
Abstract
In this work, thin layers of MoO3 were tested as potential photoanodes for water splitting. The influence of photointercalation of alkali metal cation (K+) into the MoO3 structure on the photoelectrochemical properties of the molybdenum trioxide films was investigated for the first time. MoO3 thin films were synthesized via thermal annealing of thin, metallic Mo films deposited onto the FTO substrate using a magnetron sputtering system. The Tauc and Mott–Schottky plots analysis were performed in order to determine the energy bands position of the investigated material. The photointercalation effect of K+ on photoelectrochemical properties of FTO/MoO3 photoanodes was studied using electrochemical techniques performed under simulated solar light illumination. It was proven that pristine MoO3 layers cannot act as effective photoanodes for water splitting due to the utilization of the photoexcited electrons in the intercalation process. The photochromic phenomenon related to Mo6+ centers reduction, and K+ intercalation occurs at a potential range in which the photoanode exhibits photoelectrochemical activity towards water photooxidation.

Schematic presentation of the photointercalation phenomenon influence on the photocurrent generation
Similar content being viewed by others

Introduction
Among various metal oxide semiconductors, molybdenum trioxide (MoO3) has attracted great attention because of its multifaceted functional properties [1,2,3,4]. Among crystal structures of MoO3, the thermodynamically stable α-MoO3 is commonly tested as photoactive material. It was reported that MoO3 can be successfully utilized as a photocatalyst for hazardous waste photodegradation [5, 6]. The advantage of MoO3 in comparison with commonly used TiO2, in the photoinduced organic waste decomposition process, is the possibility of visible light absorption and conversion due to the lower energy band gap of molybdenum oxide. Relatively high activity of MoO3 in photodecomposition process is related to the formation of highly reactive hydroxyl radicals during illumination [7, 8]. Besides the favorable energy band gap and the possibility of OH• radicals formation, α-MoO3 is characterized by a layered crystal structure that allows the possibility to create two-dimensional morphologies [9,10,11]. The host lattice of MoO3 provides a spacious region to accommodate atoms, in the form of intercalated alkali metal ions [12]. The intercalation process leads to the formation of colorful molybdenum bronzes, according to the reaction:
Among the many ways to induce the intercalation process, the photoinduced intercalation attracts great attention because of possible interesting applications. The photointercalation effect observed for MoO3 in contact with Na+ containing electrolyte has been utilized in a two-electrode cell for the energy storage device. The simultaneous illumination and polarization of Mo/MoO3 electrode lead to the charging (intercalation) and then energy can be released at the dark (deintercalation) [11]. The effect of molybdenum bronze formation affects the optical properties and extends the range of absorbed light. The change of MoO3 color is observed due to the charge transfer between Mo6+ and partially reduced Mo5+ centers [13]. The extension of the absorbed light range may enhance the efficiency of photoinduced processes that occur with the use of irradiated MoO3. Our previous report showed that besides energy band gap narrowing, a positive effect of photointercalation of alkali metal cations on the photocatalytic properties of molybdenum trioxide can be achieved [14]. Generally, the illumination of photocatalysts leads to the generation of e−/h+ pairs and photoexcited electrons are utilized for oxygen reduction reaction and the formation of superoxide radicals (O2˙−). The effective consumption of electrons is crucial for inhibition of adverse recombination processes and enables the use of photoexcited holes from the valence band for photooxidation phenomena. In the case of MoO3, photoexcited electrons take part in the intercalation process; thus, the photocatalytic performance is preserved even under anaerobic conditions.
There are many literature reports about the use of MoO3 as a photocatalyst for organic compounds photodecomposition [8, 15,16,17,18]. On the other hand, there are no literature reports related to the use of bare MoO3 as a photoanode for photoelectrochemical water splitting. In some reports, MoO3 is utilized only as a modifier of commonly used photoanode materials (TiO2, BiVO4) [19, 20]. The features that material has to possess to be a prospective photoanode for water photoelectrooxidation are, i.e., (i) be an n-type semiconductor, (ii) has an energy band gap allowing the visible light to be absorbed, and (iii) be characterized by the appropriate valence band location allowing water photooxidation. MoO3 has all these features.
In the present study, FTO/MoO3 electrodes were characterized as potential photoanodes for water splitting and used in PEC device for mater splitting. The energy bands position was determined on the basis of Tauc plot and Mott–Schottky analysis. The limitations of photocurrent generation are discussed taking into account the photointercalation phenomenon.
