
Abstract
Charcot-Marie-Tooth disease type 2 (CMT2) is a clinically and genetically heterogeneous inherited neuropathy. Although new causative and disease-associated genes have been identified for CMT2 in recent years, molecular diagnoses are still lacking for a majority of patients. We here studied a cohort of 35 CMT2 patients of Chinese descent, using whole exome sequencing to investigate gene mutations and then explored relationships among genotypes, clinical features, and mitochondrial DNA levels in blood as assessed by droplet digital PCR. We identified pathogenic variants in 57% of CMT2 patients. The most common genetic causes in the cohort were MFN2 mutations. Two patients with typical CMT phenotype and neuromyotonia were detected to harbor compound heterozygous variations in the HINT1 gene. In conclusion, our work supports that the molecular diagnostic rate of CMT2 patients can be increased via whole exome sequencing, and our data suggest that assessment of possible HINT1 mutations should be undertaken for CMT2 patients with neuromyotonia.



Similar content being viewed by others

References
Braathen GJ, Sand JC, Lobato A, Hoyer H, Russell MB (2010) MFN2 point mutations occur in 3.4% of Charcot-Marie-Tooth families. An investigation of 232 Norwegian CMT families. BMC Med Genet 11:48. https://doi.org/10.1186/1471-2350-11-48
Stojkovic T (2016) Hereditary neuropathies: an update. Rev Neurol (Paris) 172(12):775–778. https://doi.org/10.1016/j.neurol.2016.06.007
Bacquet J, Stojkovic T, Boyer A, Martini N, Audic F, Chabrol B, Salort-Campana E, Delmont E, Desvignes JP, Verschueren A, Attarian S, Chaussenot A, Delague V, Levy N, Bonello-Palot N (2018) Molecular diagnosis of inherited peripheral neuropathies by targeted next-generation sequencing: molecular spectrum delineation. BMJ Open 8(10):e021632. https://doi.org/10.1136/bmjopen-2018-021632
Barreto LC, Oliveira FS, Nunes PS et al (2016) Epidemiologic study of Charcot-Marie-Tooth disease: a systematic review. Neuroepidemiology. 46(3):157–165. https://doi.org/10.1159/000443706
Gemignani F, Marbini A (2001) Charcot-Marie-Tooth disease (CMT): distinctive phenotypic and genotypic features in CMT type 2. J Neurol Sci 184(1):1–9
Yoshimura A, Yuan JH, Hashiguchi A, Ando M, Higuchi Y, Nakamura T, Okamoto Y, Nakagawa M, Takashima H (2019) Genetic profile and onset features of 1005 patients with Charcot-Marie-Tooth disease in Japan. J Neurol Neurosurg Psychiatry 90(2):195–202. https://doi.org/10.1136/jnnp-2018-318839
Chandhok G, Lazarou M, Neumann B (2018) Structure, function, and regulation of mitofusin-2 in health and disease. Biol Rev Camb Philos Soc 93(2):933–949. https://doi.org/10.1111/brv.12378
Rossor AM, Polke JM, Houlden H, Reilly MM (2013) Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat Rev Neurol 9(10):562–571. https://doi.org/10.1038/nrneurol.2013.179
Pezzini I, Geroldi A, Capponi S, Gulli R, Schenone A, Grandis M, Doria-Lamba L, la Piana C, Cremonte M, Pisciotta C, Nolano M, Manganelli F, Santoro L, Mandich P, Bellone E (2016) GDAP1 mutations in Italian axonal Charcot-Marie-Tooth patients: phenotypic features and clinical course. Neuromuscul Disord 26(1):26–32. https://doi.org/10.1016/j.nmd.2015.09.008
Sitarz KS, Yu-Wai-Man P, Pyle A, et al. (2012) MFN2 mutations cause compensatory mitochondrial DNA proliferation. Brain. 135(Pt 8):e219, 211–213; author reply e220, 211–213. https://doi.org/10.1093/brain/aws049
Rouzier C, Bannwarth S, Chaussenot A, Chevrollier A, Verschueren A, Bonello-Palot N, Fragaki K, Cano A, Pouget J, Pellissier JF, Procaccio V, Chabrol B, Paquis-Flucklinger V (2012) The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy ‘plus’ phenotype. Brain. 135(Pt 1):23–34. https://doi.org/10.1093/brain/awr323
