of non-adaptive non-synonymous substitutions (ωna; in grey) between species (a), within genomes (b) and within genes (c). The R2 Pearson’s correlation coefficient is given along with significance denoted by asterisks (**P value < 0.01, ***P-value < 0.001). a Relationship between ωa and ωna with the level of species nucleotide diversity (π), used as a proxy for effective population size, obtained from Galtier (2016). Each sample point represents one species. Dots with bigger sizes correspond to D. melanogaster (data from Moutinho et al. 2019), which is the focus species of plots (b) and (c). b Relationship between ωa and ωna with the recombination rate in cM/Mb, taken from Moutinho et al. (2019). Each dot represents the mean value of ωa or ωna for each recombination rate class. c Relationship between ωa and ωna with the relative solvent accessibility (RSA), obtained from Moutinho et al. (2019). Each dot represents the mean value of ωa or ωna for each RSA class
On the other hand, if the population size decreases, α can also be strongly underestimated due to segregating slightly deleterious mutations, which will remain within the population (Eyre-Walker and Keightley 2009; Zhen et al. 2018). Such a scenario was reported to be the cause of very low rates of adaptive evolution in the human genome (Zhen et al. 2018). By considering the demography of the ancestral population, Zhen et al. (2018) revealed an α value of around 30%, higher than previous estimates for this species (Boyko et al. 2008; Eyre-Walker and Keightley 2009). Moreover, they found more strongly selected and/or more abundant advantageous mutations in humans when compared with mice and fruit flies. The authors proposed that these differences could reflect the number of traits under selection (Lourenço et al. 2013; Zhen et al. 2018). According to this hypothesis, larger long-lived organisms, such as humans, have less capacity to adapt to new environments, due to the greater number of traits under selection. Such organisms are theoretically expected to need more consecutive beneficial mutations to reach their fitness optimum, and thus a higher proportion of beneficial mutations should be accordingly detected in these species (Lourenço et al. 2013; Rousselle et al. 2018, 2019b). More studies are needed to clarify what is causing the observed differences between species.
Within-genome variation of the molecular rate adaptation
Several studies provided evidence for a substantial variation in the rate of adaptive substitutions along the genome. In this section, we summarize the factors that were found to influence the distribution of adaptive substitutions within species (Fig. 1).
-
a.
Genome-wide variables
At the genome level, recombination, mutation and gene density are important determinants of the rate of adaptive substitutions (ωa) (Marais and Charlesworth 2003; Campos et al. 2014; Castellano et al. 2016). Recombination rate is predicted to favor the fixation of adaptive substitutions (Fig. 2b) by breaking down linkage disequilibrium (Marais and Charlesworth 2003; Campos et al. 2014; Castellano et al. 2016). Advantageous mutations occurring at linked sites but in distinct individuals will interfere, so that only one will ultimately reach fixation unless a recombination event creates a haplotype carrying both of them (Hill-Robertson interference, HRi; Hill and Robertson 1966; Felsenstein 1974). As a result, genes in low recombining regions are expected to have overall lower rates of adaptive substitutions. Following a similar rationale, genes present in regions with high gene density may be subject to stronger HRi and slow rates of adaptive evolution (Castellano et al. 2016). In turn, genes with high mutation rates potentially adapt faster because they increase the levels of genetic diversity, which, consequently, increases the chance of selection operating such that adaptive processes may occur. Interestingly, Castellano et al. (2016) found that the positive correlation between mutation rate and the rate of adaptive substitutions no longer holds for genes located in regions with low recombination rate and high gene density, thus suggesting a strong effect of HRi in the presence of a large number of selected mutations with a small genetic distance between them. Similarly, Gossmann et al. (2011) observed that variations in Ne resulting from linked selection along the genome significantly impact the efficiency of natural selection in C. grandiflora and A. thaliana, where regions with larger Ne are subject to stronger purifying selection.
-
b.