Experimental
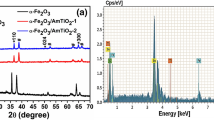
Molybdenum films were first deposited using magnetron sputtering method from the Mo target (Quorum) onto the FTO substrate (Sigma-Aldrich, 7 Ω/sq). Before the deposition of Mo films, the FTO glass substrates were subjected to ultrasonic cleaning in acetone, ethanol, and isopropyl alcohol for 15 min each, sequentially. The deposition was performed using a Quorum Q150TS coater operating at argon work pressure of 2.0 Pa at room temperature, with a discharge current of 80 mA. The thickness of the Mo layer was estimated using a quartz crystal microbalance and was equal to 100 nm. FTO/Mo was then subjected to annealing in air at 450 °C for 2 h. As a result, molybdenum trioxide (FTO/MoO3) was obtained. The layer of sputtered metal during the calcination has been completely converted into a metal oxide as it is shown on XRD patterns of samples before and after thermal treatment; see Fig. 1.

XRD patterns of (a) FTO/MoO3 and (b) FTO/Mo
Raman spectra were recorded using the micro-Raman spectrometer (Renishaw InVia) with excitation of the samples by means of an argon laser emitting at the wavelength of 514 nm, operating at 5% of its total power (~ 50 mW). The transmittance of FTO/MoO3 nanostructures was measured by UV–Vis spectrometer (Lambda 35, PerkinElmer). The spectra were registered in the range of 300–700 nm, with a scanning speed of 120 nm min−1. The energy band gap of the material was estimated using the Tauc equation assuming indirect transition.
α—absorption coefficient, hν—photon energy, Eg—energy band gap, and n—power characteristic for the type of transition (n = 0.5 for indirect transition).
The morphology of MoO3 layers was investigated using scanning electron microscopy (the Schottky field emission scanning electron microscope FEI Quanta FEG 250 with ET secondary electron detector). The beam accelerating voltage was kept at 15 kV. The chemical composition measurements before and after the photointercalation process were performed by the X-ray photoemission spectroscopy method. The XPS measurements were performed using an Argus Omicron NanoTechnology X-ray photoelectron spectrometer. The photoelectrons were excited by an Mg–Kα X-ray source. The X-ray anode was operated at 15 keV and 300 W. XPS measurements were performed at room temperature under ultra-high vacuum conditions, with pressure below 1.1 × 10−8 mbar. Data analysis was performed with the CASA XPS software package using Shirley background subtraction and a least-squares Gaussian–Lorentzian curve fitting algorithm. Obtained spectra were calibrated to give binding energy of 285.80 eV for C1s. The electrochemical and photoelectrochemical studies of materials were conducted using the AutoLab PGStat 302 N potentiostat-galvanostat system (Methrom, AutoLab) in the standard three-electrode arrangement, where FTO/MoO3 served as a working electrode (geometric surface area of 0.3 cm2). The Pt mesh was used as a counter electrode, while Ag/AgCl (3 M KCl) as a reference electrode. The electrochemical tests were carried out in deaerated 0.2 M K2SO4 (pH = 2, acidified using H2SO4). K2SO4 and H2SO4 with analytical grade were purchased from POCH. A xenon lamp equipped with AM 1.5 filter with a light intensity of 100 mW cm−2 was used as the light source.
Electrochemical impedance spectroscopy measurements were conducted at the frequency range from 20 kHz to 0.1 Hz, covering 100 points and with 10 mV amplitude of the AC signal (before and after illumination). In order to perform the Mott–Schottky analysis, impedance spectra were recorded at different potentials in the range from + 0.6 to − 0.4 V vs. Ag/AgCl (3 M KCl). Before each spectra registration, the potential was held to achieve a steady-state condition. The Mott–Schottky analysis was performed in order to determine the flat band potential.
