
Abstract
In this study, quantum mechanical calculations, such as density functional theory (DFT), have been employed to determine the active positions of nanosensor and thermodynamics functions of interaction between Ag+ and nanosensor have been calculated. HOMO and LUMO energies and energy difference between donor atoms (i) and acceptor atoms (j) have been evaluated. In addition, the effect of the number of substitution agents on the reactivity of the functional carbon nanotubes and the charge on the interacting atoms and Ag+ before and after interaction have been investigated. The geometry optimization and theoretical calculations have been carried out using B3LYP level of theory. Results show that the interaction of Ag+ with nanosensor is in terms of thermodynamically possible. The negative values of ΔG° denote a spontaneous reaction and the negative values of ΔH° represent an exothermic reaction. In addition, the nanosensor has two active positions and the product obtained through the interaction between Ag+ and oxygen of the carbonyl group is the most stable state. The interaction of Ag+ with the nanosensor is accompanied by a reduction in the energy gap (Eg) which increases the stability of the complex, causes indicating that a charge transfer occurred between the nanosensor and Ag+.
Similar content being viewed by others
Introduction
The silver ions can leak into industrial wastewater due to the corrosion of tubes and the inner surfaces of generators. SWCNT-CONH-(CH2)6NH2 is a good absorbent for silver cations. Based on the library research, 1,6-diaminohexane “NH2(CH2)6NH2”, has been used to design a nanosensor for the first time.
Over the past 25 years or so, therefore, considerable efforts have been made to utilize the unique properties of carbon nanomaterials, including fullerenes, carbon nanotubes (CNTs), and graphene, as energy materials, and tremendous progress has been made in developing high-performance energy conversion [1]. CNTs have long been recognized as the stiffest and strongest man-made material to date. In addition, their high electrical conductivity has aroused interest in the area of electrical appliances and communication-related applications [2, 3]. The strength and flexibility of CNTs have made them applicable to the other nanometric structures [4]. One of the ways to study the features of CNTs is simulation. Many of these simulations can be done through computer software or programming languages [5]. On the other hand, not all existing calculation methods are suitable for the study of a molecule, and appropriate calculation methods have to be used to investigate each molecule [6]. The first multi-walled carbon nanotube (MWCNT) and single-walled carbon nanotube (SWCNT) were built by Iijima [7] in 1991 and Donald Bethune [8] in 1993, respectively. Their discoveries were simultaneous with none of them knowing about the other’s work. When used as fillers, CNTs enable the fabrication of composites with unmatched multi-functional characteristics because of their distinctive structural and transport properties, such as excellent strength, modulus, electrical and thermal conductivities along with low density. CNTs of different forms, geometries and functionalities can be exploited. In fact, depending on preparation method and processing conditions, single-wall (SW), double-wall (DW) or multi-wall (MW) CNTs of various lengths, diameters (and, hence, aspect ratio) and chirality can be incorporated. This is while suitable chemical or physical treatments may lead to different surface morphologies, which, in turn, may improve their dispersion and adhesion in the polymeric matrix [9]. The reactivity of CNTs depends on the unbalance of the orbital π of the carbon atoms that is caused by superficial curvature. Therefore, less-diameter nanotubes are more reactive [10, 11]. Adding amine groups to lateral walls of CNTs increases the solubility of the nanotubes in water which has potential biological applications [12]. Also, given ratios on the convex surfaces of CNTs, the functionalization of CNTs by combining different reactive groups such as –OH, –NH2, –COOH and –Br has many applications in different fields. CNTs were first introduced by Britto et al. [13] for use in electrochemistry due to their small size and conductivity. They considered the smallest possible electrodes with diameters less than 1 nm [14, 15]. These compounds enable nanosensors to measure silver ions. The surface of SWCNT is oxidized more in comparison with MWCNT, and it is more negatively charged. This behavior leads to higher electrostatic absorption for SWCNT to absorb the positively charged ions [16, 17].