Protein-coding: gene-wide variables
On a gene-wide scale, it has been reported that protein function strongly influences the rate of adaptive evolution, with genes involved in the immune response presenting the highest rates of adaptation in Drosophila (Sackton et al. 2007; Obbard et al. 2009), Arabidopsis (Slotte et al. 2011), hominids (Nielsen et al. 2005; Kosiol et al. 2008) and other mammals (Kosiol et al. 2008). Sex-related genes were also reported to present higher rates of adaptive evolution in Drosophila (Pröschel et al. 2006; Haerty et al. 2007) chimpanzees (Hvilsom et al. 2012) and in plants (Gossmann et al. 2014; Crowson et al. 2017). Moreover, a recent study showed that genes involved in protein biosynthesis and signaling for protein degradation exhibit the highest rates of adaptive substitutions in Drosophila and Arabidopsis (Moutinho et al. 2019). Cytochrome P450 proteins, which are involved in defense response in plants, were also characterized by high rates of adaptation in Arabidopsis (Moutinho et al. 2019). Several studies have described that host–pathogen interactions act as key drivers of protein evolution in several taxa (Sackton et al. 2007; Obbard et al. 2009; Enard et al. 2016; Ebel et al. 2017; Mauch-Mani et al. 2017; Uricchio et al. 2019; Grandaubert et al. 2019), which could explain the observed high levels of adaptive evolution in the functions described above. Moreover, mean gene expression levels and the breadth of expression negatively impact the rate of adaptive evolution in Drosophila, where the two factors may be acting together (Duret and Mouchiroud 2000; Salvador-Martínez et al. 2018; Moutinho et al. 2019). This relationship with expression may be a consequence of stronger purifying selection in highly expressed genes, where selection acts by favoring proteins with the lowest probability of misfolding, which occurs if the protein sequence accumulates translational missense errors (Drummond et al. 2005). Additionally, the macromolecular structure of the protein was also observed to substantially impact the rate of protein adaptation in humans (Afanasyeva et al. 2018), Drosophila and Arabidopsis (Moutinho et al. 2019). In this case, proteins with a higher proportion of disordered regions (Afanasyeva et al. 2018; Moutinho et al. 2019) and/or exposed residues (Moutinho et al. 2019) are prone to accumulate more adaptive mutations, acting as important targets of positive selection.
-
c.
Protein-coding: intra-molecular factors
There is growing evidence that adaptive substitution rates also vary significantly at the intra-genic level. Studies both at the population and divergence level, have shown that the relative solvent accessibility (RSA) significantly impacts the rate of amino-acids substitutions (Fig. 2c), with exposed residues accumulating more adaptive mutations than buried ones (Goldman et al. 1998; Mirny and Shakhnovich 1999; Franzosa and Xia 2009; Liberles et al. 2012; Moutinho et al. 2019). When contrasted with the effect of residue intrinsic disorder, RSA was observed to contribute with most of the variation in ωa (95% and 87% of variance explained for A. thaliana and D. melanogaster, respectively; Moutinho et al. 2019). This suggests that solvent exposure is the main determinant of adaptive evolution at the level of protein structure, and that protein intrinsic disorder contributes with a mere additive small effect to the rate of protein adaptation (Moutinho et al. 2019). Furthermore, the type of amino-acid mutation was also reported to be an important factor affecting the rate of adaptive evolution, with more similar amino-acid changes presenting higher rates of adaptive substitutions (Grantham 1974; Miyata et al. 1979; Bergman and Eyre-Walker 2019).
-
d.
Non-coding DNA
While much attention has been given to the study of the adaptive evolution of protein-coding genes, there is increasing evidence that the non-coding regions of the genome are also key targets of positive selection. By using an MK-like approach, contrasting numbers of polymorphisms and substitutions at protein-binding and non-binding sites, Jenkins et al. (1995) reported signatures of adaptive change in the control for gene expression in D. melanogaster. Kohn et al. (2004) estimated that ~ 50% of all substitutions in the 5′ region of eight Drosophila genes were adaptively driven. By extending these approaches, Andolfatto (2005) investigated patterns of molecular evolution in multiple classes of non-coding DNA in D. melanogaster and found that around 60% and 20% of the total nucleotide divergence with D. simulans were fixed by positive selection, in UTRs and intronic/intergenic regions respectively. These findings suggest that the noncoding regions of the D. melanogaster genome are key determinants of adaptive evolution. Likewise, Haddrill et al. (2008) found signs of adaptive evolution in the non-coding regions of the D. simulans genome. These patterns go beyond the Drosophila genus since there is evidence of widespread positive selection in noncoding conserved regions along the Brassicaceae phylogeny (Williamson et al. 2014). In hominids, however, the opposite pattern is observed. Keightley et al. (2005) analyzed the downstream and upstream regions of protein-coding genes using an MK approach and found no signs of adaptive evolution. This result might reflect the overall low levels of adaptive evolution in hominid genomes due to the lower effective population sizes. With the thrive of full genome sequence data, adaptive evolution can now be more extensively studied outside the coding regions (Gronau et al. 2013), which, until now, were the focus of most studies.