Results and Discussion
Energy Band Position of MoO3
The flat band potential (Efb) was determined using the Mott–Schottky analysis. In order to estimate capacitance of the space charge region, the impedance spectra were measured at the dark conditions at different potentials (potential step = 0.025 V). Typical spectra are presented in Fig. 2a. The differences of impedance plots in the measured potential range are mainly related to the change of the electrical double layer capacitance. In order to avoid the problem with frequency dispersion of capacitance, the flat band potential was determined from 3 different frequencies (500, 1000, 1500 Hz). Obtained values are very close to each other suggesting the correctness of the analysis. The positive slope of Mott–Schottky plot presented in Fig. 2b confirmed that obtained MoO3 is an n-type semiconductor with Efb equals to about 0 ± 0.01 V vs. Ag/AgCl (3 M KCl) (0.288 V vs. NHE). The cyclic voltammetry curve (CV) of the FTO/MoO3 electrode (dark conditions) is presented also in Fig.2b. Polarization started at a rest potential equal to 0.1 V vs. Ag/AgCl (3 M KCl) towards anodic direction. There was a complex electrochemical activity with two main redox couples. There is a redox activity related to the reduction/oxidation of Mo centers [21]. Peak c1 observed at − 0.09 V and its anodic counterpart (a1) appears at 0.04 V accompanied by a shoulder (as) as at 0.075 V (E1/2 = − 0.065) is related to the Mo6+/Mo5+ redox reaction. Very similar electrochemical behavior, but shifted towards less cathodic potential, was already reported for MoO3 polarized in 1 M H2SO4 aqueous electrolyte [22]. The electrochemical reduction occurs simultaneously with cation intercalation into the MoO3 structure and clear change of the film color [1]. The excess of H+ in the electrolyte, as it was shown [22], facilitates the electrochemical reaction in comparison to K2SO4 electrolyte the difference is equal to 0.124 V indicating that insertion of protons into oxide structure is energetically favorable. The second redox couple: c2 at − 0.41 V/a2 at − 0.39 V (E1/2 = 0.40 V) is very likely to be related to the further redox reaction of Mo centers. Anbananthan et al. [22] claim that the direct conversion of Mo(V) to Mo(IV) is not possible, and it is associated with chemical reactions giving the mixture of products on lower oxidation states.

a Typical Nyquist plots recorded for FTO/MoO3 at different applied potentials. b The cyclic voltammetry curves of FTO/MoO3, recorded in 0.2 M K2SO4 (pH = 2), scan rate: 50 mV s−1 and Mott–Schottky plot of FTO/MoO3 electrode at pH adjusted to 2. c Tauc plots of FTO/MoO3 electrode (inset: absorbance spectra). d Band alignment of FTO/MoO3
The electrochromic effect was clearly seen during the polarization; thus, recorded electroactivity is observed due to the reversible formation of Mo bronzes. It was reported that the conduction band of α-MoO3 consists mainly of Mo 4d orbital [23]; thus, reduction observed on CV curve is strictly associated with the location of the conduction band. In the case of n-type semiconductors, Efb lies just below the CB [24]. This is consistent with our results, and Efb determined from MS plot is shifted towards anodic potential in comparison with the location of CB estimated from the CV curve.
In order to estimate the location of the valence band, the optical band gap was determined from the UV–Vis spectrum of the semi-transparent MoO3 layer using the Tauc plot. The spectrum is presented in Fig. 2c (inset). The clear absorption edge related to the valence to conduction band electron photoexcitation is observed. There is inconsistency in the literature reports about the type of electron transition (direct or indirect) [11, 25]. Here, two possibilities were taken into account as it is shown in Fig. 2c. Assuming direct band gap (Eg = 3.43 eV), absorption edge should be located at about 360 nm; however, pristine MoO3 layer absorbs light also at higher wavelengths. Thus, the indirect band gap equals to about 2.75 eV is also credible [26,27,28,29]. However, MoO3 tends to form oxygen vacancies; thus, the absorption in the visible range of light can be related to the presence of the defect states within the band gap [30]. The position of VB was estimated using the formula: Eg = EVB − ECB, assuming indirect transition. The location of the valence band with respect to the water oxidation potential suggests the possibility of water photooxidation on photoexcited MoO3 films. The electronic band structure of MoO3 prepared according to Mott–Schottky analysis, cyclic voltammetry, and Tauc plot is presented in Fig. 2d. Therefore, it can be concluded that despite the location of CB, which does not allow to H+ reduction and hydrogen evolution, the valence band potential is in the range that enables MoO3 to act as a visible light active photoanode for water photooxidation. The current growth at potential below − 0.6 V is related to hydrogen redox processes.
Photoelectrochemical Properties and Photointercalated Material Characterization
Photoelectrochemical properties of the prepared electrode were examined using the chronoamperometry technique recorded under intermittent illumination. The anodic potential, higher than Efb, was applied (0.8 V vs. Ag/AgCl (3 M KCl)) to provide appropriate band bending and formation of the depletion layer at space charge region near the electrode/electrolyte interface. As it is shown on the chronoamperometry curve in Fig. 3a, FTO/MoO3 electrode generates negligible photocurrent related to the water photooxidation, despite appropriate electronic bands position. It means that absorbed light cannot be efficiently converted into photocurrent (electrons flow from working to counter electrode). It is very likely that photogenerated electrons are consumed during potassium ions intercalation process for molybdenum ions valence change in MoO3 structure. Thus, molybdenum bronze can be photochemically formed, exploiting the photoexcited electrons from the conduction band as well as the diffusion of alkali metal ions into the layered oxide lattice (see Fig. 3b).

