
Abstract
The influence of the surface modification of multi-walled carbon nanotubes (MWCNT) with nitrogen-containing compounds on the performance of 40 wt% Pt/MWCNT catalysts in the oxygen electroreduction reaction (ORR) was investigated using a rotating disk electrode (RDE) at 10–35 °C in 0.1 M HClO4 as electrolyte in electrochemical cell, and in a hydrogen–oxygen polymer electrolyte membrane fuel cell (PEMFC) at 82 °C. The catalysts were characterized by low-temperature nitrogen adsorption, XPS, TEM, gas-phase CO titration, electrooxidation of the adsorbed CO monolayer, and cyclic voltammetry. It was shown that the modification of MWCNT with melamine–formaldehyde resin leads to the surface nitrogen concentration up to 8.3 at.% (CNT-MF sample). The 40 wt% Pt/CNT-MF catalyst with 0.1 mg cm−2 Pt loading on the cathode showed a good performance in PEMFC (~ 0.61 W cm−2) and a high utilization ratio (0.84) of Pt in membrane electrode assembly as compared to Pt/CNT catalyst (~ 0.37 W cm−2 and utilization of 0.29). The higher power density of nitrogen-modified catalysts was ascribed to a higher utilization of Pt in the electrode layer.
Similar content being viewed by others


Introduction
Polymer electrolyte membrane fuel cells (PEMFC) are among the most promising and rapidly developing areas of energy technologies. Platinum catalysts based on carbon supports are considered to be the most active and stable in the oxygen reduction reaction (ORR) [1, 2], while Pt/Vulcan XC-72 is widely used as model catalysts [2]. However, the wide application of the Pt/C catalysts is limited mostly by the high content of expensive platinum in the membrane electrode assembly (MEA). One approach to reduce platinum content is to increase the utilization of the active component in the cathode electrode layer of PEMFC using novel materials satisfying the following criteria [3]: (a) high specific surface area, (b) good electrical properties, and (c) high electrochemical stability under PEMFC operating conditions. Carbon nanotubes (CNT) have shown a good potential to meet these requirements, becoming, thereby, an attractive candidate electrode material [4,5,6,7,8,9].
Compared to commercial carbon blacks (e.g., Vulcan XC-72), the morphology of multi-walled CNTs contributes to both the high availability of Pt nanoparticles deposited on the surface and the improved mass transfer. However, the inert surface of CNTs inhibits immobilization of the catalyst active component—highly dispersed platinum nanoparticles [10, 11]. To increase the concentration of anchoring sites for Pt deposition, surface functionalization with nitrogen is usually performed [10, 12, 13].
With regard to platinum catalysts, the use of nitrogen-modified nanotubes as supports leads to an increased dispersion of the active component due to a higher concentration of anchoring sites for Pt adsorption. High dispersion of Pt significantly increases its utilization in the ORR [14].
Two approaches have been employed for the functionalization of the carbon nanotube surface with nitrogen-containing groups: (1) introduction of a nitrogen-containing species during the CNT synthesis; and (2) post-treatment of the CNT powder. As a rule, the first approach leads to a decreased activity of the catalysts for CNT growth, an increased diameter, the bamboo-like morphology, and a significantly reduced quality of the material [15, 16], while the second one involves additional facilities also resulting in a decreased specific surface area and large amounts of wastes [17]. Only a few papers were devoted to the investigation of nitrogen-modified carbon nanotubes (N-CNT) as supports for the platinum ORR catalysts [3, 18,19,20]. Moreover, the main efforts were aimed at studying such supports as the non-platinum ORR catalysts, where nitrogen-containing groups exhibit the catalytic properties [21,22,23,24].
As for Pt-based catalysts, 30 wt% Pt/CNT-N catalysts based on melamine-modified nanotubes that were calcined at 950 °C showed high stability in accelerated test cycles [25], as well as a more even distribution of the active component over the surface and an improved performance in PEMFC [26], which the authors attributed to the strong interaction of Pt with nitrogen groups [25]. Pt/CNT-N catalysts based on monoethanolamine-modified CNTs showed an advantage over the commercial catalyst due to the higher electrochemically active surface area of Pt [14]. The treatment of CNT in nitrogen plasma led to the introduction of 4 at.% surface nitrogen, which resulted in an improved performance in PEMFC as compared to unmodified Pt/CNT [27]. The catalysts containing both the carbon nanotubes doped with nitrogen (N-CNT) as supports and the monodisperse platinum nanoparticles obtained using N-containing templates as the active component were shown to have a high activity with an increased current density due to a decrease in mass transfer [28]. The Pt/CNT-N catalyst based on CNT modified with ethylenediamine also showed a high performance in PEMFC [29]. The prospect of Pt/N-CNT catalysts in PEMFC was reported in [30], where the catalysts showed high stability and power density. Nevertheless, “the optimal modification” of the carbon nanotube surface with nitrogen-containing groups is still challenging.
In this paper, a series of 40 wt% Pt catalysts based on multi-walled carbon nanotubes modified with nitrogen was prepared and studied. Two approaches were used for the CNT surface functionalization with nitrogen: polymerization of adsorbed melamine and melamine–formaldehyde resin in the presence of CNT. CNT modified by the second approach was recently shown to be promising for the Pt-free ORR catalyst in alkaline media [31]. Performance of the catalysts in the ORR was evaluated in electrochemical cell and in PEMFC. The results showed that the use of nitrogen-modified nanotubes makes it possible to increase significantly the utilization of Pt and the specific power density in PEMFC.
Experimental
Preparation of carbon supports
The CNT samples, represented by multi-walled carbon nanotubes, were synthesized by the catalytic chemical vapor deposition (CCVD) at 670 °C in Ar/C2H4 1:1, 400 sccm flow using the Fe2Co/Al2O3 catalyst [32]. This catalyst exhibits high activity (> 120 gMWCNT g −1cat h ) and provides high purity (no amorphous carbon, catalyst impurities less than 2% by weight), low concentration of defects in CNT [33] and a narrow size distribution of nanotubes diameter (dmean ~ 9.4 nm). Before depositing platinum, the nanotubes were treated with nitric acid (conc.), as described in [34]. The concentration of surface carboxyl groups estimated by titration was 0.8 group nm−2 [33].
