
Abstract
Intestinal fibrosis leading to strictures remains a significant clinical problem in inflammatory bowel diseases (IBD). The role of bacterial components in activating intestinal mesenchymal cells and driving fibrogenesis is largely unexplored. Tamoxifen-inducible α-SMA promoter Cre mice crossed with floxed MyD88 mice were subjected to chronic dextran sodium sulfate colitis. MyD88 was deleted prior to or after induction of colitis. Human intestinal myofibroblasts (HIMF) were exposed to various bacterial components and assessed for fibronectin (FN) and collagen I (Col1) production. RNA sequencing was performed. Post-transcriptional regulation was assessed by polysome profiling assay. Selective deletion of MyD88 in α-SMA-positive cells prior to, but not after induction of, experimental colitis decreased the degree of intestinal fibrosis. HIMF selectively responded to flagellin with enhanced FN or Col1 protein production in a MyD88-dependent manner. RNA sequencing suggested minimal transcriptional changes induced by flagellin in HIMF. Polysome profiling revealed higher proportions of FN and Col1 mRNA in the actively translated fractions of flagellin exposed HIMF, which was mediated by eIF2 alpha and 4EBP1. In conclusion, selectivity of flagellin-induced ECM secretion in HIMF is post-transcriptionally regulated. The results may represent a novel and targetable link between the gut microbiota and intestinal fibrogenesis.
Similar content being viewed by others

Introduction
Organ fibrosis is a frequent complication of many chronic inflammatory diseases, including both forms of inflammatory bowel disease (IBD), Crohn’s disease (CD), and ulcerative colitis (UC).1,2 More than 30% of CD patients develop fibrosis-induced intestinal strictures and eventually require surgery.3,4 Fibrosis also occurs in UC, but is restricted to the mucosa and submucosa.5 Immunosuppressive and biological agents can control intestinal inflammation,6 but specific anti-fibrotic therapies are not available yet because a mechanistic understanding of intestinal fibrogenesis is limited.
Fibrosis represents an excessive accumulation of extracellular matrix (ECM) components, such as fibronectin (FN) and Collagen 1 (Col1), that leads to structural and functional abnormalities.7 The main effector cell-mediating fibrosis in all organs is the mesenchymal cell, which exists in three distinct but interrelated forms: the fibroblast, the myofibroblast, and the smooth muscle cell,7 the latter two being α-smooth muscle actin positive. In active IBD these cells are exposed to microbes, immune cells, non-immune cells and their products, many of which activate the myofibroblasts and cause them to produce excess ECM.8
IBD pathogenesis involves an abnormal immune response to the gut microbiota. Essentially all intestinal immune and non-immune cells sense microbe-derived, pathogen-associated molecular patterns (PAMPs) through pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) and Nod-like receptors (NLRs), resulting in innate and adaptive immune responses.9 All TLRs, except TLR3, signal through the adaptor molecule MyD88.10 Although these microbial-sensing pathways have been extensively studied in IBD from an immunological perspective, only few reports suggest a link between the microbiota and fibrosis. CD patients with high levels of anti-bacterial antibodies have the greatest frequency of strictures,11 experimental animals develop intestinal fibrosis when challenged with commensal or infectious agents12 and intestinal myofibroblasts can be activated by lipopolysaccharide (LPS) and lipoteichoic acid,13,14 Two recent reports further strengthened the link between the microbiota and tissue remodeling in the intestine of experimental animals.15,16
Information on specific mechanisms underlying a cause-and-effect relationship between the gut microbiota and human intestinal myofibroblasts (HIMF) are currently lacking. Based on the hypothesis that microbes can trigger a pro-fibrogenic response, we investigated whether bacterial products could convert intestinal α-smooth muscle actin (α-SMA)-positive HIMF into an activated pro-fibrogenic phenotype in vivo and in vitro, and which mechanisms may be involved. Deletion of MyD88 selectively in α-SMA-positive cells prior to induction of experimental fibrosis ameliorated intestinal ECM deposition. Although intestinal mesenchymal cells express multiple TLRs and NLRs, a pro-fibrogenic phenotype was triggered exclusively by flagellin, a broad activator of innate and adaptive immunity.17 Such events involved MyD88-dependent pathways that are post-transcriptionally regulated and resulted in excessive ECM production. Deletion of MyD88 selectively in α-SMA-positive cells after induction of experimental fibrosis did not change ECM deposition. These findings provide direct evidence for a novel mechanistic link between the gut microbiota and intestinal fibrogenesis.