Current limitations and future perspectives
In the last two decades, numerous methods have been developed to detect and quantify adaptive evolution. This, together with the availability of datasets spanning many genes and species, increased our knowledge of the factors underlying the heterogeneity of rates of molecular adaptation within genomes and between species. However, existing methods rely on several assumptions that can create biases in the estimates of adaptive evolution when not met. For instance, the methods reviewed here assume that synonymous mutations are neutral, which may not always be a valid approximation, especially in species with large effective population sizes (Lawrie et al. 2013). Several studies have documented that selection for codon usage also affects the rate of synonymous substitutions in several species, including Drosophila (Akashi 1994; Comeron et al. 1999), the European aspen (Ingvarsson 2010) and non-model animals (Galtier et al. 2018), mammals and birds (Rousselle et al. 2019a). Finding a proper neutral reference remains a challenging goal. Yet, a similar approach to that used in codon models (Yang and Nielsen 2008; Spielman and Wilke 2016; Rodrigue and Lartillot 2017) could, in principle, be considered for methods inferring the rate of adaptive evolution by accounting for the evolution of synonymous sites. This would lead to a more realistic null model of neutral evolution and, consequently, less biased estimates of the molecular rate of adaptation (Rodrigue and Lartillot 2017).
Another challenge consists of better accounting for the confounding effects of demography. Some methods fit a simplified demographic model (DFE-alpha, DoFE) while others correct for demography by adding extra parameters, one per frequency category of the SFS (Grapes, polyDFE). The number of such parameters, therefore, increases with the sample size and can quickly lead to model overparameterization issues. Extending the methods to use a continuous SFS constitutes one perspective to accommodate increasingly larger datasets. Alternatively, the demography of the population could also be estimated from the currently available coalescent methods (i.e. the SMC ++, Terhorst et al. 2017; or ∂a∂i, Gutenkunst et al. 2009).
Besides, current models often assume a constant DFE across the whole genome. This can lead to a bad model fit because selection varies within and between genomic regions. Such an assumption can be relaxed by allowing DFE parameters to vary along the genome. Moreover, the use of an outgroup species to infer the ancestral allele (polymorphism orientation) can lead to biases in the estimates of adaptive evolution, whether the outgroup is a very closely-related species or a very distantly-related one (Hernandez et al. 2007). This can be alleviated by using multiple outgroup species and probabilistic ancestral allele reconstructions (e.g. Keightley and Jackson 2018). Furthermore, by using only one outgroup sequence, these methods are estimating divergence on the total branch separating the focal and the outgroup species. Using a second outgroup species and a phylogenetic approach, however, would allow restricting the estimation of the divergence parameters to the branch of the study species.
Furthermore, these methods assume that all sites are equally sampled in all individuals and do not intrinsically account for the possibility of missing data. Pre-processing of the data is therefore required, which can introduce biases if too many sites have to be discarded. Finally, methods relying on patterns of polymorphism cannot track positively selected mutations of individual sites, limiting the power of these analyses in detecting positive selection at the site level. Combing such population genetics approaches with mutation accumulation experiments is a promising avenue to further understand the fitness effect of particular mutations. This, however, would have to be done across several generations so that enough mutations could be generated.
Conclusions
The development of statistical approaches based on the pioneering work of McDonald and Kreitman (1991), together with the increasing availability of genome sequences at the population level, paved the way for the qualitative and quantitative assessment of rates of adaptive evolution, both between species and within genomes. Growing evidence suggests a substantial variation of the molecular adaptive rate at distinct levels of molecular evolution, emphasizing the multitude of factors that can influence the rate of adaptation. These studies introduced a conceptual and theoretical framework that, we posit, will serve as a basis for increasingly realistic models that will strengthen our understanding of the fitness effect of new mutations and, therefore, the molecular basis of adaptation.