The CNT-M sample was synthesized by polymerization of adsorbed melamine. A colloidal dispersion of CNT (100 mg) in an aqueous solution of melamine (20.5 mg) was obtained via sonication (15 min, 22 kHz, 400 W) to provide a suspension and disentangle the agglomerates of carbon nanotubes [35]. The water was, then, evaporated at 80 °C for 24 h. The following treatment in Ar flow (400 sccm) at 320 °C for 4 h led to ~ 10% decrease in mass due to the polycondensation of melamine with the formation of melam (Fig. 1-1) [36, 37].

Synthesis scheme of nitrogen-containing polymers on the surface of CNTs. 1 Polymerization of melamine (CNT-M), 2 synthesis of a melamine–formaldehyde resin (CNT-MF)
The CNT-MF sample was synthesized via the polymerization of a melamine–formaldehyde resin on the CNT surface by the reaction shown in Fig. 1-2. For this purpose, a CNT dispersion (1000 mg) was prepared in an aqueous solution (150 ml) of melamine (210 mg) and formaldehyde (0.32 ml) and subjected to ultrasonic treatment (22 kHz, 400 W) for 15 min. The dispersion was then heated to 70 °C with constant stirring. Continuous sonication was used to prevent the secondary agglomeration and sedimentation deposition of CNTs from the dispersion. The MF polymerization was initiated by adding a KOH solution to achieve the pH level of about 8–9. The reaction time was of 3 h. After cooling, the excess of water was removed by a rotary vacuum evaporator. The resulting gelled sample was completely dried in an oven at 60 °C for 24 h.
Synthesis of Pt/CNT catalysts
Pt/C catalysts were prepared using a procedure similar to that described in [38, 39]. The carbon support was suspended in an H2PtCl6 solution, and the suspension was heated to 80 °C. Next, a NaOOCH + Na2CO3 solution was dosed into the suspension under vigorous stirring and the suspension was then aged at pH 6–7 and 80° C for 1 h. The completeness of platinum deposition was checked by performing a qualitative HCl + SnCl2 test for platinum in the mother solution. Thereafter, a NaOOCH + Na2CO3 solution was added until pH 8–9 and the mixture was left standing for 30 min, cooled, and filtered. The substance collected on the filter was washed with distilled water until free Cl− ions were determined by AgNO3 tests. The resulting solid was dried in vacuum at 80 °C, and the catalyst was finally reduced with flowing H2 at 150 °C.
Physico-chemical characterizations
Texture of the carbon supports was characterized by low-temperature nitrogen sorption using an ASAP 2400 instrument (Micromeritics). Surface area and pore size distribution of the samples were calculated using Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) equations from the desorption branch of the isotherms. Carbon samples were treated at 300 °C up to a residual pressure of 10−3 torr. The initial branch of the N2 adsorption isotherm in a range of P/P0 = 0.05–0.2 was used to calculate the BET specific surface area (SBET), while a value of the isotherm at P/P0 = 0.98, to determine the total pore volume (V∑) having a diameter less than 110 nm. Pore diameter (D) was calculated by the BET model using the formula D = 4V∑/SBET. Relative errors in determination of specific surface area of the carbon support were 10%. High-resolution transmission electron microscopy (HRTEM) studies were performed using an electron microscope JEM-2010 (JEOL, Japan) with a resolution of 0.14 nm and lattice accelerating potential of 200 kV. Size distributions of Pt nanoparticles were obtained using HRTEM data. The average particle size (d TEMN ) and the surface average particle size (d TEMs ) were calculated using the following equation [40]:
where d represents the average particle size (d TEMN at k = 1) and the surface average particle size (d TEMs at k = 3). The number of particles used for the calculations was not less than 500.
XPS experiments were performed using a photoelectron spectrometer KRATOS ES 300. To assess the qualitative composition of the surface and determine the presence of impurities, the spectra were obtained in the range of 0–1150 eV with a 1 eV step at an energy transmission analyzer with hν = 50 eV. The quantitative composition of the surface and the charge state of elements were determined by obtaining precision photoelectron spectra of individual lines in increments of 0.1 eV at an energy transmission analyzer with hν = 25 eV. The primary radiation was the magnesium Kα line (hv = 1253.6 eV). The dispersion (DPt) of Pt nanoparticles in Pt/C catalysts was determined by pulsed CO chemisorption at 20 °C (gas-phase CO titration, GPT) as a ratio of the total Pt surface atoms to the total number of metal atoms in the sample (NΣ), suggesting that the Pt surface atoms bind CO to a ratio of CO/Pt = 1 (mol mol−1) [41]. Generally, for Pt/C catalyst with a spherical shape of Pt nanoparticles, the average volume-surface diameter (ds) of platinum crystallites can be calculated using the formula d GPTs (nm) = 1.08/DPt [41]. This method was described in the literature [41, 42] and used for Pt dispersion determination in Pt/C catalysts [39, 40, 43]. The relative errors were 10%. Theoretical Pt surface area (S GPTPt , m2 g −1Pt ) was determined as S GPTPt = 6/ρd GPTs .
Electrochemical measurements
A given amount of a catalyst (1 mg ml−1) was dispersed in a mixture of isopropanol and Milli-Q water (3:2 v/v) and treated in an ultrasonic bath until a stable suspension was obtained (approximately 30 min). An aliquot of the suspension of a catalyst powder was pipetted on the clean and dry surface of the polished GC rod (Alfa Aesar, type 1 with the diameter of 5 mm) and dried under Ar flow at ambient temperature for 40 min. The Pt content was 36 μg cm−2. Before measurements, the prepared working electrodes Pt/C/GC were cycled at a sweep rate of 50 mV s−1 in the supporting electrolyte (Ar purged 0.1 M HClO4) to obtain a stable cyclic voltammetry (CV) curve. Generally, up to five CV pretreatment cycles were performed to achieve a stable CV curve.
Electrochemical measurements were performed in a temperature-controlled three-electrode glass cell using a potentiostat Autolab PGSTAT100 and a rotating disk electrode Autolab RDE at 25 °C in a solution of 0.1 M HClO4. Reference electrode served as a RHE: Pt/H2(g)/H+ (electrolyte). The counter electrode was a Pt foil. All potentials are given relative to RHE. Electrochemically active surface of platinum (EASPt) was determined in electrochemical cell (EC) or in membrane electrode assembly (MEA) from the charge (QCO) obtained by integrating the CO-adsorbed electrooxidation peak using specific charge of CO monolayer platinum (qCO = 420 μC cm−2 [44]), mass of platinum (m) and the following equation:
Pt utilization in electrochemical cell (EC) or in MEA was determined using the following equation:
The electrocatalytic activities of Pt/C catalysts in the ORR were determined in electrochemical cell using the RDE method. The electrolyte was saturated with pure oxygen for 20 min, and the current–potential curves were recorded (1.10–0.30 V vs. RHE; 10 mV s−1) at various rotation rates of the disk electrode, from 400 to 3600 rpm. Before the measurements, the electrode was held at 1.10 V during 120 s to equilibrate the system. Diffusion current (ID) was determined at the electrode potential of 0.4 V. Kinetic current (Ik) was calculated at 0.90 V according to the following equation:
where ID is the diffusion current at 0.4 V (A), I is the measured current (A), and Ik is the kinetic current (A).