Results
Specific deletion of MyD88 in α-SMA-positive cells prior to induction of intestinal fibrosis
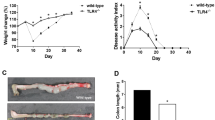
We first assesses the effect of MyD88 deletion in vivo selectively in α-SMA-positive cells in experimental intestinal fibrosis using tamoxifen-inducible α-SMA Cre mice (α-SMA CreERT2) crossed with MyD88 “floxed” mice (Supplementary Fig. 1a). This was accomplished by (1) confirmation of genetic recombination via PCR on genotyping; (2) active expression of Cre-recombinase in the muscularis mucosa and subepithelial myofibroblasts after exposing the α-SMA CreERT2 mice to tamoxifen (Supplementary Fig. 1c); and (3) reduced MyD88 gene expression in intestinal tissue after tamoxifen administration (Supplementary Fig. 1d). Clinical activity score: Mice fed with dextran sodium sulfate (DSS) developed clinical signs of colitis as evidenced by a clinical score >0.5 as early as day 3 (Fig. 1a). There was no clinically meaningful difference in clinical score throughout the experiment when comparing DSS wild type (WT) and DSS α-SMA CreERT2/MyD88 F/F (MyD88 deletion) mice (Fig. 1a). There was no significant difference between WT and MyD88 deleted mice not administered DSS and animals developed no clinical signs of colitis. At the end of the experiment (day 38, after the second cycle recovery) all animals had a similar overall clinical score and no evidence of colitis. When individual clinical parameters (weight loss, rectal bleeding, and stool consistency) were scored separately, no differences were noted between DSS WT and DSS MyD88 deletion or no DSS WT and no DSS MyD88 deletion. Colon length: No significant differences were noted among all groups regardless of DSS administration (Fig. 1b). Inflammatory score: Histologic evaluation of the distal colon in DSS-fed mice revealed erosions and inflammatory cell infiltrates, exposing the submucosa to luminal content (Fig. 1c). At the end of the second cycle of DSS (day 38) mice with MyD88 deletion that had received DSS displayed a significantly lower inflammatory score compared to WT animals given DSS. (Fig. 1c). Fibrosis location and severity score: In animals given DSS fibrosis was consistently more pronounced in the distal colon than the proximal colon and the terminal ileum (Fig. 1d, e). In these same animals that received DSS, the distal colon of MyD88 deleted mice exhibited significantly lower fibrosis scores compared to the colon of WT animals. (Fig. 1d, e). These results were confirmed by automatic quantification of Sirius red integrated density in the submucosa (Fig. 1f). To inquire which type of ECM was reduced in mice with MyD88 deletion, we stained colon cross-sections for ColI and FN and found a decreased amount of both molecules in the DSS MyD88 deleted mice compared to DSS WT mice (Fig. 1g) as shown by immunohistochemistry and integrated density analysis. We additionally measured the thickness of the layers of the murine intestinal wall and found a decrease in thickness of the muscularis mucosa, submucosa and muscularis propria in DSS-given MyD88 deleted mice compared to the DSS WT mice (Fig. 1h).


a Clinical score (ranges from 0 to 4). Overall, there is no clinically meaningful difference in between the two DSS and the two no DSS strains over the course of the experiment. b Colon length decreased in DSS wild type (WT) compared to no DSS WT mice, but there was no difference between DSS WT and DSS after α-SMA-specific deletion of MyD88 (MyD88 KO) mice. c Histopathologic inflammation score. There was increase in inflammation in DSS WT compared to no DSS WT mice, and a difference between DSS WT and DSS MyD88 KO mice. Representative images of murine colonic sections stained with hematoxylin and eosin and indicating epithelial barrier disruption with exposure of the submucosa to the lumen after treatment with DSS are shown in the panel. d, e Fibrosis score location and grade. There was an increase in fibrosis grade and location within the intestinal bowel wall in DSS WT compared to no DSS WT mice. α-SMA-specific deletion of MyD88 decreased fibrosis location and grade in DSS WT compared to DSS MyD88 KO mice. f In parallel with the decrease in pathologist graded fibrosis we found a reduction in the sirius red integrated density in the submucosa and g the amount of FN and ColI on immunostaining and as determined by integrated density measurements. h Measurement of wall thickness. There was a decrease in thickness of the muscularis mucosa, submucosa, and muscularis propria in the DSS MyD88 KO compared to the DSS WT mice. N = 10 for DSS per group and n = 5 for no DSS per group. *p < 0.05 or **p < 0.01 for DSS WT compared to DSS MyD88 KO; #p < 0.05 or ##p < 0.01 for DSS WT compared to no DSS WT. WT wild type, DSS dextran sodium sulfate.