a The chronoamperometry curves recorded at 0.8 V vs. Ag/AgCl (3 M KCl) under intermittent illumination (100 mW cm−2, AM1.5) in 0.2 M K2SO4 (pH = 2) for FTO/MoO3. b The schematic diagram of photoexcited charge transfer in FTO/MoO3 photoanode system
The photointercalation process was confirmed by long-term chronoamperometry test recorded for both non-illuminated and illuminated electrodes. In the case of the test performed in the dark, there was a little decrease of anodic current from 18.5 to 15 μA/cm2 and no change of the film color was observed (Fig. 4a). Thus, the intercalation does not take place under the given conditions (E = 0.8 V). On the other hand, the anodic current increases from 18.9 to 26 μA/cm2 during the experiment performed under simulated solar light illumination. Simultaneously, the semi-transparent MoO3 film, turned from light gray to blue, proved that light illumination causes the formation of molybdenum bronzes. Generally, MoO3 exhibits electrochromic performance related to the Mo6+ centers reduction and cation intercalation induced by applied cathodic potential [30]. In the case of illuminated FTO/MoO3 electrode characterized in this work, the effect of film colorization is observed even at an anodic potential (see photo in the inset of Fig. 4a). The clear increase of current after about 60 min of illumination is related to the photocurrent generation. The possibility of photocurrent generation by molybdenum bronze is probably related to the (i) energy band gap narrowing and (ii) almost complete occupation of host lattice by K+. The photoelectrochemical reaction that results in the photochromic response of MoO3 can be described by considering the injection of a quantity (x) of univalent cations (M+) and utilization of an equal quantity of photogenerated electrons (e−). The reaction can be summarized as follows (assuming formation of reduced Mo centers at valence not lower than 5):

a Change in the value of current (E = 0.8 V) with time: during illumination (red line) and dark (black line). Electrolyte: 0.2 M K2SO4 (pH = 2). b Mo:K ratio changes with time during photointercalation
As a result, the film turns blue due to the intervalence-charge transfer from the newly formed Mo5+ (valence band-like) to adjacent Mo6+ (conduction band) (Eq. (5)) [31].
The EDX measurements were performed in order to estimate the amount of intercalated K+ into MoO3 lattice during the long-illumination. Atomic Mo:K ratio depending on the photointercalation duration is presented in Fig. 4b. The linear increase of the intercalant amount was observed. After 2 h, the estimated composition of the film can be represented by the formula \( {\mathrm{K}}_{0.19}{\mathrm{Mo}}_{0.19}^{\mathrm{V}}{\mathrm{Mo}}_{0.81}^{\mathrm{V}\mathrm{I}}{\mathrm{O}}_3 \).
The effect of H+ adsorption and intercalation during photoelectrochemical tests could be also taken into account. According to the literature reports, protons can interact with lattice corner-sharing oxygen atoms and for mover doped, unstable HxMoO3. As considered by Borgschulte et al., the “theoretical” H2O groups may be released from the structure forming MoO3−x rich in oxygen vacancies [32]. It leads to the same effect of generation of new bands in the absorbance spectrum and color change [33]. Nevertheless, the EDX analysis showed significant amounts of intercalated K+ ions; thus, photointercalation may be treated as the main reason for observed photoelectrochemical behavior.
The XPS measurements were performed in order to investigate the oxidation state of Mo before and after photointercalation. The XPS spectrum of the Mo 3d binding energy region for an as-prepared sample of FTO/MoO3 with two well-resolved spectral lines which after curve fitting were identified as the characteristic Mo 3d spin-orbit doublet peaks of MoO3: Mo 3d5/2 6+ at 233.4 eV and Mo 3d3/2 6+ at 236.6 eV [21, 34, 35] is presented in Fig. 5. This spectrum confirmed the purity of the α-MoO3 phase of the as-manufactured electrode and provided a useful baseline against which the effect of illumination could be assessed. The spectrum of the sample that was stored for 2 h in the acidified K2SO4 (pH = 2) electrolyte without illumination was registered for comparison. All bands in Mo 3d binding energy region for both samples overlapped; thus, the effect of intercalation does not occur in the dark, as it is shown in Fig. 5. For the FTO/MoO3 electrode after photointercalation, new bands at the spectrum appeared, suggesting a change of Mo oxidation state. For the electrode after illumination, spin-orbit components were assigned as follows: 232.9 eV (Mo 3d5/2 5+), 233.4 eV (Mo 3d5/2 6+), 236.1 eV (Mo 3d3/2 5+) and 236.6 eV (Mo 3d3/2 6+).