Mass (MA) and surface (SA) catalytic activities were determined at 1600 rpm by dividing the kinetic current obtained at 0.9 V by mass and surface of platinum, respectively. Effective activation energy was estimated in the 10–35 °C temperature interval using Arrhenius equation:
where Ik is the kinetic current at a given overpotential, CO2 (T) is the oxygen concentration in electrolyte, which depends on the temperature, A is the constant, and E effact is the effective activation energy.
Fuel cell
Before using, Nafion 212 NRE (Sigma-Aldrich Co.) membranes were treated at 80 °C for 1 h sequentially in 1 M H2SO4 (high purity), 1 M H2O2 and distilled water. Commercial 20 wt% Pt/Vulcan XC-72 was used as the anode catalyst. 40% Pt/CNT (Pt/CNT), 40% Pt/CNT-M (Pt/CNT-M) and 40% Pt/CNT-MF (Pt/CNT-MF) were used as the cathode catalysts. Carbon Toray paper (TGP-H-120) and Sigracet 25BS (SGL Technol.) were used as the cathode and anode gas diffusion layers (GDL), respectively. The choice of such GDL is based on the experimental data found in our group, which showed that MEA based on these types of GDL results in the highest specific power in the PEMFC. Pt loading on the anode was 0.2 mgPt cm−2. Pt and Vulcan XC-72 loading on the cathode was 0.1 mgPt cm−2 and 0.6 mg cm−2, as it was reported in [43]. The addition of carbon support to the electrode can improve significantly the performance of the MEA, as was pointed out in [43]. Nafion content in the electrode layer was 25 wt% with respect to the total weight of the layer.
Catalyst ink was prepared by the following procedure. A weighed sample of the catalyst powder was added to 300 μl of a 50 vol.% solution of isopropyl alcohol in water. The solution was shaken intensively and treated for 25 min in an ultrasonic bath. This procedure was repeated two times. The required amount of Nafion® (5 mass.% dispersion in water, Aldrich) was added to the ink and treated in an ultrasonic bath for another 25 min.
Preparation of MEA was as follows. First, the membrane was covered with Nafion® binder at both sides by spraying 82 μl of Nafion® dispersion in water, followed by baking at 130 °C during 40 min. Then the catalyst ink was sprayed by airbrush on the membrane heated to 60–80 °C. To provide better contact, the membrane coated with cathode and anode electrode layers was pressed between two Teflon sheets at a temperature of 130 °C and a pressure of 5 atm during 1.5 min. Before measurements, the catalytic membrane was sandwiched between the anode and cathode GDL and placed in a fuel cell (Electrochem. Inc.). The geometric surface area of the working electrodes was 5 cm2. The polarization curves were recorded at 82 °C in two modes. The first one (Mode 1) was a galvanostatic mode with a scan rate of 0.001 A s−1 (at 0–0.02 A cm−2 in the current density range) and 0.01 A s−1 (at a current density higher than 0.02 A cm−2). In the second mode (Mode 2), the potential was stepped from 0.3 to –0.9 V with every 0.05 V. The current was measured after 2 min of the potential established indicating the reversible conditions.
The streams of hydrogen (100% RH) and oxygen (70% RH) were supplied to the anode and cathode at rates of 2.4 and 4.8 ml s−1, respectively. Polarization curves were recorded using potentiostat Autolab PGStat302N and booster Autolab BSTR20A. Electrochemically active surface area of platinum in the Pt/C catalysts was determined in a fuel cell at 75 °C using electrooxidation of adsorbed CO. In this experiment, the anode was supplied with hydrogen (100% RH), while the cathode was first purged with CO during 15 min at a potential of 0.05 V, and then CO was replaced by He flow at the same potential for another 15 min. CV of CO electrooxidation were collected in the 0.05–1.2 V potential range with a 0.05 V s−1 scan rate.
Impedance measurements (EIS) were performed during Mode 2 potentiostatic experiments in the 10−1–105 Hz frequency region with 0.01 V of potential amplitude.
Results and discussion
Nitrogen-modified CNT supports
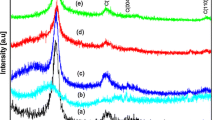
Melamine (Fig. 1-1, CNT-M) and melamine–formaldehyde resin (Fig. 1-2, CNT-MF) were employed as a coating for the CNT surface due to the high content of N-containing groups (the nitrogen content in the melamine–formaldehyde resin is ~ 55.6 wt%), an aromatic nature and a planar structure. The last two characteristics are of particular interest for providing uniform coating and high adhesion of N-containing polymers to the CNT surface. According to the XPS data (Fig. 2), the modification leads to the appearance of nitrogen-containing groups on the surface of nanotubes with a content of atoms of 1 and 8.3 at.% for CNT-M and CNT-MF samples, respectively.

XPS spectra of the N1s line with inelastically scattered electrons background subtracted using the Shirley model and deconvolution using the pseudo-Voigt function deconvolution for samples CNT-M (left) and CNT-MF (right)
According to the low-temperature nitrogen adsorption data (Table 1), the specific surface area of the samples is reduced compared to the original CNT. This is due to the fact that the mass of the modified nanotube was increased, while the surface did not change much.
XPS data (Fig. 2) indicate the presence of three nitrogen components on the surface: 398.6 eV—pyridinic nitrogen (45–52%), 399.8 eV—pyrrolic or nitrile (43%), and 401 eV—within graphitic structure (5–12%) [45, 46]. These data are in well agreement with the types of nitrogen groups that could be formed in the polymerization reactions (Fig. 1). Polymerization of melamine on the surface of MWCNTs (CNT-M) leads to a lower concentration of pyridinic and pyrrolic groups and a higher content of nitrogen in graphitic structure (Table 1). High surface concentration of pyridinic and pyrrolic groups, which are the anchoring sites for platinum ions, allows us to expect a homogeneous distribution of the dispersed active component.