In this study, we used SWCNT-COOH [18, 19] and “NH2(CH2)6NH2” to simulate the nanosensor. This nanosensor can be used for the measurement of Ag+ and the treatment of water and wastewater. The studied nanosensor consists of one oxygen atom and two nitrogen atoms. However, molecular electrostatic potential (MEP) chart shows that there are two active sites for the interaction of silver cations with nanosensor. The lone pairs on these two atoms can interact with Ag+. Therefore, we examined these two atoms as the active points of the nanosensor. This research investigates the type of atoms in nanosensor (SWCNT-CONH-(CH2)6NH2) interacting with Ag+, the thermodynamics functions such as change in the standard Gibbs free energy (ΔG°) and change in the standard enthalpy energy (ΔH°) of these interactions, charge-transfer transition energy (E(2)), electron density, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energies, and the effect of the number of substitution agents on nanosensor reactivity in a gaseous environment. Analyzing the natural bond orbital is done through examining the NBO [20]. NBOs are natural bonds with maximum electron density [21, 22].
Methods
In this paper, theoretical calculations were performed using the Lee–Yang–Parr correlation functional (B3LYP) level of theory, DFT and LanL2DZ basis set. All frequency calculations include thermochemical analysis of the system. By default, this analysis is carried out at 298.15 K and 1 atmosphere of pressure with the Gaussian 03w’s sanction [23] in the gaseous environment. Nanotube modeler [24] and Gauss View 5.0 [25] graphical software were used to model nanotubes and interact with the structures [26], respectively. The density of state (DOS) diagrams were plotted by GaussSum v3.0 [27] software. Considering that arm-chair single-walled carbon nanotubes are more conductive than the zig-zag [28] and chiral nanotubes, arm-chair SWCNTs were employed in this part of the study. The nanostructure used in this study is N-(6-aminohexyl) carboxamide, SWCNT-CONH-(CH2)6NH2.
Generation of nanosensor
Nanosensor is produced by the reaction of 1,6-diaminohexane “NH2(CH2)6NH2” and the carboxylated single-walled carbon nanotube (SWCNT-COOH) according to the Eq. 1.
The optimization of the nanostructure
The initial structure for the optimization nanostructure is a model of SWCNT, drawn by nanotube Modeler software (Fig. 1a), and then, modeled by Gauss View 5.0 software. To design an interacting complex, we chose SWCNT-COOH (Fig. 1b) and “NH2(CH2)6NH2” (Fig. 1c) was added to a carboxylic agent in the next step. It was then optimized at B3LYP/LanL2DZ level of theory. The formation energy for the optimized system is calculated in Hartree, which is converted into kcal mol−1 [29]; each Hartree is 627.5095 kcal per mol.

Single-walled carbon nanotube (SWCNT) (a), Carboxylated carbon nanotube (SWCNT-COOH) (b), 1-6 diamino hexane (c)
Results and discussion
Effect of the number of substitution agents on the reactivity of nanostructure
The nanostructure (SWCNT-CONH-(CH2)6NH2) is produced by the reaction between 1,6-diaminohexane and SWCNTS-COOH. To investigate the effect of the number of substitution agents on the reactivity, the stability of two nanostructures with arm-chair single-walled carbon nanotubes of equal carbon atoms (16 carbon atoms) and different number of substitution agents was compared. The formation energy for the nanostructure with one substitution agent (Fig. 2a) was − 673.88 × 103 kcal mol−1, and the corresponding value for the nanostructure with two substitution agents (Fig. 2b) was − 987.11 × 103 kcal mol−1. Therefore, as the number of substrate factors on CNTS increases, they become more stable and the resulting reactivity decreases. This is because of the space obstruction that makes the nanostructure stable against the reaction.