XPS spectra in the a Mo 3d and b K 2p binding energy region of FTO/MoO3 electrodes
As can be seen in Fig. 6b, potassium is present only in the sample after illumination in the acidified K2SO4 electrolyte. The peaks at 293.1 and 295.9 eV are assignable to K 2p of K+ [36]. The presence of potassium ions and change of Mo oxidation state confirm the formation of molybdenum bronze.

Raman spectra of FTO/MoO3 before and after illumination in K2SO4 (pH = 2)
In order to characterize the molybdenum bronze formed during photointercalation, Raman and UV–Vis spectroscopy measurements were performed. Raman spectra of the sample before and after illumination are shown in Fig. 6. Spectra exhibit typical Raman scattering bands of orthorhombic α-MoO3 [37, 38]. The bands in the range of 600–1000 cm−1, 400–600 cm−1, and below 200 cm−1 correspond to the stretching, deformation, and lattice modes of α-MoO3, respectively. The band at 996 cm−1can be assigned to terminal oxygen (Mo=O) stretching vibrations, the peak at 818 cm−1 is attributed to the doubly coordinated bridging oxygen (Mo2–O) stretching mode, whereas the signal recorded at 665 cm−1comes from the triply coordinated oxygen (Mo3–O) stretching mode [39]. In the case of the film after CA test, additional peaks appeared. The intercalation caused the crystal transformation from the original α-MoO3 into the mixed structure of molybdenum bronze and substoichiometric MoO3 [40, 41].
The comparison of UV–Vis spectra of the MoO3 film before and after 2 h illumination in 0.2 M K2SO4 (pH = 2) electrolyte is presented in Fig. 7. The spectrum of the sample after photointercalations is characterized by new bands in the visible range of electromagnetic radiation due to the presence of inter bandgap states and valence band-like Mo5+ states [42]. Such a phenomenon is advantageous in the case of photocatalyst materials because the higher absorbance ability could enhance the generation of photocurrent and efficiency of organic pollutant photodegradation. Thus, it has been confirmed that the process of photoexcited electrons consumption for photointercalation occurs even at an applied anodic potential.

Absorbance spectra of FTO/MoO3 before and after illumination in K2SO4 (pH = 2)
Since photoexcited electrons are consumed directly on FTO/MoO3 layer, thus it is evident that photocurrent of water oxidation is not observed. However, photoexcited holes from the valence band of MoO3 can take part in the photooxidation of water according to the reaction (Eq. 6):
Nevertheless, photogenerated holes may be also responsible for the photocorrosion of MoO3. The reaction can lead to the MoO3 dissolution according to reaction present as Eq. 7 [31].
The effect of MoO3 dissolution was tracked using SEM/EDX. The morphology of FTO/MoO3 samples after keeping them in K2SO4 (pH = 2) solution in the dark as well as under illumination was investigated and is presented in Fig. 8. The morphology of the pristine MoO3 film is presented in Fig. 8 inset for comparison. The effect of illumination is clearly seen. The morphology of the sample kept in the dark is preserved; thus, MoO3 dissolution does not occur in the absence of light. In the case of the MoO3 layer exposed to visible light illumination, the MoO3 film is characterized by the new phase of Mo-bronze with a relatively high content of K+ ions (measured using EDX). However, the average diameter of MoO3 grains is clearly smaller in the case of the pre-illuminated sample. It is probably related to the photocorrosion and formation of soluble molybdate ions.