The morphology of Pt nanoparticles
The TEM data (Fig. 3) show that platinum nanoparticles are distributed uniformly over the surface of the catalysts studied. The shape of the particles in all the samples is close to spherical, allowing the estimation of the average particle size (Table 2). In some samples (Pt/CNT and Pt/CNT-M), extended worm-like structures are occasionally observed, which consist also of spherical particles. Compared to the Pt/CNT catalyst, the average size of platinum nanoparticles in the nitrogen-modified samples is lower, presumably due to the high specific surface area and concentration of defective centers for the adsorption of platinum induced by polymerized resins. Nitrogen modification of CNT surface decreases d TEMN from 2.8 (Pt/CNT) to 2.3 (Pt/CNT-M) and 1.8 nm (Pt/CNT-MF). This is in line with the literature data, where a smaller Pt size was obtained on N-modified CNT [47] and correlates with the TEM data—Pt nanoparticles are localized mainly on N-containing coatings (Fig. 4). At the same time, the smallest average size of platinum nanoparticles is observed for the Pt/CNT-MF sample, in which the maximum concentration of surface nitrogen groups is observed (see Table 1).

TEM micrographs of the catalysts: Pt/CNT (1), Pt/CNT-M (2) and Pt/CNT-MF (3). Insets: Pt particle size distribution average size (d TEMN , black) and surface average size (d TEMS , gray)

TEM micrographs with high magnification of the catalysts: Pt/CNT-M (1) and Pt/CNT-MF (2). Arrows show the film of melam (1) and melamine–formaldehyde resin (2). Pt nanoparticles are attached to the modified layers
A good agreement is observed between the surface-averaged size of Pt nanoparticles in the Pt/CNT sample obtained by gas-phase CO titration (d GPTs ) and TEM data (d TEMs ), indicating that the TEM data describe adequately the entire surface of these catalysts. For the modified samples, higher d GPTs values are found as compared to d TEMs . This implies that a part of the active component is in the form of coarse particles with a large diameter, which were not taken into account when calculating the particle size distribution. All the catalysts except Pt/CNT have a narrow size distribution.
Electrochemical properties of Pt/C catalysts
According to the cyclic voltammetry analysis (CVA) data (Fig. 5-1), the Pt surface in Pt/C catalysts is typical of polycrystalline platinum. Peaks in the hydrogen region (0.05–0.4 V) and in the oxygen compound region (0.6–1.2 V) are well observed. Electrochemical oxidation of a monolayer of adsorbed CO (Fig. 6-2) is represented by the main oxidation peak of COads at potentials of 0.82–0.85 V in all the catalysts. An additional band is found in the lower potentials (0.65–0.74 V), which is most pronounced for the Pt/CNT sample. According to the literature, it refers to the oxidation of CO adsorbed either at points of contact between platinum particles or on the surface of large platinum particles [48, 49]. The presence of two peaks in CO electrooxidation was also observed for the Pt catalyst supported on carbon nanofilaments, which was attributed to a better metal-support interaction [50]. Electrochemically active surface of Pt (EAS ECPt ) was obtained from the CO electrooxidation data, and utilization factors were obtained as a ratio of EASPt to the total Pt surface determined from the gas-phase CO titration (Table 2). It is found that EAS ECPt obtained for nitrogen-modified samples are higher than that found for Pt/CNT. This effect is undoubtedly due to the nitrogen influence: the smaller Pt nanoparticle size as well as the higher utilization ratio of active component (Table 2). The ORR curves of the catalysts are shown in Fig. 5-3. Good linear dependences in the Koutetsky–Levich coordinates were found for all the catalysts (Fig. 5-4).

1 CV curves of the Pt/C catalysts, scan rate—0.05 V s−1; 2 CV curves of CO monolayer electrooxidation, scan rate—0.05 V s−1; 3 ORR curves for Pt/C catalysts at 0.9 V, rotating rate—1600 rpm, scan rate—0.01 V s−1, anode scan—closed symbols, cathode scan—opened symbols, inset—Koutecky–Levich plots for Pt/C catalysts; 4 Arrhenius plot obtained at 0.9 V (η = − 0.329 V) for anode (closed symbols) and cathode (open symbols) scans. Electrolyte 0.1 M HClO4, T = 25 °C

Polarization (open symbols, left axes) and power (filled symbols, right axes) characteristics of the MEAs based on Pt/C catalysts. 1 Mode 1 (galvanostatic); 2 Mode 2 (potentiostatic). Experimental conditions: 82 °C, anode—H2 (2.4 ml s−1, 100% RH), cathode—O2 (4.8 ml s−1, 70% RH). Pt loading: anode—0.2 mg cm−2, cathode—0.1 mg cm−2 for all the catalysts
The mass and surface activities as well as effective activation energies of the catalysts were obtained at 0.9 V from the ORR curves for both the anode and cathode scans (Table 3). Interestingly, the catalysts based on nitrogen-modified supports demonstrate a much higher Pt utilization ratio (0.8–0.9), as compared to that of Pt/CNT sample (only 0.6) (Table 2). Such a difference could be due to the effect of nitrogen modification of the supports. The analysis of Table 3 reveals that Pt/CNT-MF with the N content of 8.3 at.% has the highest mass activities in both the anode and cathode scans, which could be due to the high utilization of Pt nanoparticles. Interestingly, the Pt-free CNT sample with the nitrogen content of 8.4 at.% showed the best performance in the ORR in the series of CNT with the 0–10.7 at.% N content. This was ascribed to the high concentration of pyridinic groups in this sample [51]. However, the surface activity of this sample in the anode scan (245 μA cm −2Pt ) is lower than that of Pt/CNT sample (288 μA cm −2Pt ), which is due apparently to the size effect. The mass and surface activities of the Pt/CNT-MF sample in the cathode scan were found to be the highest ones among the catalysts. The activities obtained in this work are higher than that reported in the literature for Pt/C@PANI (30%) catalysts (68 mA mg −1Pt ) [52].
The lower activity of the catalysts measured in the cathode scan direction, as compared to the anode scan direction, is well known in the literature [53, 54] and caused by blocking of the active sites by oxygenated compounds adsorbed on platinum [54, 55]. The higher mass and surface cathodic activities of the nitrogen-modified Pt/CNT-MF sample are possibly associated with a larger surface area of Pt nanoparticles that is not blocked by oxygen groups when a potential of 0.9 V is reached in the cathode scan [56,57,58]. The effective activation energies obtained in this work at overpotential η = 0.323 V (37.3–40.8 kJ mol−1 in anodic scan) well correspond to the literature values of 38–49 kJ mol−1 at η = 0.3 V and 20–33 kJ mol−1 at η = 0.35 V [59] obtained for Pt/C catalysts. Modification of CNT with nitrogen virtually does not change the effective activation energy in the anode scan, whereas a noticeable decrease is observed on the cathode scan. Most probably, the higher activity and the lower effective activation energy observed at the cathode scan in N-modified CNT are explained by the interaction of Pt with N-centers of the CNT surface, which was reported in the literature [47].