Characterization of α-SMA-positive cells in the human intestinal mucosa
Given the profound effects of MyD88 deletion in murine experimental intestinal fibrosis, we examined the location and number of α-SMA-positive mesenchymal cells in control and IBD-involved human colon. In controls cells were located primarily adjacent to epithelial crypts, while others were interspersed in the lamina propria in the form of vascular smooth muscle cells (Fig. 2a). In active UC and CD mucosa localization was similar, but with a marked increase of α-SMA-positive cells throughout the lamina propria (Fig. 2a). These results demonstrate the intimate proximity of mesenchymal cells to gut luminal contents, so that any epithelial barrier defect could readily expose them to multiple and varied microbial components. We also investigated the potential expression pattern of PRRs by the intestinal mesenchymal cells. All HIMF expressed mRNA for nucleotide oligomerization domain NOD1 and NOD2 as well as toll-like receptor (TLR) 1 to 9, irrespective of their CD, UC, or control origin (Fig. 2b).

a Immunohistochemistry of the intestinal mucosa of IBD and control patients. Subepithelial and lamina propria α-SMA-positive mesenchymal cells are separate from the lumen by a single layer of epithelial cells. An increase of α-SMA-positive cells is noted in CD and UC compared to normal mucosa. Red: α-SMA-myofibroblasts; green: E-cadherin-positive epithelial cells; blue: DAPI-positive nuclei. Magnification ×100. Image representative of three samples per condition. b mRNA expression levels of Nod-like receptor 1 and 2 (NOD) and Toll-like receptors 1–9 (TLR) by HIMF measured by RT-PCR. No differences were noted among control, CD, and UC HIMF. Image representative of n = 3 for control, CD, and UC HIMF, respectively. c ECM secretion by HIMF in response to TLR and NLR ligands. Subconfluent HIMF were cultured for 72 h in the presence or absence of DAP, FSL-1, LPS, and FLA-ST alone or in combination. FN was measured via ELISA and Col1 via RIA. N = 8 for control and 11 for IBD HIMF. *p < 0.05, **p < 0.01 compared to untreated (Unst.) HIMF. d The results in a were confirmed with immunocytochemistry using antibodies to FN and Col1. Magnification ×100. Image representative of n = 4 control and 5 IBD HIMF. e FN secretion by HIMF in response to heat-inactivated wild-type S. typhimurium (WT; SL1344 aroA) and a flagellin-negative variant (ΔfliC/fljB). FN was measured via ELISA (n = 8). *p < 0.05, **p < 0.01. *p < 0.05; **p < 0.01 compared to untreated (Unt.).
Mesenchymal cell activation and ECM secretion in response to PRR ligands
We next exposed HIMF to ligands specific for TLR2/6, TLR4, TLR5 (synthetic diacylated lipoprotein (FSL-1), LPS, flagellin from S. typhimurium (FLA-ST); all three MyD88-dependent signaling) and NOD1 (γ-D-Glu-m dipeptide (DAP); MyD88 independent signaling), alone or in combination, and assessed ECM production by measuring secretion of FN and Col1 because both are abundantly produced in gut inflammation-induced fibrogenesis8 and were reduced in the MyD88 murine deletion model. HIMF spontaneously secreted FN and Col1, which increased only after stimulation with FLA-ST, alone or in combination with other ligands, while none of these alone was able to do so (Fig. 2c, d).
Because bacteria express a wide variety of PRR ligands, we confirmed the flagellin-dominant effect on ECM production by exposing HIMF to heat-inactivated S. typhimurium. Flagellin-positive WT S. typhimurium significantly increased FN secretion by HIMF, while the flagellin-negative S. typhimurium ΔfliC/fljB mutant did so to a significantly lower degree (Fig. 2e). No significant differences were detected between recombinant ligand- or whole bacteria-activated control, CD and UC derived HIMF (data not shown).