SEM images of the FTO/MoO3: a after keeping in the electrolyte (2 h) (inset: the morphology of the pristine MoO3 film) and b after photointercalation (2 h). EDX spectrum of the FTO/MoO3: c after keeping in electrolyte and d after photointercalation
An additional electrochemical test was performed to confirm that the reactions (6) and (7) occur on the illuminated FTO/MoO3 electrode immersed in an aqueous electrolyte. In order to detect dissolved oxygen, the cyclic voltammetry curves were recorded during Pt disc electrode polarization mounted in the front of an FTO/MoO3 before and after the photoelectrochemical test. The CV curves are presented in Fig. 9. After 2 h of the polarization and illumination of MoO3 film, the oxygen reduction peak was clearly registered and it disappeared after electrolyte purging with Ar. The presence of oxygen in the electrolyte is likely to be related to two phenomena: (i) photoexcited holes from the valence band of MoO3 can take part in photooxidation of water (Eq. 6) and (ii) photogenerated holes may be also responsible for the photocorrosion of MoO3 with simultaneous oxygen evolution (Eq. 7).

Cyclic voltammetry curves recorded on the Pt electrode in the electrolyte before and after the FTO/MoO3 photoelectrochemical test
The influence of K+ photointercalation into MoO3 films was investigated using Mott–Schottky analysis. The comparison of Mott–Schottky plots for material before and after 2 h of illumination is shown in Fig. 10. Both electrodes exhibited a positive slope of C−2 f(E) function confirming n-type semiconducting properties. The shift of flat-band potential towards cathodic direction for the pre-illuminated sample was observed. The similar effect was observed for, e.g., reduced TiO2 nanotubes [43]. Analysis of the Mott–Schottky plot allows the donor density to be determined. The lower slope of C−2 f(E) suggests a significantly higher charge carrier concentration for the photointercalated sample. The electron donor concentration was calculated using the following equation:
where e is the electron charge, ε the dielectric constant of MoO3 (ε = 18) [44], ε0 is the permittivity of vacuum, ND is the charge carrier density, and V is the applied voltage. The calculated donor densities of MoO3 (before illumination) and MoO3 (after illumination) are 3.45 × 1019 and 1.50 × 1020 cm−3, respectively.

The Mott–Schottky plot for FTO/MoO3 before and after illumination
As oxygen vacancy serving as electron donor has been accepted generally as the main cause for the n-type conductivity of semiconductors [45], one can assume that the lower slope of Mott–Schottky plot is related to the formation of them during photointercalation. Thus, the photointercalation may lead to an increase of oxygen vacancy concentration, which is responsible for the enhanced photoelectrocatalytic activity of MoO3. Generally, the increase of oxygen vacancies density leads to the enhancement of photoelectrochemical properties of photoanodes [46, 47].
Thus, photoelectrochemical properties of the FTO/MoO3 electrodes (pristine and photointercalated) were compared using chronoamperometry recorded under intermittent illumination. (E = 0.8 V, 0.2 M K2SO4, pH = 2); see Fig. 11a. It is clearly seen that illumination pretreatment leads to the improvement of generated photocurrent from 0.12 to 1.55 μA cm−2. Photogenerated electrons are not used anymore in the intercalation process, because the molybdenum bronze is already formed during the previous stage. The potassium cation intercalation and/or oxygen vacancies formation can introduce a defect band that decreases effective bandgap of MoO3, and material can naturally absorb more light in the visible range hence increasing the photoelectrocatalytic activity. The clear enhancement of photoactivity may result from the increased concentration of electron donors (oxygen vacancies) as it was shown in Fig. 10. However, such sub-stoichiometric\( {\mathrm{M}}_x{\mathrm{M}\mathrm{o}}_x^{\mathrm{V}}{\mathrm{M}\mathrm{o}}_{1-x}^{\mathrm{V}\mathrm{I}}{\mathrm{O}}_3 \) has been shown to have low photostability [48]. The same effect was observed in this work, and the photocurrent density decrease with time (see Fig. 11b).

a The comparison of CA curves of MoO3 after and before illumination at 0.8 V vs. Ag/AgCl (3 M KCl). b The long-term chronoamperometry test (E = 0.8 V vs. Ag/AgCl (3 M KCl)) recorded during illumination of FTO/MoO3 after pre-illumination in 0.2 M K2SO4 (pH = 2) electrolyte
Conclusions
In summary, we report the influence of alkali metal cation intercalation into MoO3 on its photoelectrochemical properties. It was proven that obtained material cannot act as effective photoanodes for water splitting due to the fact that photogenerated electrons are consumed for the potassium ions photointercalation process. Thus, molybdenum bronze can be photochemically formed, exploiting the photoexcited electrons from the conduction band as well as the diffusion of metal ions into the crystal lattice structure. The photochromic phenomenon related to Mo6+ centers reduction and K+ intercalation occurs at a potential range in which the photoanode exhibits photoelectrochemical activity towards water photooxidation. Thus, the utilization of electrons in the bronze forming reaction prevents the flow of electrons to the counter electrode, and hence, the generation of photocurrent. Pre-illumination of FTO/MoO3 leads to the formation of more effective photoanode; however, the stability of photointercalated MoO3 is not satisfactory. Achieved visible light absorption of photochromic bronze is seen as a potential feature giving rise in photoelectrocatalytic activity of thin films under Vis irradiation. In the present work, we have focused on the investigation of the reason of poor photoactivity of MoO3 films in a photoelectrochemical water oxidation process.