Kalmar B, Innes A, Wanisch K, Kolaszynska AK, Pandraud A, Kelly G, Abramov AY, Reilly MM, Schiavo G, Greensmith L (2017) Mitochondrial deficits and abnormal mitochondrial retrograde axonal transport play a role in the pathogenesis of mutant Hsp27-induced Charcot Marie Tooth disease. Hum Mol Genet 26(17):3313–3326. https://doi.org/10.1093/hmg/ddx216
Gentil BJ, Cooper L (2012) Molecular basis of axonal dysfunction and traffic impairments in CMT. Brain Res Bull 88(5):444–453. https://doi.org/10.1016/j.brainresbull.2012.05.003
Campbell PD, Shen K, Sapio MR, Glenn TD, Talbot WS, Marlow FL (2014) Unique function of kinesin Kif5A in localization of mitochondria in axons. J Neurosci 34(44):14717–14732. https://doi.org/10.1523/jneurosci.2770-14.2014
Eschbach J, Sinniger J, Bouitbir J, Fergani A, Schlagowski AI, Zoll J, Geny B, René F, Larmet Y, Marion V, Baloh RH, Harms MB, Shy ME, Messadeq N, Weydt P, Loeffler JP, Ludolph AC, Dupuis L (2013) Dynein mutations associated with hereditary motor neuropathies impair mitochondrial morphology and function with age. Neurobiol Dis 58:220–230. https://doi.org/10.1016/j.nbd.2013.05.015
Bomont P, Cavalier L, Blondeau F, Hamida CB, Belal S, Tazir M, Demir E, Topaloglu H, Korinthenberg R, Tüysüz B, Landrieu P, Hentati F, Koenig M (2000) The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet 26(3):370–374. https://doi.org/10.1038/81701
Wong YC, Ysselstein D, Krainc D (2018) Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature. 554(7692):382–386. https://doi.org/10.1038/nature25486
Jacquier A, Delorme C, Belotti E, Juntas-Morales R, Solé G, Dubourg O, Giroux M, Maurage CA, Castellani V, Rebelo A, Abrams A, Züchner S, Stojkovic T, Schaeffer L, Latour P (2017) Cryptic amyloidogenic elements in mutant NEFH causing Charcot-Marie-Tooth 2 trigger aggresome formation and neuronal death. Acta neuropathologica communications 5(1):55. https://doi.org/10.1186/s40478-017-0457-1
Berciano J, Garcia A, Gallardo E et al (2017) Intermediate Charcot-Marie-Tooth disease: an electrophysiological reappraisal and systematic review. J Neurol 2017 264:1655–1677. https://doi.org/10.1007/s00415-017-8474-3
Fridman V, Bundy B, Reilly MM, Pareyson D, Bacon C, Burns J, Day J, Feely S, Finkel RS, Grider T, Kirk CA, Herrmann DN, Laurá M, Li J, Lloyd T, Sumner CJ, Muntoni F, Piscosquito G, Ramchandren S, Shy R, Siskind CE, Yum SW, Moroni I, Pagliano E, Zuchner S, Scherer SS, Shy ME (2015) CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J Neurol Neurosurg Psychiatry 86(8):873–878. https://doi.org/10.1136/jnnp-2014-308826
Memon AA, Zoller B, Hedelius A et al (2017) Quantification of mitochondrial DNA copy number in suspected cancer patients by a well optimized ddPCR method. Biomolecular detection and quantification 13:32–39. https://doi.org/10.1016/j.bdq.2017.08.001
Skuratovskaia D, Zatolokin P, Vulf M, Mazunin I, Litvinova L (2019) Interrelation of chemerin and TNF-alpha with mtDNA copy number in adipose tissues and blood cells in obese patients with and without type 2 diabetes. BMC Med Genet 12(Suppl 2):40. https://doi.org/10.1186/s12920-019-0485-8
Guerrini F, Paolicchi M, Ghio F, Ciabatti E, Grassi S, Salehzadeh S, Ercolano G, Metelli MR, del Re M, Iovino L, Petrini I, Carulli G, Cecconi N, Rousseau M, Cervetti G, Galimberti S (2016) The droplet digital PCR: a new valid molecular approach for the assessment of B-RAF V600E mutation in hairy cell leukemia. Front Pharmacol 7(363). https://doi.org/10.3389/fphar.2016.00363
Di Meglio C, Bonello-Palot N, Boulay C et al (2016) Clinical and allelic heterogeneity in a pediatric cohort of 11 patients carrying MFN2 mutation. Brain and Development 38(5):498–506. https://doi.org/10.1016/j.braindev.2015.11.006
Fridman V, Bundy B, Reilly MM, Pareyson D, Bacon C, Burns J, Day J, Feely S, Finkel RS, Grider T, Kirk CA, Herrmann DN, Laurá M, Li J, Lloyd T, Sumner CJ, Muntoni F, Piscosquito G, Ramchandren S, Shy R, Siskind CE, Yum SW, Moroni I, Pagliano E, Zuchner S, Scherer SS, Shy ME, Inherited Neuropathies Consortium (2015) CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J Neurol Neurosurg Psychiatry 86(8):873–878. https://doi.org/10.1136/jnnp-2014-308826