Performance in PEMFC
The catalysts obtained were studied in PEMFC with 0.1 mg cm−2 of Pt loading on cathode added with Vulcan XC-72 carbon. Figure 6 shows the polarization curves and the specific power densities obtained for the Pt/C catalysts prepared here. Analysis of the results shows that the performance of the catalysts in PEMFC, measured by the both galvanostatic (Mode 1) and potentiostatic (Mode 2) methods virtually coincide (Table 4), indicating that the results were obtained under equilibrium conditions. The performance of the catalysts is in line with nitrogen content in the samples: Pt/CNT-MF > Pt/CNT-M > Pt/CNT, which is consistent with the literature data for Pt-free catalysts [23].
To evaluate the effect of various factors affecting the performance of the catalysts, an electrooxidation of adsorbed CO monolayer in PEMFC (Table 4) as well as impedance spectroscopic studies were conducted. The dependences of the real part (Z′) of the impedance on the imaginary part (Z″) measured at different potentials are displayed in Fig. 7.

Nyquist plots for the catalysts measured at potentials 0.8 V (1), 0.6 V (2), 0.4 V (3) and 0.3 V (4). Real and imaginary parts of impedance were corrected for the electrochemically active surface of Pt in MEA (Table 4). In the inset, the high-frequency regions are presented without EAS correction
Table 4 presents the maximum specific power density and utilization of Pt in electrochemical cell (U ECPt ) and in PEMFC (U MEAPt ) for all the catalysts studied. The U MEAPt correlates well with the U ECPt . As in the first case, the largest values (0.77–0.91) are shown by the catalysts with N-functionalized carbons (Table 4).
The impedance represented by the Nyquist diagram (Fig. 7) is the sum of anode and cathode impedances. Since the anode impedance is much smaller than the cathode one due to a higher activity in the hydrogen oxidation reaction as compared to that of the ORR, the impedance obtained can be regarded as cathodic. The membrane resistance determined at a high frequency (see insets in Fig. 7) for all the catalysts lies in the range of 50–75 mΩ cm 2geo and depends little on the applied potential. The semicircle diameter at medium frequency (Rct) is the resistance of charge transfer in the ORR and is determined by the activity of the catalyst. From the data presented, it can be concluded that at a potential of 800 mV, for all the catalysts Rct is approximately the same and lies in the range of 43–48 Ω cm 2Pt . With a decrease in the potential to 600 mV, a decrease in Rct and appearance of the second low-frequency semicircle is observed at 400 mV for all the catalysts; this is known in the literature as a characteristic of transport restrictions. According to the data presented, Pt/CNT catalysts show the best performance at 400 mV (Rct = 6.3 Ω cm 2Pt ). Catalysts based on functionalized nanotubes also show close but little higher values of Rct, 13.5 Ω cm 2Pt . At 300 mV, all the catalysts show an increase in resistance. This is most likely due to flooding of the catalyst layer, since under these conditions a maximum current is achieved, producing much water. Thus, the following conclusions can be drawn from the data obtained.
-
1.
At low overvoltages (E = 800 mV), the catalysts show similar Rct values, indicating close values of the specific catalytic activity of Pt in the ORR in PEMFC.
-
2.
At high overvoltages (E = 400 mV), the specific activities of nitrogen-containing (Pt/CNT-M and Pt/CNT-MF) catalysts are similar and lower than that of Pt/CNT sample.
-
3.
With a further increase in overvoltage (E = 300 mV), the transport constraints, possibly flooding, are found for all the catalysts, with the strongest effect of such limitations being observed for the Pt/CNT-M sample.
Discussion
It should be noted that the catalysts based on nitrogen-functionalized carbon nanotubes with a low-Pt loading (0.1 mg cm−2) showed the specific power density values (5.5–6.2 W mg −1Pt ) higher than those obtained in the literature for Pt/CNT and Pt/N-CNT catalysts (Table 5).
Modification of carbon nanotubes with nitrogen-containing polymers leads to a lower Pt average size, which is related to a higher concentration of the anchoring sites for Pt ion adsorption during the synthesis of Pt/C catalysts. This, in turn, might result in a higher utilization ratio of the active Pt component observed in electrochemical cell as well as in the PEMFC. The lower activity of the catalysts based on nitrogen-modified (N-CNT) nanotubes in the ORR, found in electrochemical cell, can be related to the size effect, which has been well documented in the literature [60]. In fact, it can be seen from Fig. 3 that these catalysts have a maximum amount of ca. 2 nm particles, which show minor activities in the ORR [60]. The lower activities of Pt/CNT-M(F) catalysts are corroborated by the higher Rct found by impedance measurements. Nevertheless, these catalysts show higher specific power densities in PEMFC as compared to Pt/CNT catalyst; this is caused by the high utilization of Pt (up to 0.91) in the cathode electrode (Table 4).
Variation of the methods for modifying the surface of carbon nanotubes by nitrogen-containing polymers makes it possible to control the content of surface nitrogen groups. The highest concentration (8 at.%) was obtained using the surface polymerization of the melamine–formaldehyde resin. The higher activity values in the ORR and specific power density in PEMFC are observed for Pt/CNT-MF catalyst with a high content of nitrogen surface groups. This is especially noticeable when comparing the activities obtained in the cathode scans (see Table 3), where the platinum surface is covered mainly with oxygen compounds.
Conclusions
A series of multi-walled carbon nanotubes (CNT) modified with nitrogen-containing groups was synthesized. The modification was carried out using two surface polymerization approaches: (1) melamine adsorbed on the surface of nanotubes (CNT-M), and (2) melamine–formaldehyde resin (CNT-MF). According to XPS, the observable nitrogen content in CNTs was of 1 and 8.3 at.%, respectively. According to CO gas-phase titration and TEM data, 40 mass. % Pt/C catalysts synthesized using nitrogen-modified carbon nanotubes showed almost uniform distribution of metal nanoparticles with the surface average size of 3–4 nm. However, the nitrogen-modified catalysts showed a greater part of Pt nanoparticles with the 0–2 nm dimension. Electrochemical studies of the catalysts revealed a high utilization degree of the active component (0.8–0.9) in nitrogen-modified catalysts, while Pt/CNT catalysts showed a lower value (0.6).