References
I.A. de Castro, R.S. Datta, J.Z. Ou, A. Castellanos-Gomez, S. Sriram, T. Daeneke, K. Kalantar-Zadeh, Adv. Mater. 1701619, 1 (2017)
N.A. Chernova, M. Roppolo, C. Dillon, M.S. Whittingham, RSC Adv. 19, 2526 (2009)
D.R. Pereiraa, C. Díaz-Guerra, M. Peres, S. Magalhaesa, J.G. Correia, J.G. Marques, A.G. Silva, E. Alves, K. Lorenz, Acta Mater. 169, 15 (2019)
C.G. Granqvist, Handbook of Inorganic Electrochromic Materials (Elsevier Science B.V, Amsterdam, 2002), p. 207
A. Arfaoui, S. Touihri, A. Mhamdi, A. Labidi, T. Manoubi, Appl. Surf. Sci. 357, 1089 (2015)
X. Yuan, B. Yang, X. Hu, X. Dong, Y. Wei, J. Zhu, Appl. Surf. Sci. 357, 968 (2015)
Y. He, L. Zhang, X. Wang, Y. Wu, H. Lin, L. Zhao, W. Weng, H. Wan, M. Fan, RSC Adv. 4, 13610 (2014)
A. Chithambararaj, N.S. Sanjini, S. Velmathi, A.C. Bose, Phys. Chem. Chem. Phys. 15, 14761 (2013)
H. Zhang, L. Gao, Y. Gong, Electrochem. Commun. 52, 67 (2015)
K. Kalantar-Zadeh, J. Tang, M. Wang, K.L. Wang, A. Shailos, K. Galatsis, R. Kojima, V. Strong, A. Lech, W. Wlodarski, R.B. Kaner, Nanoscale. 2, 429 (2010)
S.N. Lou, N. Yap, J. Scott, R. Amal, Y.H. Ng, Sci. Rep. 4, 7428 (2014)
M. Wang, K.J. Koski, ACS Nano 9, 3226 (2015)
K. Ajito, L.A. Nagahara, D.A. Tryk, K. Hashimoto, A. Fujishima, J. Phys. Chem. 99, 16383 (1995)
M. Szkoda, K. Trzciński, M. Klein, K. Siuzdak, A. Lisowska-Oleksiak, Sep. Purif. Technol. 197, 382 (2018)
Y. Liu, P. Feng, Z. Wang, X. Jiao, F. Akhtar, Sci. Rep. 7, 1845 (2017)
L. Mai, F. Yang, Y. Zhao, X. Xu, L. Xu, B. Hu, Y. Luo, H. Liu, Mater. Today 14, 346 (2011)
J. Papp, S. Soled, K. Dwight, A. Wold, Chem. Mater. 6, 496 (1994)
S. Lam, J. Sin, A.Z. Abdullah, A.R. Mohamed, J. Mol. Catal. A, Chem. 370, 123 (2013)
M. Yang, L. Zhang, B. Jin, L. Huang, Y. Gan, Appl. Surf. Sci. 364, 410 (2016)
H. He, Y. Zhou, G. Ke, X. Zhong, M. Yang, L. Bian, K. Lv, F. Dong, Electrochim. Acta 257, 181 (2017)
B. Mendoza-sánchez, T. Brousse, C. Ramirez-Castro, V. Nicolosi, P.S. Grant, Electrochim. Acta 91, 253 (2013)
N. Anbananthan, K.N. Rao, V.K. Venkatesan, J. Electroanal. Chem. 207, 374 (1994)
Q. Qu, W. Zhang, K. Huang, H. Chen, Comput. Mater. Sci. 130, 242 (2017)
A.W. Bott, D. Ph, Electrochem. Semicond. 3, 87 (1998)
A. Bouzidi, N. Benramdane, H. Tabet-derraz, C. Mathieu, B. Khelifa, Mater. Sci. and Eng.: B. 97, 6 (2003)
M.B. Rahmani, S.H. Keshmiri, J. Yu, A.Z. Sadek, L. Al-Mashat, A. Moafi, K. Latham, Y.X. Li, W. Wlodarski, K. Kalantar-Zadeh, Sensors Actuators B Chem. 145, 13 (2010)
E.B. Santos, J.M.D.S. Silva, I.O. Mazali, Vib. Spectrosc. 54, 89 (2010)
Y. Liu, S. Yang, Y. Lu, V. Podval, W. Chen, G.S. Zakharova, Appl. Surf. Sci. 359, 114 (2015)