The optimized structure of arm-chair carbon nanotube (2, 2) with one substitution agent (a) and two substitution agents (b) in the gaseous environment
The active positions of the nanostructure
We can analyze the amount of occupancy in single bonds, double bonds, and delocalized electron pairs using the natural bond orbital (NBO) method. NBO command is performed on the final output file of the optimized molecule. Lone pair (LP) is the electron with a non-bonding pair [30, 31]. Table 1 shows the oxygen of carbonyl group (oxygen no. 25) with two lone pairs, nitrogen atoms in the amide group (nitrogen no. 27) and amine group (nitrogen no. 44), with one lone pair each (Fig. 2a). To calculate the active positions of the nanosensor, the chk extension file was opened with the Gauss View 5.0 software after optimizing the nanostructure. Surfaces/contours were selected from the results menu, and the molecular electrostatic potential (MEP) chart was plotted for the nanostructure (Fig. 3a, b). Different amounts of electrostatic potential are shown in various colors. Negative electrostatic potential (for an electrophylic attack in red) shows a region where the electron density is high. Positive electrostatic potential (for a nucleophilic attack in blue) shows a region where the electron density is low [32,33,34,35]. Figure 3a, b show that two active sites can be considered for the interaction of silver cations with SWCNT-CONH-(CH2)6NH2. Atom number 1 is oxygen of carbonyl group and atom number 2 is nitrogen of amine group (Fig. 4). Silver ions were arranged near the mentioned active sites, at identical distances from positions 1 and 2 and then, these structures were optimized individually in the gaseous environment. The energy for the interaction of Ag+ with each of the active positions of nanostructure was then calculated. The optimization results of the nanostructure–silver ion system are presented in Table 2. The table shows that the product obtained from the interaction of silver cation with the oxygen of the carbonyl group (Fig. 5a) (less formation energy) is the most stable state and the one obtained from the interaction of Ag+ with the nitrogen of the amine group (Fig. 5b) (more formation energy) is the most unstable due to the steric hindrance. Due to the fact that the oxygen is more electronegative compared to the nitrogen atom, and the number of non-bonded electrons pairs on the oxygen atom of the carbonyl group is higher than that of the amine nitrogen atom, the oxygen of the carbonyl group is more reactive relative to the nitrogen of the amine group. Figure 5a, b show the most stable and unstable interactions of silver cations with nanostructure in the gaseous environment. Figure 5c demonstrates that there is no interaction between Ag+ and the nitrogen atom of the amide group related to the nanosensor in the gaseous environment.

The molecular electrostatic potential (MEP) chart (transparent) (a) and (mesh) (b) for the nanosensor

Two active positions of the nanosensor

The optimal structure of the interaction of Ag+ with the oxygen atom of the carbonyl group (a), the nitrogen atom of the amine group (b) and lack of interaction between the Ag+ and the nitrogen atom of the amide group related to the nanosensor (c) in the gaseous environment
Formation energy calculation for Ag+/nanosensor system
To calculate the formation energy of Ag+/nanosensor complex, the interacting complexes were modeled through locating Ag+ at a certain distance from the oxygen of the carbonyl group and the nitrogen of the amine (Fig. 5a, b) of nanosensor in Gauss View 5.0 software and then optimized using B3LYP method. The output file shows the formation energy of complex (Ag+—SWCNT-CO-NH-(CH2)6NH2) in Hartree (Table 2).
The charge-transfer energy (E(2))
After performing NBO analysis on the optimized structures resulting from Ag+ interaction with the active points of nanostructure, the amount of charge-transfer energy (E(2)) between electron donor and electron acceptor, energy difference between donor atoms (i) and acceptor atoms (j) ((Ej–Ei)), and the interaction energy between two bonds (Fj,i) can be measured. Table 3 only shows the results of the interaction between nanosensor active sites and Ag+. Intramolecular interactions are ignored due to the size of the nanosensor. High temperatures as well as positive particles lead to electrical conduction in semiconductors. The transfer of electrons from valance level of the oxygen atom of carbonyl group and amine nitrogen atoms to valance level of Ag+ creates the cavity in valance level of the oxygen atom and nitrogen atoms, and also causes the electrons transition in valance level of the oxygen and the nitrogen atoms (in the same strip). This creates electrical conductivity. Electrons transfer is done from the delocalized oxygen lone pairs to the unoccupied orbital of the silver cation (LP (2) O 25 → LP* (6) Ag 49 and LP (1) O 25) → LP* (6) Ag 49), for which the stabilization energy is 28.58 kcal mol−1. Electrons transfer are done from the delocalized nitrogen lone pair of amine nitrogen atom to the unoccupied orbital of silver cation ((LP (1) N 44) → LP* (6) Ag 49) with the charge-transfer energy of 30.96 kcal.mol−1. It is worth mentioning that the energy difference between the donor–acceptor atoms in a bond of (Ej–Ei) is calculated based on the atomic unit (a.u) system (each 1 a.u. of energy is 627.509 kcal mol−1). Electrons transfer are done from the delocalized oxygen atom lone pair and the delocalized pairs of amine nitrogen atom to the unoccupied orbital of the silver cation and the energy is used to transfer electrons. Since the number of non-bonded electrons pairs on the oxygen atom of the carbonyl group is higher than that of the amine nitrogen atom, it has more potential energy than the nitrogen atom in the amine group. The energy difference between Ag+ (acceptor) and the oxygen atom (donor) (Ej–Ei) is the most and the energy difference between the amine nitrogen (donor) and Ag+ (acceptor) is the least. Therefore, less energy is used to transfer non-bonded electrons pairs on the oxygen atom of the carbonyl group to Ag+ (less E(2)) than the nitrogen atom in the amine group. In Table 3, intermolecular interactions have been ignored.