Manganelli F, Tozza S, Pisciotta C, Bellone E, Iodice R, Nolano M, Geroldi A, Capponi S, Mandich P, Santoro L (2014) Charcot-Marie-Tooth disease: frequency of genetic subtypes in a Southern Italy population. J Peripher Nerv Syst 19(4):292–298. https://doi.org/10.1111/jns.12092
Nam SH, Hong YB, Hyun YS, Nam da E, Kwak G, Hwang SH, Choi BO, Chung KW (2016) Identification of genetic causes of inherited peripheral neuropathies by targeted gene panel sequencing. Mol Cell 39(5):382–388. https://doi.org/10.14348/molcells.2016.2288
Murphy SM, Laura M, Fawcett K, Pandraud A, Liu YT, Davidson GL, Rossor AM, Polke JM, Castleman V, Manji H, Lunn MPT, Bull K, Ramdharry G, Davis M, Blake JC, Houlden H, Reilly MM (2012) Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry 83(7):706–710. https://doi.org/10.1136/jnnp-2012-302451
Vaeth S, Christensen R, Duno M et al (2019) Genetic analysis of Charcot-Marie-Tooth disease in Denmark and the implementation of a next generation sequencing platform. European journal of medical genetics 62(1):1–8. https://doi.org/10.1016/j.ejmg.2018.04.003
Wang R, He J, Li JJ, Ni W, Wu ZY, Chen WJ, Wang Y (2015) Clinical and genetic spectra in a series of Chinese patients with Charcot-Marie-Tooth disease. Clin Chim Acta 451:263–270. https://doi.org/10.1016/j.cca.2015.10.007
Casasnovas C, Banchs I, Cassereau J, Gueguen N, Chevrollier A, Martinez-Matos JA, Bonneau D, Volpini V (2010) Phenotypic spectrum of MFN2 mutations in the Spanish population. J Med Genet 47(4):249–256. https://doi.org/10.1136/jmg.2009.072488
Calvo J, Funalot B, Ouvrier RA, Lazaro L, Toutain A, de Mas P, Bouche P, Gilbert-Dussardier B, Arne-Bes MC, Carrière JP, Journel H, Minot-Myhie MC, Guillou C, Ghorab K, Magy L, Sturtz F, Vallat JM, Magdelaine C (2009) Genotype-phenotype correlations in Charcot-Marie-Tooth disease type 2 caused by mitofusin 2 mutations. Arch Neurol 66(12):1511–1516. https://doi.org/10.1001/archneurol.2009.284
Choi BO, Nakhro K, Park HJ et al (2015) A cohort study of MFN2 mutations and phenotypic spectrums in Charcot-Marie-Tooth disease 2A patients. Clin Genet 87(6):594–598. https://doi.org/10.1186/s12883-015-0430-1
Iapadre G, Morana G, Vari MS, Pinto F, Lanteri P, Tessa A, Santorelli FM, Striano P, Verrotti A (2018) A novel homozygous MFN2 mutation associated with severe and atypical CMT2 phenotype. Eur J Paediatr Neurol 22(3):563–567. https://doi.org/10.1016/j.ejpn.2017.12.020
Sivera R, Sevilla T, Vilchez JJ, Martinez-Rubio D, Chumillas MJ, Vazquez JF, Muelas N, Bataller L, Millan JM, Palau F, Espinos C (2013) Charcot-Marie-Tooth disease: genetic and clinical spectrum in a Spanish clinical series. Neurology. 81(18):1617–1625. https://doi.org/10.1212/WNL.0b013e3182a9f56a
Zuchner S, De Jonghe P, Jordanova A et al (2006) Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol 59(2):276–281. https://doi.org/10.1002/ana.20797
Zhao H, Race V, Matthijs G, de Jonghe P, Robberecht W, Lambrechts D, van Damme P (2014) Exome sequencing reveals HINT1 mutations as a cause of distal hereditary motor neuropathy. European journal of human genetics : EJHG 22(6):847–850. https://doi.org/10.1038/ejhg.2013.231
Horga A, Cottenie E, Tomaselli PJ, Rojas-García R, Salvado M, Villarreal-Pérez L, Gamez J, Márquez-Infante C, Houlden H, Reilly MM (2015) Absence of HINT1 mutations in a UK and Spanish cohort of patients with inherited neuropathies. J Neurol 262(8):1984–1986. https://doi.org/10.1007/s00415-015-7851-z
Jerath NU, Shy ME, Grider T, Gutmann L (2015) A case of neuromyotonia and axonal motor neuropathy: a report of a HINT1 mutation in the United States. Muscle Nerve 52(6):1110–1113. https://doi.org/10.1002/mus.24774