References
Afanasyeva A, Bockwoldt M, Cooney CR et al (2018) Human long intrinsically disordered protein regions are frequent targets of positive selection. Genome Res 28(7):975–982
Akashi H (1994) Synonymous codon usage in drosophila melanogaster: natural selection and translational accuracy. Genet Soc Am 136:927–935
Akashi H (1996) Molecular evolution between drosophila melanogaster and D. sirnulam reduced codon bias, faster rates of amino acid substitution, and larger proteins in D. melarwgaster. DNA Seq 144:1297–1307
Andolfatto P (2005) Adaptive evolution of non-coding DNA in Drosophila. Nature 437(7062):1149–1152
Andolfatto P (2007) Hitchhiking effects of recurrent beneficial amino acid substitutions in the. Genome Res 17(12):1755–1762
Barnaud A, Trigueros G, McKey D, Joly HI (2008) High outcrossing rates in fields with mixed sorghum landraces: how are landraces maintained? Heredity 101(5):445–452
Barrier M, Bustamante CD, Yu J, Purugganan MD (2003) Selection on rapidly evolving proteins in the arabidopsis genome. Genetics 163(2):723–733
Barroso GV, Moutinho AF, Dutheil JY (2019) A population genetics lexicon. In: Dutheil JY (ed) Statistical population genomics. Springer, Berlin
Barton NH (1995) Linkage and the limits to natural selection. Genetics 140(2):821–841
Barton NH (2000) Estimating linkage disequilibria. Heredity 84(2):373–389
Bataillon T (2003) Shaking the “deleterious mutations” dogma? Trends Ecol Evol 18(7):315–317
Bataillon T, Bailey SF (2014) Effects of new mutations on fitness: insights from models and data. Ann N Y Acad Sci 1320(1):76–92
Bergman J, Eyre-Walker A (2019) Does adaptive protein evolution proceed by large or small steps at the amino acid level? Mol Biol Evol 36(5):990–998
Bierne N, Eyre-Walker A (2004) The genomic rate of adaptive amino acid substitution in Drosophila. Mol Biol Evol 21(7):1350–1360
Bijlsma R, Allard RW, Kahler AL (1986) Non random mating in an open-pollinated maize population. Genetics 112(3):669–680
Boyko AR, Williamson SH, Indap AR et al (2008) Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet 4(5):e1000083
Brookfield JF, Sharp PM (1994) Neutralism and selectionism face up to DNA data. Trends Genet 10(4):109–111
Bustamante CD, Wakeley J, Sawyer S, Hartl DL (2001) Directional selection and the site-frequency spectrum. Genetics 159(4):1779–1788
Bustamante CD, Nielsen R, Sawyer SA et al (2002) The cost of inbreeding in arabidopsis. Nature 416(6880):531–534
Bustamante CD, Fledel-Alon A, Williamson S et al (2005) Natural selection on protein-coding genes in the human genome. Nature 437(7062):1153–1157
Campos JL, Halligan DL, Haddrill PR, Charlesworth B (2014) The relation between recombination rate and patterns of molecular evolution and variation in drosophila melanogaster. Mol Biol Evol 31(4):1010–1028
Carneiro M, Albert FW, Melo-Ferreira J et al (2012) Evidence for widespread positive and purifying selection across the european rabbit (oryctolagus cuniculus) genome. Mol Biol Evol 29(7):1837–1849
Castellano D, Coronado-Zamora M, Campos JL et al (2016) Adaptive evolution is substantially impeded by hill-Robertson interference in drosophila. Mol Biol Evol 33(2):442–455
Castellano D, Macià MC, Tataru P et al (2019) Comparison of the full distribution of fitness effects of new amino acid mutations across great apes. Genetics 213:696971
Charlesworth B (1994) The effect of background selection against deleterious mutations on weakly selected, linked variants. Genet Res 63(3):213–227
Charlesworth B (2012) The effects of deleterious mutations on evolution at linked sites. Genetics 190(1):5–22
Charlesworth B, Charlesworth D (2010) Elements of evolutionary genetics. Roberts and Company Publishers, Englewood
Charlesworth J, Eyre-Walker A (2006) The rate of adaptive evolution in enteric bacteria. Mol Biol Evol 23(7):1348–1356
Charlesworth B, Morgan MT, Charlesworth D (1993) The effect of deleterious mutations on neutral molecular variation. Genetics 134(4):289–303
Comeron JM, Kreitman M, Aguadé M (1999) Natural selection on synonymous sites is correlated with gene length and recombination in drosophila. Genetics 151(1):239–249
Crowson D, Barrett SCH, Wright SI (2017) Purifying and positive selection influence patterns of gene loss and gene expression in the evolution of a plant sex chromosome system. Mol Biol Evol 34(5):1140–1154