H. Yan, P. Song, S. Zhang, J. Zhang, Z. Yang, Q. Wang, Sensors Actuators B Chem. 236, 201 (2016)
M.M.Y.A. Alsaif, A.F. Chrimes, T. Daeneke, S. Balendhran, D.O. Bellisario, Y. Son, M.R. Field, W. Zhang, H. Nili, E.P. Nguyen, K. Latham, J. Van Embden, M.S. Strano, J.Z. Ou, K. Kalantar-Zadeh, Adv. Funct. Mater. 26, 91 (2016)
T. He, J. Yao, J. Photochem. Photobiol. C Photochem. Rev. 4, 125 (2003)
A. Borgschulte, O. Sambalova, R. Delmelle, S. Jenatsch, R. Hany, F. Nüesch, Sci. Rep. 7, 40761 (2017)
M. Dieterle, G. Weinberg, G. Mestl, Raman spectroscopy of molybdenum oxides. Phys. Chem. Chem. Phys. 4, 812 (2002)
X.K. Hu, Y.T. Qian, Z.T. Song, J.R. Huang, R. Cao, J.Q. Xiao, Chem. Mater. 20, 1527 (2008)
Z. Hu, C. Zhou, M. Zheng, J. Lu, B. Varghese, H. Cheng, C.H. Sow, J. Phys. Chem. C 116, 3962 (2012)
H. Liu, D. Chen, Z. Wang, H. Jing, Applied Catal. B, Environ. 203, 300 (2016)
Y. Chen, C. Lu, L. Xu, Y. Ma, W. Hou, J.-J. Zhu, Cryst. Eng. Comm. 12, 3740 (2010)
S. Alizadeh, S.A. Hassanzadeh-Tabrizi, Ceram. Int. 41, 10839 (2015)
K. Ajito, L.A. Nagahara, D.A. Tryk, K. Hashimoto, A. Fujishima, J. Phys. Chem. 99, 16388 (1995)
J.Z. Ou, J.L. Campbell, D. Yao, W. Wlodarski, K. Kalantar-Zadeh, J. Phys, Chem. C. 115, 10757 (2011)
D. Di Yao, J.Z. Ou, K. Latham, S. Zhuiykov, A.P.O. Mullane, K. Kalantar-Zadeh, Crystal Growth and Design. 12, 1865 (2012)
H.A. Tahini, X. Tan, S.N. Lou, J. Scott, R. Amal, Y.H. Ng, S.C. Smith, ACS Appl. Mater. Interfaces 8, 10911 (2016)
Z. Dong, D. Ding, RSC Adv. 8, 5652 (2018)
S.K. Deb, J.A. Chopoorian, J. App. Phys. 37, 4818 (2014)
A. Janotti, J.B. Varley, P. Rinke, N. Umezawa, G. Kresse, C.G. Van de Walle, Phys. Rev. B 81, 085212 (2010)
A. Pu, J. Deng, M. Li, J. Gao, H. Zhang, Y. Hao, J. Zhong, X. Sun, J. Mater.Chem. A. 2, 2491 (2014)
Y. Zhang, X. Zhang, D. Wang, F. Wan, Y. Liu, Appl. Surf. Sci. 403, 389 (2017)
H. Li, K. Yu, Z. Tang, H. Fu, Z. Zhu, Phys. Chem. Chem. Phys. 18, 14074 (2016)
Acknowledgments
M.S. is supported by the Foundation for Polish Science. Acknowledgments for the Institute of Fluid-Flow Machinery Polish Academy of Sciences for providing a magnetron sputter.
Funding
MS received financial support from the National Science Centre of Poland, NCN, under contract 2016/23/N/ST5/02071.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Szkoda, M., Trzciński, K., Łapiński, M. et al. Photoinduced K+ Intercalation into MoO3/FTO Photoanode—the Impact on the Photoelectrochemical Performance. Electrocatalysis 11, 111–120 (2020). https://doi.org/10.1007/s12678-019-00561-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12678-019-00561-2