A comparison of the performance of the catalysts in PEMFC with 0.1 mg cm−2 Pt loading on the cathode and the impedance data showed that the Pt/CNT catalyst was the most active one, with, however, the lower specific power of 0.34–0.39 W cm−2, which is due to the low electrochemically active surface of the latter in MEA (24 m2 g −1pt for Pt/CNT) as found from the CO electrooxidation. On the other hand, nitrogen-modified Pt/CNT catalysts showed high values of specific power (0.54–0.55 W cm−2 for Pt/CNT-M with 1 at.% of nitrogen content, and 0.60–0.62 W cm−2 for Pt/CNT-MF with 8.3 at.% of nitrogen content). Since their activity according to the impedance method is similar, the difference in specific power is attributed to differences in the utilization ratio of Pt in MEA (0.79 and 0.84 for Pt/CNT-M and Pt/CNT-MF, respectively).
References
Rabis, A., Rodriguez, P., Schmidt, T.J.: Electrocatalysis for polymer electrolyte fuel cells: recent achievements and future challenges. ACS Catal. 2, 864–890 (2012)
Brouzgoua, A., Song, S.Q., Tsiakaras, P.: Low and non-platinum electrocatalysts for PEMFCs: current status, challenges and prospects. Appl. Catal. B: Environ. 127, 371–388 (2012)
Zhang, W., Sherrell, P., Minett, A.I., Razal, J.M., Chen, J.: Carbon nanotube architectures as catalyst supports for proton exchange membrane fuel cells. Energy Environ. Sci. 3, 1286–1293 (2010)
Hasche, F., Oezaslana, M., Strasser, P.: Activity, stability and degradation of multi walled carbon nanotube (MWCNT) supported Pt fuel cell electrocatalysts. Phys. Chem. Chem. Phys. 12, 15251–15258 (2010)
Sharma, S., Pollet, B.G.: Support materials for PEMFC and DMFC electrocatalysts—a review. J. Power Sources 208, 96–119 (2012)
Zhai, Y., Zhu, Z., Dong, S.: Carbon-based nanostructures for advanced catalysis. Chem. Cat. Chem. 7, 2806–2815 (2015)
Trogadas, P., Fuller, T.F., Strasser, P.: Carbon as catalyst and support for electrochemical energy conversion. Carbon 75, 5–42 (2014)
Guo, L., Jiang, W.-J., Zhang, Y., Hu, J.-S., Wei, Z.-D., Wan, L.-J.: Embedding Pt nanocrystals in N-doped porous carbon/carbon nanotubes toward highly stable electrocatalysts for the oxygen reduction reaction. ACS Catal. 5, 2903–2909 (2015)
Shahgaldi, S., Hamelin, J.: Improved carbon nanostructures as a novel catalyst support in the cathode side of PEMFC: a critical review. Carbon 94, 705–728 (2015)
Yu, X., Ye, S.: Recent advances in activity and durability enhancement of Pt/C catalytic cathode in PEMFC Part I. Physico-chemical and electronic interaction between Pt and carbon support, and activity enhancement of Pt/C catalyst. J. Power Sources 172, 133–144 (2007)
Rajalakshmi, N., Ryu, H., Shaijumon, M.M., Ramaprabhu, S.: Performance of polymer electrolyte membrane fuel cells with carbon nanotubes as oxygen reduction catalyst support material. J. Power Sources 140, 250–257 (2005)
Karthikeyan, N., Vinayan, B.P., Rajesh, M., Balaji, K., Subramani, A.K., Ramaprabhu, S.: Highly durable platinum based cathode electrocatalysts for PEMFC application using oxygen and nitrogen functional groups attached nanocarbon supports. Fuel Cells 15, 278–287 (2015)
Golovin, V.A., Maltseva, N.V., Gribov, E.N., Okunev, A.G.: New nitrogen-containing carbon supports with improved corrosion resistance for proton exchange membrane fuel cells. Int. J. Hydrog. Energy 42, 11159–11165 (2017)
Chiang, Y.-C., Hsieh, M.-K., Hsu, H.-H.: The effect of carbon supports on the performance of platinum/carbon nanotubes for proton exchange membrane fuel cells. Thin Solid Films 570, 221–229 (2014)
Nxumalo, E.N., Coville, N.J.: Nitrogen doped carbon nanotubes from organometallic compounds: a review. Materials 3, 2141–2171 (2010)
Ayala, P., Arenal, R., Rümmeli, M., Rubio, A., Pichler, T.: The doping of carbon nanotubes with nitrogen and their potential applications. Carbon 48, 575–586 (2010)
Ma, P.C., Siddiqui, N.A., Marom, G., Kim, J.-K.: Dispersion and functionalization of carbon nanotubes for polymer-based nanocomposites: a review. Compos. A Appl. Sci. Manuf. 41, 1345–1367 (2010)
Cao, M., Wu, D., Cao, R.: Recent advances in the stabilization of platinum electrocatalysts for fuel–cell reactions. Chem. Cat. Chem. 6, 26–45 (2014)
Fang, B., Kim, M.S., Kim, J.H., Song, M.Y., Wang, Y.J., Wang, H., Wilkinson, D.P., Yu, J.S.: High Pt loading on functionalized multiwall carbon nanotubes as a highly efficient cathode electrocatalyst for proton exchange membrane fuel cells. J. Mater. Chem. 21, 8066–8073 (2011)
Hernández-Fernández, P., Montiel, M., Ocón, P., Gómez de la Fuente, J.L., García-Rodríguez, S., Rojas, S., Fierro, J.L.G.: Functionalization of multi-walled carbon nanotubes and application as supports for electrocatalysts in proton-exchange membrane fuel cell. Appl. Catal. B Environ. 99, 334–352 (2010)
Chen, Z., Higgins, D., Tao, H., Hsu, R.S., Chen, Z.: Highly active nitrogen-doped carbon nanotubes for oxygen reduction reaction in fuel cell applications. J. Phys. Chem. C 113, 21008–21013 (2009)
Xiong, W., Du, F., Liu, Y., Perez Jr., A., Supp, M., Ramakrishnan, T.S., Dai, L., Jiang, L.: 3-D carbon nanotube structures used as high performance catalyst for oxygen reduction reaction. J. Am. Chem. Soc. 132, 15839–15841 (2010)