Dong S, Stam R, Cano LM et al (2014) Effector specialization in a lineage of the Irish potato famine pathogen. Science 343(6170):552–555
Drummond DA, Bloom JD, Adami C et al (2005) Why highly expressed proteins evolve slowly. Proc Natl Acad Sci 102(40):14338–14343
Duret L, Mouchiroud D (2000) Determinants of substitution rates in mammalian genes: expression pattern affects selection intensity but not mutation rate. Mol Biol Evol 17(1):68–74
Ebel ER, Telis N, Venkataram S et al (2017) High rate of adaptation of mammalian proteins that interact with Plasmodium and related parasites. PLoS Genet 13(9):e1007023
Enard D, Messer PW, Petrov DA (2014) Genome-wide signals of positive selection in human evolution. Genome Res 24(6):885–895
Enard D, Cai L, Gwennap C, Petrov DA (2016) Viruses are a dominant driver of protein adaptation in mammals. Elife. 5:e12469
Eyre-Walker A (2002) Changing effective population size and the McDonald-Kreitman test. Genetics 162(4):2017–2024
Eyre-Walker A (2006) The genomic rate of adaptive evolution. Trends Ecol Evol 21:569–575
Eyre-Walker A, Keightley PD (2009) Estimating the rate of adaptive molecular evolution in the presence of slightly deleterious mutations and population size change. Mol Biol Evol 26(9):2097–2108
Eyre-Walker A, Woolfit M, Phelps T (2006) The distribution of fitness effects of new deleterious amino acid mutations in humans. Genetics 173(2):891–900
Fay JC, Wyckoff GJ, Wu CI (2001) Positive and negative selection on the human genome. Genetics 158:1227–1234
Fay J, Wyckoff G, Wu C (2002) Testing the neutral theory of molecular evolution with genomic data from Drosophila. Nature 415(6875):1024–1026
Foxe JP, Dar VUN, Zheng H et al (2008) Selection on amino acid substitutions in Arabidopsis. Mol Biol Evol 25(7):1375–1383
Franzosa EA, Xia Y (2009) Structural determinants of protein evolution are context-sensitive at the residue level. Mol Biol Evol 26(10):2387–2395
Galtier N (2016) Adaptive protein evolution in animals and the effective population size hypothesis. PLoS Genet 12(1):e1005774
Galtier N, Roux C, Rousselle M et al (2018) Codon usage bias in animals: disentangling the effects of natural selection, effective population size, and GC-biased gene conversion. Mol Biol Evol 35(5):1092–1103
Gillespie JH (2000) Genetic drift in infinite populations: the pseudohitchhiking model. Genetics 155:909–919
Goldman N, Thorne JL, Jones DT (1998) Assessing the impact of secondary structure and solvent accessibility on protein evolution. Genetics 149:445–458
Gossmann TI, Song BH, Windsor AJ et al (2010) Genome wide analyses reveal little evidence for adaptive evolution in many plant species. Mol Biol Evol 27(8):1822–1832
Gossmann TI, Woolfit M, Eyre-Walker A (2011) Quantifying the variation in the effective population size within a genome. Genetics 189(4):1389–1402
Gossmann TI, Keightley PD, Eyre-Walker A (2012) The effect of variation in the effective population size on the rate of adaptive molecular evolution in eukaryotes. Genome Biol Evol 4(5):658–667
Gossmann TI, Schmid MW, Grossniklaus U, Schmid KJ (2014) Selection-driven evolution of sex-biased genes is consistent with sexual selection in arabidopsis thaliana. Mol Biol Evol 31(3):574–583
Grandaubert J, Dutheil JY, Stukenbrock EH (2019) The genomic determinants of adaptive evolution in a fungal pathogen. Evolution Letters 3(3):299–312
Grantham R (1974) Amino acid difference formula to help explain protein evolution. Science 185(4154):862–864
Gronau I, Arbiza L, Mohammed J, Siepel A (2013) Inference of natural selection from interspersed genomic elements based on polymorphism and divergence. Mol Biol Evol 30(5):1159–1171
Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD (2009) Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet 5:e1000695
Haddrill PR, Bachtrog D, Andolfatto P (2008) Positive and negative selection on noncoding DNA in Drosophila simulans. Mol Biol Evol 25(9):1825–1834
Haddrill PR, Loewe L, Charlesworth B (2010) Estimating the parameters of selection on nonsynonymous mutations in Drosophila pseudoobscura and D. miranda. Genetics 185(4):1381–1396
Haerty W, Jagadeeshan S, Kulathinal RJ et al (2007) Evolution in the fast lane: rapidly evolving sex-related genes in Drosophila. Genetics 177(3):1321–1335