Charge distribution and distance between the silver cation and interacting atoms
The charges on the oxygen atom of the carbonyl group and the nitrogen of the amine group, before the interaction with Ag+ (Fig. 6a), and the charge on the silver cation after the interaction with each of the active positions of the nanostructure were measured (Fig. 6b, c). The results presented in Table 4 show that the positive charge on Ag+ decreased in both interacting systems. Therefore, the interaction between the silver cation and each of the active nanostructures was completed. The electrons flow from electronegative atoms to the cation. The positive charge of Ag+ after interacting with the oxygen of the carbonyl group is greater than that of the interaction with the other active position (Fig. 5a, b). Therefore, the electron cloud density surrounding the Ag+ that interacts with the oxygen atom decreased, and so its volume subsequently. As a result, the distance between the silver atom and oxygen atom of the carbonyl group becomes shorter and its stability is increased. Therefore, the interaction between Ag+ and the oxygen atom of the carbonyl group is the most stable interaction. Comparison of interaction distance between silver ion and the oxygen of the carbonyl group (2.13075 Å) with the corresponding value for the interaction of Ag+ with the nitrogen of the amine group (2.23011 Å) shows that the interaction of Ag+ with the oxygen of carbonyl group is the most stable interaction (Fig. 7a, b). The results of calculations of atoms in molecule (AIM) of Ag+/nanosensor system show that the distance between some of the atoms of Fig. 5a is in accordance with Table 5.

The charges on the oxygen of the carbonyl group and nitrogen of the amine group, before interaction with Ag+ (a), the charges on Ag+ after interaction with the oxygen of the carbonyl group (b) and after interaction with the nitrogen of the amine group (c), in the gaseous environment

Distance between Ag+ and the oxygen of the carbonyl group (a) distance between silver cation and nitrogen of the amine group (b)
Calculation of thermodynamics functions for the interaction of Ag+ with nanosensor
To evaluate the thermodynamic functions including Gibbs free energy change, enthalpy and entropy for the interaction of silver cations with nanostructures, vibrational frequency calculations were carried out following a geometric optimization of the fragments, Ag+ and nanosensor, and their interacting systems at B3LYP/LanL2DZ level of theory. We can obtain the thermodynamic functions related to the interaction of Ag+ with nanosensor using the relationships (2), (3) and (4), and the output of thermochemistry [23].
In this part of the research, there is a schematic of a nanostructure in which the arm-chair SWCNTs (2, 2) with 16 carbon atoms has been explored. The thermodynamic functions related to the interaction of Ag+ with two active positions of nanosensor (Fig. 4) in the gaseous environment are presented in Table 6. In the Gaussian 03w’s sanction output file, the quantities for thermodynamics energies are obtained in terms of Hartree. The result of this Table 6 shows that the interaction of Ag+ with two positions SWCNT-CO-NH-(CH2)6NH2 is thermodynamically possible. The negative values of ΔH° show that the reaction is exothermic. In addition, thermodynamic calculations show that the interaction of Ag+ with the oxygen atom of the carbonyl group in the gaseous environment is thermodynamically more favorable. In addition, the negative change in the standard entropy (ΔS°) is scientifically justified.
Energy levels of HOMO and LUMO
The energy gap is an important parameter to determine the amount of electron transfer. After optimization procedure, the energy of the HOMO and LUMO can be extracted. The energy gap is obtained by subtracting the energy values of HOMO and LUMO [36,37,38].
(Energy gap = LUMO − HOMO). The results for HOMO and LUMO energy values related to Ag+, nanosensor, and their interacting systems are presented in Table 7. The energy gap between the HOMO of nanosensor (− 0.8319 × 10−23 kcal) and LUMO of the silver cation (− 1.4484 × 10−23 kcal) is low (0.6165 × 10−23 kcal) and this quantity proves the interaction between nanosensor and Ag+. The energy gap in the interaction of the silver cation with the oxygen of carbonyl group (0.0630 × 10−23 kcal) is lower than the energy gap in the interaction of the Ag+ with the amine nitrogen (0.0858 × 10−23 kcal).