P1 L, Brožková DŠ, Krůtová M, Neupauerová J, Haberlová J, Mazanec R, Dvořáčková N, Goldenberg Z, Seeman P (2015) Mutations in HINT1 are one of the most frequent causes of hereditary neuropathy among Czech patients and neuromyotonia is rather an underdiagnosed symptom. Neurogenetics 16(1):43–54. https://doi.org/10.1007/s10048-014-0427-8
Rauchenzauner M, Fruhwirth M, Hecht M, Kofler M, Witsch-Baumgartner M, Fauth C (2016) A novel variant in the HINT1 gene in a girl with autosomal recessive axonal neuropathy with neuromyotonia: thorough neurological examination gives the clue. Neuropediatrics. 47(2):119–122. https://doi.org/10.1055/s-0035-1570493
Veltsista D, Chroni E (2016) A first case report of HINT1-related axonal neuropathy with neuromyotonia in a Greek family. Clin Neurol Neurosurg 148:85–87. https://doi.org/10.1016/j.clineuro.2016.07.012
Dohrn MF, Glockle N, Mulahasanovic L et al (2017) Frequent genes in rare diseases: panel-based next generation sequencing to disclose causal mutations in hereditary neuropathies. J Neurochem 143(5):507–522. https://doi.org/10.1111/jnc.14217
Meng L, Fu J, Lv H, Zhang W, Wang Z, Yuan Y (2018) Novel mutations in HINT1 gene cause autosomal recessive axonal neuropathy with neuromyotonia in two cases of sensorimotor neuropathy and one case of motor neuropathy. Neuromuscul Disord 28(8):646–651. https://doi.org/10.1016/j.nmd.2018.05.003
Wang Z, Lin J, Qiao K, Cai S, Zhang VW, Zhao C, Lu J (2018) Novel mutations in HINT1 gene cause the autosomal recessive axonal neuropathy with neuromyotonia. European journal of medical genetics 62:190–194. https://doi.org/10.1016/j.ejmg.2018.07.009
Peeters K, Chamova T, Tournev I, Jordanova A (2017) Axonal neuropathy with neuromyotonia: there is a HINT. Brain. 140(4):868–877. https://doi.org/10.1093/brain/aww301
Pareyson D, Saveri P, Sagnelli A, Piscosquito G (2015) Mitochondrial dynamics and inherited peripheral nerve diseases. Neurosci Lett 596:66–77. https://doi.org/10.1016/j.neulet.2015.04.001
Arribat Y, Broskey NT, Greggio C, Boutant M, Conde Alonso S, Kulkarni SS, Lagarrigue S, Carnero EA, Besson C, Cantó C, Amati F (2018) Distinct patterns of skeletal muscle mitochondria fusion, fission and mitophagy upon duration of exercise training. Acta Physiol (Oxford) 225:e13179. https://doi.org/10.1111/apha.13179
Acknowledgments
We would like to thank all the participants for their help and willingness to participate in this study. We thank the reviewers for the comments. This work was supported by the grant 81500980 and U1505222 from the National Natural Science Foundation of China, grant 81870929 from the National Natural Science Foundation of China, and grant 2018-CX-25 from Medical Innovation Project for Research Talents of Fujian Province.
Author information
Authors and Affiliations
Contributions
Study concept and design (Jin He and Yi Lin); acquisition of data (Jin He, Shan Lin, Liuqing Xu, Guorong Xu, Ling-Ling Guo); analysis and interpretation of data (Jin He, Shan Lin, Liuqing Xu, Ling-Ling Guo); drafting of the manuscript (Jin He, Shan Lin, Bi-Juan Lin); critical revision of the manuscript for important intellectual content (Jin He and Yi Lin); obtaining of funding (Jin He and Yi Lin); study supervision (Ning Wang and Wanjin Chen).
Corresponding authors
Ethics declarations
Written informed consent was obtained from all the patients included in this study. This study was approved by the Ethics Committee of the First Affiliated Hospital of Fujian Medical University.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
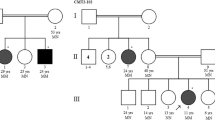
Supplementary Fig.S1
The pedigree charts of CMT2 patients. (PNG 587 kb)
ESM 1
(DOCX 20 kb)
Rights and permissions
About this article

Cite this article
Lin, S., Xu, LQ., Xu, GR. et al. Whole exome sequencing reveals a broader variant spectrum of Charcot-Marie-Tooth disease type 2. Neurogenetics 21, 79–86 (2020). https://doi.org/10.1007/s10048-019-00591-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-019-00591-4