Chen, Z., Higgins, D., Chen, Z.: Nitrogen doped carbon nanotubes and their impact on the oxygen reduction reaction in fuel cells. Carbon 48, 3057–3065 (2010)
Alexeyeva, N., Shulga, E., Kisand, V., Kink, I., Tammeveski, K.: Electroreduction of oxygen on nitrogen-doped carbon nanotube modified glassy carbon electrodes in acid and alkaline solutions. J. Electroanal. Chem. 648, 169–175 (2010)
Chen, Y., Wang, J., Liu, H., Li, R., Sun, X., Ye, S., Knights, S.: Enhanced stability of Pt electrocatalysts by nitrogen doping in CNTs for PEM fuel cells. Electrochem. Commun. 11, 2071–2076 (2009)
Chen, Y., Wang, J., Liu, H., Banis, M.N., Li, R., Sun, X., Sham, T.-K., Ye, S., Knights, S.: Nitrogen doping effects on carbon nanotubes and the origin of the enhanced electrocatalytic activity of supported pt for proton-exchange membrane fuel cells. J. Phys. Chem. C 115, 3769–3776 (2011)
Jafri, R.I., Rajalakshmi, N., Ramaprabhu, S.: Nitrogen-doped multi-walled carbon nanocoils as catalyst support for oxygen reduction reaction in proton exchange membrane fuel cell. J. Power Sources 195, 8080–8083 (2010)
Vijayaraghavan, G., Stevenson, K.J.: Synergistic assembly of dendrimer-templated platinum catalysts on nitrogen-doped carbon nanotube electrodes for oxygen reduction. Langmuir 23, 5279–5282 (2007)
Higgins, D.C., Meza, D., Chen, Z.: Nitrogen-doped carbon nanotubes as platinum catalyst supports for oxygen reduction reaction in proton exchange membrane fuel cells. J. Phys. Chem. C 114, 21982–21988 (2010)
Zhang, Q., Zhang, Y., Cai, W., Yu, X., Ling, Y., Yang, Z.: Nitrogen doped carbon layer coated platinum electrocatalyst supported on carbon nanotubes with enhanced stability. Int. J. Hydrog. Energy 42, 16773–16781 (2017)
Zhang, X., Huang, Y., Chen, X., Gao, Q., Zhang, W.: Nitrogen-doped carbon nanotubes based on melamine-formaldehyde resin as highly efficient catalyst for oxygen reduction reaction. J. Colloid Interface Sci. 509, 1–9 (2018)
Kuznetsov, V.L., Krasnikov, D.V., Schmakov, A.N., Elumeeva, K.V.: In situ and ex situ time resolved study of multi-component Fe–Co oxide catalyst activation during MWNT synthesis. Phys. Status Solidi B 249, 2390–2394 (2012)
Kuznetsov, V.L., Bokova-Sirosh, S.N., Moseenkov, S.I., Ishchenko, A.V., Krasnikov, D.V., Kazakova, M.A., Romanenko, A.I., Tkachev, E.N., Obraztsova, E.D.: Raman spectra for characterization of defective CVD multi-walled carbon nanotubes. Phys. Status Solidi B 251, 2444–2450 (2014)
Mazov, I.N., Kuznetsov, A.M., Simonova, I.A., Stadnichenko, A.I., Ischenko, A.V., Romanenko, A.I., Tkachev, E.N., Anikeeva, O.B.: Oxidation behavior of multiwall carbon nanotubes with different diameters and morphology. Appl. Surf. Sci. 258, 6272–6280 (2012)
Kazakova, M.A., Kuznetsov, V.L., Semikolenova, N.V., Moseenkov, S.I., Krasnikov, D.V., Matsko, M.A., Ischenko, A.V., Zakharov, V.A., Romanenko, A.I., Anikeeva, O.B., Tkachev, E.N., Suslyaev, V.I., Zhuravlev, V.A., Dorozkin, K.V.: Comparative study of multiwalled carbon nanotube/polyethylene composites produced via different techniques. Phys. Status Solidi B 251, 2437–2443 (2014)
Li, X.F., Kong, L.N., Yang, J.H., Gao, M., Hu, T.J., Wu, X.T., Li, M.: Synthesis and characterization of nitrogen-doped carbon nanotubes by pyrolysis of melamine. Appl. Phys. A Mater. Sci. Process. 113, 735–739 (2013)
Zhao, Y.C., Yu, D.L., Zhou, H.W., Tian, Y.J., Yanagisawa, O.: Turbostratic carbon nitride prepared by pyrolysis of melamine. J. Mater. Sci. 40, 2645–2647 (2005)
Voropaev, I.N., Simonov, P.A., Romanenko, A.V.: Formation of Pt/C catalysts on various carbon supports. Russ. J. Inorg. Chem. 54, 1531–1536 (2009)
Kaiser, J., Simonov, P.A., Zaikovskii, V.I., Hartnig, C., Jörissen, L., Savinova, E.R.: Influence of carbon support on the performance of platinum based oxygen reduction catalysts in a polymer electrolyte fuel cell. J. Appl. Electrochem. 37(12), 1429–1437 (2007)
Rao, V., Simonov, P.A., Savinova, E.R., Plaksin, G.V., Cherepanova, S.V., Kryukova, G.N., Stimming, U.: The influence of carbon support porosity on the activity of PtRu/Sibunit anode catalysts for methanol oxidation. J. Power Sources 145, 178–187 (2005)
Hicks, R.H., Yen, Q.-J., Bell, A.T.: Effects of metal-support interactions on the chemisorption of H2 and CO on PdSiO2 and PdLa2O3. J. Catal. 89(2), 498–510 (1984)
Bacaud, R., Blanchard, G., Charcosset, H., Tournayan, L.: Determination of the dispersity of supported platinum catalysts by hydrogen, oxygen or carbon monoxided chemisorption and hydrogen-oxygen titrations, using volumetric, gravimetric and catharometric techniques. React. Kinet. Catal. Lett. 12, 357–363 (1979)
Gribov, E.N., Zinovieva, A.Y., Voropaev, I.N., Simonov, P.A., Romanenko, A.V., Okunev, A.G.: Activities of Pt/Sibunit-1562 catalysts in the ORR in PEMFC: effect of Pt content and Pt load at cathode. Int. J. Hydrog. Energy 37, 11894–11903 (2012)
Trasatti, S., Petrii, O.A.: Real surface area measurements in electrochemistry. Pure Appl. Chem. 63(5), 711–734 (1991)