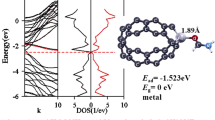
The electron density of state diagrams for the nanosensor and the Ag+/nanosensor system has been plotted by GaussSum v3.0 package (Fig. 8a–c). In these diagrams, HOMO is green and LUMO is red. In addition, the energy gap, which is the distance between the last green line and the first red line, is indicated in this graph. The DOS diagrams show that the energy gap in the interaction of the Ag+ with Ag+/nanosensor system is lower than that of the nanosensor. It can be concluded that after the interaction of the Ag+ with the nanosensor, the electron properties and conductivity of the nanosensor are affected according to the following relationship:

The density of state (DOS) diagrams for the nanosensor (a), the product obtained from the interaction of Ag+ with the oxygen of carbonyl group (b) and the product obtained from the interaction of Ag+ with nitrogen of amine group the interaction (c)
Here, σ is the electrical conductivity and k is Boltzmann’s constant. According to this equation, less Eg values at a given temperature will result in greater electrical conductivity. Generally, the electron density of state diagram represents the analysis of the electron population in each orbital and it also represents a molecular orbital arrangement in a specific energy range. The level of Fermi energy is considered zero. In Fig. 8a, which indicates the electron density of state diagram for the nanosensor, the electron density between HOMO (the last green line) and LUMO (the first red line) is zero, and the energy gap (the distance between the last green line and the first red line) is the highest. Because there is no interaction between the nanosensor and Ag+, the electron is not exchanged between the nanosensor and the silver cation. However, in Fig. 8b, c which indicate the electron density of state diagrams for Ag+/nanosensor system, the distance between the last green line and the first red line decreased more than the amount in Fig. 8a and the electron density is not zero. Figure 8c has the least energy gap and the highest electron density please see the distance between the last green line and the first red line (Eg) and above the distance between the last green line and the first red line (the electron density in the Fig. 8a–c). Therefore, the interaction of Ag+ with the oxygen of the carbonyl group is more stable. Figures 9, 10, 11 and 12 show the orbitals of HOMO and LOMO of silver cation, nanosensor and interaction products.

HOMO (a) and LUMO (b) orbitals of Ag+

HOMO (a) and LUMO (b) orbitals of SWCNT-CO-NH-(CH2)6 NH2

HOMO (a) and LUMO (b) orbitals of the interaction of Ag+ and oxygen of carbonyl group interacting system

HOMO (a) and LUMO (b) orbitals of the Ag+ and nitrogen of amine group
Conclusion
The SWCNT-CONH-(CH2)6NH2 is produced by the reaction of “NH2(CH2)6NH2” and SWCNT-COOH. Calculations of DFT are carried out on nanosensor SWCNT-CONH-(CH2)6NH2 in interaction with Ag+ in a gaseous environment. All calculations were performed using B3LYP as DFT functional and LanL2DZ basis set at 298.15 K and 1 atmospheric pressure in the gaseous environment. Quantum mechanical calculations using density functional theory (DFT) on the nanosensor (SWCNT-CONH-(CH2)6NH2) show that the oxygen of carbonyl group and nitrogen of amine group are two active sites for the interaction of silver cations with SWCNT-CONH-(CH2)6NH2. The type of nanosensor atoms interacting with silver cation affects the thermodynamic functions. The number of substitutions on carbon nanotubes has an impact on their reactivity. That is, as the number of substitutions rises, the carbon nanotubes become more stable, and therefore less reactive. In Ag+—SWCNT-CO-NH-(CH2)6NH2, the energy gap (Eg) has decreased relative to SWCNT-CO-NH-(CH2)6NH2. Reducing the energy gap increases the stability of the complex. Thus, smaller Eg values at a given temperature will result in greater electrical conductivity. Decrement in the positive charge on the silver cation after interaction indicates that there was a charge transfer between nanosensor and Ag+. The positive charge of Ag+ after interacting with the oxygen of the carbonyl group is higher than the charge after the interaction of the Ag+ with other active position. Thus, the electron cloud density around Ag+ that interacts with the oxygen atom decreases and so does its volume. As a result, the interaction of Ag+ with the oxygen of the carbonyl group is the most stable interaction. The distance between the Ag+ and oxygen of the carbonyl group is lower (2.13075 Å) than the distance between the silver cation and nitrogen of the amine group (2.23011 Å). Therefore, the interaction between Ag+ and oxygen of the carbonyl group is more stable. The negativeness of ΔG° indicates that the interaction of Ag+ with nanosensor is spontaneous. The negativeness of ΔH° indicates that the interaction of Ag+ with nanosensor is exothermic. The interaction of Ag+ with a functional single-walled carbon nanotube in the gaseous phase is accompanied by a reduction of irregularities (ΔS° < 0). Consequently, the SWCNT-CONH-(CH2)6NH2 is a good absorbent for silver cations.