Halligan DL, Oliver F, Eyre-Walker A et al (2010) Evidence for pervasive adaptive protein evolution in wild mice. PLoS Genet 6(1):e1000825
Hamblin MT, Casa AM, Sun H et al (2006) Challenges of detecting directional selection after a bottleneck: lessons from Sorghum bicolor. Genetics 173(2):953–964
Haudry A, Laurent S, Kapun M (2019) Statistical population genomics of fruit flies. In: Dutheil JY (ed) Statistical population genomics. Springer, Berlin
Hernandez RD, Williamson SH, Bustamante CD (2007) Context dependence, ancestral misidentification, and spurious signatures of natural selection. Mol Biol Evol 24(8):1792–1800
Hey J (1999) The neutralist, the fly and the selectionist. Trends Ecol Evol 14:35–38
Hill WG, Robertson A (1966) The effect of linkage on limits to artificial selection. Genet Res 8(3):269–294
Hilson P, Allemeersch J, Altmann T et al (2004) Versatile gene-specific sequence tags for Arabidopsis functional genomics: transcript profiling and reverse genetics applications. Genome Res 14:2176–2189
Hoekstra HE, Hirschmann RJ, Bundey RA et al (2006) A single amino acid mutation contributes to adaptive beach mouse color pattern. Science 313(5783):101–104
Hudson RR, Kreitman M, Aguadé M (1987) A test of neutral molecular evolution based on nucleotide data. Genetics 116:153–159
Hvilsom C, Qian Y, Bataillon T et al (2012) Extensive X-linked adaptive evolution in central chimpanzees. Proc Natl Acad Sci 109(6):2054–2059
Ingvarsson PK (2010) Natural selection on synonymous and nonsynonymous mutations shapes patterns of polymorphism in populus tremula. Mol Biol Evol 27(3):650–660
Jenkins DL, Ortori CA, Brookfield JFY (1995) A test for adaptive change in DNA sequences controlling transcription. Proc R Soc B Biol Sci 261(1361):203–207
Jensen JD, Payseur BA, Stephan W et al (2019) The importance of the neutral theory in 1968 and 50 years on: a response to Kern and Hahn 2018. Evolution 73(1):111–114
Jones MR, Mills LS, Alves PC et al (2018) Adaptive introgression underlies polymorphic seasonal camouflage in snowshoe hares. Science 360(6395):1355–1358
Keightley PD, Eyre-Walker A (2007) Joint inference of the distribution of fitness effects of deleterious mutations and population demography based on nucleotide polymorphism frequencies. Genetics 177(4):2251–2261
Keightley PD, Jackson BC (2018) Use of transgene-induced rnai to regulate endogenous gene expression. Genetics 209:897–906
Keightley PD, Lercher MJ, Eyre-Walker A (2005) Evidence for widespread degradation of gene control regions in hominid genomes. PLoS Biol 3(2):0282–0288
Kern AD, Hahn MW (2018) The neutral theory in light of natural selection. Mol Biol Evol 35:1366–1371
Kimura M (1968) Evolutionary rate at the molecular level. Nature 217:624–626
Kohn MH, Fang S, Wu CI (2004) Inference of positive and negative selection on the 5′ regulatory regions of drosophila genes. Mol Biol Evol 21(2):374–383
Kosakovsky Pond SL, Murrell B, Fourment M et al (2011) A random effects branch-site model for detecting episodic diversifying selection. Mol Biol Evol 28(11):3033–3043
Kosiol C, Vinař T, Da Fonseca RR et al (2008) Patterns of positive selection in six mammalian genomes. PLoS Genet 4(8):e1000144
Lawrie DS, Messer PW, Hershberg R, Petrov DA (2013) Strong purifying selection at synonymous sites in D. melanogaster. PLoS Genet 9:33–40
Li W-H, Wu C-I, Luo C-C (1985) A new method for extimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon usage. Mol Biol Evol 2(2):150–174
Liberles DA, Teichmann SA, Bahar I et al (2012) The interface of protein structure, protein biophysics, and molecular evolution. Protein Sci 21:769–785
Liti G, Carter DM, Moses AM et al (2009) Population genomics of domestic and wild yeasts. Nature 458(7236):337–341
Loire E, Chiari Y, Bernard A et al (2013) Population genomics of the endangered giant Galápagos tortoise. Genome Biol 14(12):R136
Lourenço JM, Glémin S, Galtier N (2013) The rate of molecular adaptation in a changing environment. Mol Biol Evol 30(6):1292–1301
Macpherson JM, Sella G, Davis JC, Petrov DA (2007) Genomewide spatial correspondence between nonsynonymous divergence and neutral polymorphism reveals extensive adaptation in Drosophila. Genetics 177(4):2083–2099
Marais G, Charlesworth B (2003) Genome evolution: recombination speeds up adaptive evolution. Curr Biol 13(2):68–70