Kundu, S., Xia, W., Busser, W., Becker, M., Schmidt, D.A., Havenith, M., Muhler, M.: The formation of nitrogen-containing functional groups on carbon nanotube surfaces: a quantitative XPS and TPD study. Phys. Chem. Chem. Phys. 12, 4351–4359 (2010)
Pels, J.R., Kapteijn, F., Moulijn, J.A., Zhu, Q., Thomas, K.M.: Evolution if nitrogen functionalities in carbonaceous materialos during pyrolysis. Carbon 33, 1641–1653 (1995)
Ning, X., Yu, H., Peng, F., Wang, H.: Pt nanoparticles interacting with graphitic nitrogen of N-doped carbon nanotubes: effect of electronic properties on activity for aerobic oxidation of glycerol and electro-oxidation of CO. J. Catal. 325, 136–144 (2015)
Cherstiouk, O.V., Simonov, P.A., Zaikovskii, V.I., Savinova, E.R.: CO monolayer oxidation at Pt nanoparticles supported on glassy carbon electrodes. J. Electroanal. Chem. 554, 241–251 (2003)
Maillard, F., Savinova, E.R., Stimming, U.: CO monolayer oxidation on Pt nanoparticles: further insights into the particle size effects. J. Electroanal. Chem. 599, 221–232 (2007)
Sebastián, D., Calderón, J.C., González-Expósito, J.A., Pastor, E., Martínez-Huerta, M.V., Suelves, I., Moliner, R., Lázaro, M.J.: Influence of carbon nanofiber properties as electrocatalyst support on the electrochemical performance for PEM fuel cells. Int. J. Hydrog. Energy 35(18), 9934–9942 (2010)
Rao, C.V., Cabrera, C.R., Ishikawa, Y.: In search of the active site in nitrogen-doped carbon nanotube electrodes for the oxygen reduction reaction. J. Phys. Chem. Lett. 1, 2622–2627 (2010)
Chen, S., Wei, Z., Qi, X., Dong, L., Guo, Y.-G., Wan, L., Shao, Z., Li, L.: Nanostructured polyaniline-decorated Pt/C@PANI core–shell atalyst with enhanced durability and activity. J. Am. Chem. Soc. 134, 13252–13255 (2012)
Shinozaki, J.I., Zack, J.W., Richards, R.M., Pivovar, B.S., Kocha, S.S.: Oxygen reduction reaction measurements on platinum electrocatalysts utilizing rotating disk electrode technique I. Impact of impurities, measurement protocols and applied corrections. J. Electrochem. Soc. 162(10), 1144–1158 (2015)
Zalitis, C.M., Kramer, D., Kucernak, A.R.: Electrocatalytic performance of fuel cell reactions at low catalyst loading and high mass transport. Phys. Chem. Chem. Phys. 15, 4329–4340 (2013)
Tanaka, H., Sugawara, S., Shinohara, K., Ueno, T., Suzuki, S., Hoshi, N., Nakamura, M.: Infrared reflection absorption spectroscopy of OH adsorption on the low index planes of Pt. Electrocatalysis 6, 295–299 (2015)
Sugawara, S., Suzuki, Y., Kocha, S.S., Shinohara, K.: Electrocatalytic activity analysis of PEFC cathode by 1-D macrohomogeneous model of catalyst layer. Electrochemistry 79(5), 404–413 (2011)
Nagai, T., Murata, H., Morimoto, Y.: Analysis of the relation between oxidation state and ORR activity of Pt by linear sweep voltammetry. ECS Trans. 33(1), 125–130 (2010)
Sugawara, S., Tsujita, K., Mitsushima, S., Shinohara, K., Ota, K.: Simultaneous electrochemical measurement of oxygen reduction and Pt oxide formation/reduction on pt nanoparticle surface. Electrocatalysis 2, 60–68 (2011)
Neyerlin, K.C., Gu, W., Jorne, J., Gasteiger, H.A.: Determination of catalyst unique parameters for the oxygen reduction reaction in a PEMFC. J. Electrochem. Soc. 153(10), A1955–A1963 (2006)
Sui, S., Wang, X., Zhou, X., Su, Y., Riffat, S., Liu, C.-J.: A comprehensive review of Pt electrocatalysts for the oxygen reduction reaction: nanostructure, activity, mechanism and carbon support in PEM fuel cells. J. Mater. Chem. A 5, 1808–1825 (2017)
Zhang, W., Chen, J., Swiegers, G.F., Ma, Z.-F., Wallace, G.G.: Microwave-assisted synthesis of Pt/CNT nanocomposite electrocatalysts for PEM fuel cells. Nanoscale 2, 282–286 (2010)
Fang, B., Chaudhari, N.K., Kim, M.-S., Kim, J.H., Yu, J.-S.: Homogeneous deposition of platinum nanoparticles on carbon black for proton exchange membrane fuel cell. J. Am. Chem. Soc. 131, 15338 (2009)
Jafri, R.I., Rajalakshmi, N., Dhathathreyan, K.S., Ramaprabhu, S.: Nitrogen doped graphene prepared by hydrothermal and thermal solid state methods Jafras catalyst supports for fuel cell. Int. J. Hydrog. Energy 40, 4337–43438 (2015)
Jung, D.H., Bae, S.J., Kim, S.J., Nahm, K.S., Kim, P.: Effect of the Pt precursor on the morphology and catalytic performance of Pt-impregnated on Pd/C for the oxygen reduction reaction in polymer electrolyte fuel cells. Int. J. Hydrog. Energy 36, 9115–9122 (2011)
Esfandiari, A., Kazemeini, M., Bastani, D.: Synthesis, characterization and performance determination of an Ag@Pt/C electrocatalyst for the ORR in a PEM fuel cell. Int. J. Hydrog. Energy 41, 20720–20730 (2016)
Acknowledgements
This work was supported by Ministry of Science and Higher Education of the Russian Federation. The authors would like to thank Dr. R.V. Gulyaev (XPS studies), Dr. A.V. Ischenko and Dr. E.Yu. Gerasimov (TEM studies).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gribov, E.N., Kuznetsov, A.N., Golovin, V.A. et al. Effect of modification of multi-walled carbon nanotubes with nitrogen-containing polymers on the electrochemical performance of Pt/CNT catalysts in PEMFC. Mater Renew Sustain Energy 8, 7 (2019). https://doi.org/10.1007/s40243-019-0143-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40243-019-0143-2