References
Snook, G.A.M., Kao, P., Best, A.S.: Conducting-polymer-based supercapacitor devices and electrodes. J. Power Sources 196(1), 1–12 (2011)
Spitalsky, Z., Tasis, D., Papagelis, K., Galiotis, C.: Carbon nanotube-polymer composites: chemistry, processing, mechanical and electrical properties. Prog. Polym. Sci. 35(3), 357–401 (2010)
Singh, V., Joung, D., Das, S., Khondaker, S.L., Zhai, L.: Graphene based materials: past, present and future. Prog. Mater. Sci. 56(8), 1178–1271 (2011)
Abdalla, S., Al-Marzouki, F., Abdel-Daiem, A., Al-Ghamdi, A.A.: Different technical applications of carbon nanotubes. Nanoscale Res. Lett. 10(20), 358 (2015)
Sankar, P.A.G., Kumar, K.U.: Mechanical and electrical properties of single walled carbon nanotubes: a computational study. Eur. J. Sci. Res. 60(3), 342–358 (2011)
Boul, P.J., Turner, K., Li, J., Pulikkathara, M.X., Dwivedi, R.C., Sosa, E.D., Lu, Y., Kuznetsov, O.V., Moloney, P., Wilkins, R., O’Rourke, M.J., Khabashesku, V.N., Arepalli, S., Yowell, L.: Single wall carbon nanotube response to proton radiation. J. Phys. Chem. C 113(32), 14467–14473 (2009)
Iijima, S.: Helical microtubules of graphitic carbon. Nature 354, 56–58 (1991)
Iijima, S., Yudasaka, M., Kataura, H., Ichihashi, T., Qin, L.C., Kar, S.: Diameter enlargement of HiPco single-wall carbon nanotubes by heat treatment. Nano Lett. 1(9), 487–489 (2001)
Guadagno, L., De Vivo, B., Di Bartolomeo, A., Lamberti, P., Sorrentino, A., Tucci, V., Vittoria, V., Vertuccio, L.: Effect of functionalization on the thermo-mechanical and electrical behavior of multi-wall carbon nanotube/epoxy composites. J. Sci. Dir. 49(6), 1919–1930 (2011)
Futaba, D.N., Goto, J., Yasuda, S., Yamada, T., Yumura, M., Hata, K.: A background level of oxygen-containing aromatics for synthetic control of carbon nanotube structure. J. Am. Chem. Soc. 131(44), 15992–15993 (2009)
Pastine, S.J., Okawa, D., Kessler, B., Rolandi, M., Llorente, M., Zettl, A., Jean, M.J.: A facile and patternable method for the surface modification of carbon nanotube forests using perfluoroarylazides. J. Am. Chem. Soc. 130(13), 4238–4239 (2008)
Foldvari, M., Bagonluri, M.: Carbon nanotubes as functional excipients for nanomedicines: I. Pharmaceutical properties. J. Sci. Dir. Nanomed. Nanotech. Bio Med. 4(3), 173–182 (2008)
Britto, P.J., Santhanam, K.S.V., Ajayan, P.M.: Carbon nanotube electrode for oxidation of dopamine. Biol. Chem. 41(1), 121–125 (1996)
Bernholc, J., Brenner, D., Buongiorno Nardelli, M., Meunier, V., Roland, C.: Mechanical and electrical properties of nanotubes. J. Ann. Rev. Mater. Res. 32, 347–375 (2002)
Giahi, M., Arvand, M., Mirzaei, M., Bagherinia, M.A.: Determination of pseudoephedrine hydrochloride in some pharmaceutical drugs by potentiometric membrane sensor based on pseudoephedrine–phosphotungstate ion pair. J. Anal. Lett. 42(6), 870–880 (2009)
Li, Y.H., et al.: Lead adsorption on carbon nanotubes. J. Chem. Phys. Lett. 357(3), 263–266 (2002)
Gadhave, A., Waghmare, J.: Removal of heavy metal ions from wastewater by carbon nanotubes (CNTs). Int. J. Chem. Sci. Appl. 5(2), 56–67 (2014)
Chen, C.Y., Jafvert, C.T.: Photoreactivity of carboxylated single-walled carbon nanotubes in sunlight: reactive oxygen species production in water. J. Am. Chem. Soc. 44(17), 6674–6679 (2010)
Liu, X., Kane, A.B., Hurt, R.H.: Biodurability of single-walled carbon nanotubes depends on surface functionalization. J. Sci. Dir. 48(7), 1961–1969 (2010)