Mauch-Mani B, Baccelli I, Luna E, Flors V (2017) Defense priming: an adaptive part of induced resistance. Annu Rev Plant Biol 68(1):485–512
McDonald J, Kreitman M (1991) Adaptive evolution at the Adh locus in Drosophila. Nature 351:652–654
Messer PW, Petrov DA (2012) Frequent adaptation and the McDonald-Kreitman Test. Proc Natl Acad Sci 110(21):8615–8620
Mikkelsen TS, Hillier LW, Eichler EE et al (2005) Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437(7055):69–87
Mirny LA, Shakhnovich EI (1999) Universally conserved positions in protein folds: reading evolutionary signals about stability, folding kinetics and function. J Mol Biol 291:177–196
Miyata T, Miyazawa S, Yasunaga T (1979) Two types of amino acid substitutions in protein evolution. J Mol Evol 12:219–236
Moutinho AF, Trancoso FF, Dutheil JY (2019) The impact of protein architecture on adaptive evolution. Mol Biol Evol 36(9):2013–2028
Nielsen R, Yang Z (1998) Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936
Nielsen R, Yang Z (2003) Estimating the distribution of selection coefficients from phylogenetic data with applications to mitochondrial and viral DNA. Mol Biol Evol 20(8):1231–1239
Nielsen R, Bustamante C, Clark AG et al (2005) A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol 3(6):0976–0985
Obbard DJ, Welch JJ, Kim KW, Jiggins FM (2009) Quantifying adaptive evolution in the Drosophila immune system. PLoS Genet 5(10):e1000698
Ohta T (1992) The nearly neutral theory of molecular evolution. Annu Rev Ecol Syst 23:263–286
Ometto L, Glinka S, De Lorenzo D, Stephan W (2005) Inferring the effects of demography and selection on Drosophila melanogaster populations from a chromosome-wide scan of DNA variation. Mol Biol Evol 22:2119–2130
Pröschel M, Zhang Z, Parsch J (2006) Widespread adaptive evolution of Drosophila genes with sex-biased expression. Genetics 174(2):893–900
Racimo F, Schraiber JG (2014) Approximation to the distribution of fitness effects across functional categories in human segregating polymorphisms. PLoS Genet 10(11):e1004697
Rodrigue N, Lartillot N (2017) Detecting adaptation in protein-coding genes using a Bayesian site-heterogeneous mutation-selection codon substitution model. Mol Biol Evol 34(1):204–214
Ross-Ibarra J, Tenaillon M, Gaut BS (2009) Historical divergence and gene flow in the genus Zea. Genetics 181(4):1399–1413
Rousselle M, Mollion M, Nabholz B et al (2018) Overestimation of the adaptive substitution rate in fluctuating populations. Biol Lett 14(5):20180055
Rousselle M, Laverré A, Figuet E et al (2019a) Influence of recombination and GC-biased gene conversion on the adaptive and nonadaptive substitution rate in mammals versus birds. Mol Biol Evol 36:458–471
Rousselle M, Simion P, Tilak M-K et al (2019b) Is adaptation limited by mutation? A timescale dependent effect of genetic diversity on the adaptive substitution rate in animals. bioRxiv 64:3619
Roux J, Privman E, Moretti S et al (2014) Patterns of positive selection in seven ant genomes. Mol Biol Evol 31:1661–1685
Rutter MT, Roles A, Conner JK et al (2012) Fitness of Arabidopsis thaliana mutation accumulation lines whose spontaneous mutations are known. Evolution 66(7):2335–2339
Sackton TB, Lazzaro BP, Schlenke TA et al (2007) Dynamic evolution of the innate immune system in Drosophila. Nat Genet 39(12):1461–1468
Salvador-Martínez I, Coronado-Zamora M, Castellano D et al (2018) Mapping selection within drosophila melanogaster Embryo’s Anatomy. Mol Biol Evol 35(1):66–79
Sawyer S, Hartl D (1992) Population genetics of polymorphism and divergence. Genetics 132:1161–1176
Sawyer SA, Kulathinal RJ, Bustamante CD, Hartl DL (2003) Bayesian analysis suggests that most amino acid replacements in drosophila are driven by positive selection. J Mol Evol 57:154–164
Schneider A, Charlesworth B, Eyre-Walker A, Keightley PD (2011) A method for inferring the rate of occurrence and fitness effects of advantageous mutations. Genetics 189:1427–1437
Schraiber JG, Akey JM (2015) Methods and models for unravelling human evolutionary history. Nat Rev Genet 16(12):727–740
Sella G, Petrov DA, Przeworski M, Andolfatto P (2009) Pervasive natural selection in the Drosophila genome? PLoS Genet 5(6):e1000495
Shapiro JA, Huang W, Zhang C et al (2007) Adaptive genic evolution in the Drosophila genomes. Proc Natl Acad Sci 104(7):2271–2276