Kavitha, E., Sundaraganesan, N., Sebastian, S., Kurt, S.M.: Molecular structure, anharmonic vibrational frequencies and NBO analysis of naphthalene acetic acid by density functional theory calculations. J. Sci. Dir. 77(3), 612–619 (2010)
Glendening, E.D., Landis, C.R., Weinhold, F.: Natural bond orbital methods. J. Comput. Molecular. Sci. 2(1), 1–42 (2012)
Dadkhah, A.: Editorial-a new trend to rehabilitation. Iran. Rehabil. J. 12(19), 4–4 (2014)
Foresman, J.B., Frisch, Æ.: Exploring Chemistry with Electronic Structure Methods, 3rd edn. Gaussian, Inc, Wallingford (2015)
Yoshida, M.: Nanotube modeler—generation of nano-geometries, version 1.8, J. Cryst. Soft (2005–2018)
Introduction Gaussian 09 and How to GaussView 5 Programs Version 1. 2 (2010). http://www.gaussian.com/g_tech/1.htm
Frisch, E., Hratchian, H.P., Dennington II, R.D., et al.: GaussView, Version 5.0. 8. Gaussian, Inc, Wallingford (2009)
O’Boyle, N.M., Tenderholt, A.L., Langner, K.M.: Cclib: a library for package-independent computational chemistry algorithms. J. Comput. Chem. 29(5), 839–845 (2008)
Hirsch, A.: Functionalization of single-walled carbon nanotubes. Angew. Chem. Int. Ed. 41(11), 1853–1859 (2002)
Foresman, J.B., Frisch, Æ.: Exploring Chemistry with Electronic Structure Methods. A Guide to Using Gaussian. Gaussian, Inc, Wallingford (1996)
Reed, A.E., Weinstock, R.B., Weinhold, F.: Natural population analysis. J. Chem. Phys. 83(2), 735–746 (1985)
Reed, A.E., Curtiss, L.A., Weinhold, F.: Intermolecular interactions from a natural bond orbital, donor–acceptor viewpoint. J. Chem. Rev. 88(6), 899–926 (1988)
Politzer, P., Murray, J.S., Bulat, F.A., Riley, K.E.: Perspectives on halogen bonding and other σ-hole interactions: Lex parsimoniae (Occam’s Razor). J. Comput. Chem. 998(15), 2–8 (2012)
Luque, F., et al.: SCRF calculation of the effect of water on the topology of the molecular electrostatic potential. J. Phys. Chem. 97(37), 9380–9384 (1993)
Politzer, P., Murray, J.S.: The fundamental nature and role of the electrostatic potential in atoms and molecules. J. Chem. Acc. 108(3), 134–142 (2002)
Murray, J.S., Politzer, P.: The electrostatic potential. Wiley Online Libr. 1(2), 153–163 (2011)
Mosquera, M.A.: Support for the existence of invertible maps between electronic densities and non-analytic 1-body external potentials in non-relativistic time-dependent quantum mechanics. J. Chem. Phys. 147(13), 134110 (2017)
Karabacak, M., Cinar, M., Kurt, M.: Molecular structure and vibrational assignments of hippuric acid: a detailed density functional theoretical study. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. J. Sci. Dir. 74(5), 1197–1203 (2009)
Bonness, S., et al.: Theoretical investigation on the second hyperpolarizabilities of open-shell singlet systems by spin-unrestricted density functional theory with long-range correction: range separating parameter dependence. J. Chem. Phys. Lett. 493(1), 195–199 (2010)
Acknowledgements
We would like to thank Iran Nanotechnology Initiative Council for their assistance in the collection of data.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mehdizadeh, K., Giahi, M. A DFT study on N-6-amino-hexylamide functionalized single-walled carbon nanotubes in interaction with silver ion in a gaseous environment. J Nanostruct Chem 9, 39–51 (2019). https://doi.org/10.1007/s40097-019-0296-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40097-019-0296-7