Shaw FH, Geyer CJ, Shaw RG (2002) A comprehensive model of mutations affecting fitness and inferences for Arabidopsis thaliana. Evolution 56:453–463
Slotte T, Foxe JP, Hazzouri KM, Wright SI (2010) Genome-wide evidence for efficient positive and purifying selection in capsella grandiflora, a plant species with a large effective population size. Mol Biol Evol 27(8):1813–1821
Slotte T, Bataillon T, Hansen TT et al (2011) Genomic determinants of protein evolution and polymorphism in arabidopsis. Genome Biol Evol 3:1210–1219
Smith NGC, Eyre-Walker A (2002) Adaptive protein evolution in Drosophila. Nature 415:1022–1024
Spielman SJ, Wilke CO (2016) Extensively parameterized mutation-selection models reliably capture site-specific selective constraint. Mol Biol Evol 33(11):2990–3001
Stoletzki N, Eyre-Walker A (2011) Estimation of the neutrality index. Mol Biol Evol 28:63–70
Strasburg JL, Kane NC, Raduski AR et al (2011) Effective population size is positively correlated with levels of adaptive divergence among annual sunflowers. Mol Biol Evol 28(5):1569–1580
Stukenbrock EH, Bataillon T, Dutheil JY et al (2011) The making of a new pathogen: insights from comparative population genomics of the domesticated wheat pathogen Mycosphaerella graminicola and its wild sister species. Genome Res 21(12):2157–2166
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tataru P, Bataillon T (2019) PolyDFEv2.0: testing for invariance of the distribution of fitness effects within and across species. Bioinformatics 3:1–2
Tataru P, Mollion M, Glémin S, Bataillon T (2017) Inference of distribution of fitness effects and. Genetics 207:1103–1119
Terhorst J, Kamm JA, Song YS (2017) Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat Genet 49:303–309
Tsagkogeorga G, Cahais V, Galtier N (2012) The population genomics of a fast evolver: high levels of diversity, functional constraint, andmolecular adaptation in the tunicate Ciona intestinalis. Genome Biol Evol 4:740–749
Uricchio LH, Petrov DA, Enard D (2019) Exploiting selection at linked sites to infer the rate and strength of adaptation. Nat Ecol Evol. 3(6):977
Voight BF, Kudaravalli S, Wen X, Pritchard JK (2006) A map of recent positive selection in the human genome. PLoS Biol 4:0446–0458
Welch JJ (2006) Estimating the genomewide rate of adaptive protein evolution in drosophila. Genetics 173(2):821–837
Williamson S (2003) Adaptation in the env gene of HIV-1 and evolutionary theories of disease progression. Mol Biol Evol 20(8):1318–1325
Williamson RJ, Josephs EB, Platts AE et al (2014) Evidence for widespread positive and negative selection in coding and conserved noncoding Regions of Capsella grandiflora. PLoS Genet 10(9):e1004622
Yang Z, Nielsen R (1998) Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J Mol Evol 46(4):409–418
Yang Z, Nielsen R (2002) Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol 19(2):908–917
Yang Z, Nielsen R (2008) Mutation-selection models of codon substitution and their use to estimate selective strengths on codon usage. Mol Biol Evol 25(3):568–579
Yang Z, Nielsen R, Goldman N, Pedersen AK (2000) Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155(1):431–449
Yang Z, Wong WSW, Nielsen R (2005) Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol 22(4):1107–1118
Zhang L, Li WH (2005) Human SNPs reveal no evidence of frequent positive selection. Mol Biol Evol 22(12):2504–2507
Zhang J, Nielsen R, Yang Z (2005) Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol 22(12):2472–2479
Zhen Y, Huber CD, Davies RW, Lohmueller KE (2018) Stronger and higher proportion of beneficial amino acid changing mutations in humans compared to mice and flies. https://doi.org/10.1101/427583
Acknowledgements
Open access funding provided by Max Planck Society. The authors thank Adam Eyre-Walker for fruitful discussions. JYD acknowledges funding from the Max Planck Society.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No conflict of interest is to be declared.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Moutinho, A.F., Bataillon, T. & Dutheil, J.Y. Variation of the adaptive substitution rate between species and within genomes. Evol Ecol 34, 315–338 (2020). https://doi.org/10.1007/s10682-019-10026-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10682-019-10026-z