Pro-fibrotic effect of flagellin and other fibrogenic mediators
To determine the strength of flagellin’s pro-fibrotic effect we compared it to that of known mediators of fibrosis, namely interleukin-1β (IL-1β), IL-13, tumor necrosis factor-α (TNF-α), basic fibroblast growth factor (bFGF), and transforming growth factor-β1 (TGF-β1) in optimal concentrations (Supplementary Fig. 3a). A significant increase in FN secretion was observed when HIMF were exposed to TNF-α, bFGF and TGF-β1, but not IL-13 and IL-1β. IL-13, bFGF, IL-1β, and TGF-β1 significantly augmented Col1 secretion, the strongest effect being induced by TGF-β1. FLA-ST also significantly enhanced FN and Col1 secretion, with a further increase when combined to the TLR2/6 ligand.
Distinct regulation of inflammation and ECM production in PRR-activated human intestinal mesenchymal cells
Intestinal inflammation and fibrosis are intimately intertwined. To investigate whether the flagellin-specific response resulting in increased ECM production is also involved in the secretion of proinflammatory mediators, we compared FN secretion to the prototypical proinflammatory cytokine IL-6 in the same HIMF cultures. FLA-ST alone increased FN secretion far more than IL-6 secretion, and did so even in combination with DAP or LPS stimulation, while it was similarly effective in inducing FN and IL-6 in combination with FSL-1 (Supplementary Fig. 3b). In contrast, all other ligands and their combinations only increased IL-6 secretion, unless combined with FLA-ST (Supplementary Fig. 3b).
The pro-fibrotic response of flagellin is TGF-β1-independent
Intestinal mesenchymal cells are a major source of TGF-β1 and its potential induction via flagellin could explain their enhanced ECM secretion via autocrine TGF-β1 pathways. We therefore inhibited TGF-β1 signaling with the activin receptor-like kinase (ALK)5 inhibitor SB431542 before and during flagellin stimulation of HIMF. ALK5 blockade abrogated the TGF-β1-induced secretion of FN, but failed to inhibit the FN-inducing activity of FLA-ST, alone or in combination with other ligands (Supplementary Fig. 3c). Similar results were obtained using neutralizing anti-TGF-β1 antibodies in the supernatants of FLA-ST-activated HIMF (not shown). Additionally, we measured total TGF-β1 in the supernatants of FLA-ST-treated and untreated cell cultures. HIMF spontaneously secreted low amounts of TGF-β1 that did not increase upon stimulation with FLA-ST (data not shown). Interestingly, exposure of HIMF to the combination of flagellin and TGF-β1 led to higher secretion of FN compared to either flagellin or TGF-β1 alone (Supplementary Fig. 3d).
Lack of mesenchymal cell proliferation, migration, and α-SMA expression upregulation in response to bacterial components
The observed increase in ECM secretion by human intestinal mesenchymal cells could be related to differences in cell growth in response to flagellin. To investigate this possibility, we assessed the proliferation of HIMF after exposure to DAP, FSL-1, LPS, or FLA-ST using the same conditions used for the ECM production experiments. None of the ligands significantly increased proliferation by HIMF, while the positive control bFGF (10 ng/ml) did so (data not shown). A heightened activation state of mesenchymal cells is usually associated with an increased α-SMA expression. As measured by immunoblot, exposure to DAP, FSL-1, LPS, or FLA-ST for up to 72 h did not increase α-SMA expression in HIMF derived from IBD or control tissues, while TGF-β1 did so (data not shown). These results were confirmed by immunocytochemistry (data not shown). We additionally assessed, whether FLA-ST increased migration of HIMC with or without FN. While HIMF showed an increased migration towards FN, FLA-ST did not influence migration in the presence or absence of FN (data not shown).
Involvement of the MyD88 and MAPK signaling pathways in flagellin-induced ECM secretion by intestinal mesenchymal cells
Multiple pathways are involved in signaling downstream of activated TLRs.9 We therefore investigated whether flagellin leads to the activation of the canonical TLR signaling molecules MyD88, TRAF6, IRAK1, MAPK, and NFκB in HIMF, and whether HIMF FN secretion was dependent on this pathway. Incubation with FLA-ST rapidly induced binding of MyD88 and TRAF6 to IRAK1 in pull down experiments, suggesting that flagellin activates prototypical TLR signaling pathways in HIMF (Fig. 3a). In addition, FLA-ST led to the phosphorylation of extracellular-signal regulated kinase (ERK), p38 mitogen-activated protein kinase (MAPK), and NFκB p65 (Fig. 3b). This activation pattern was not unique to FLA-ST as FSL-1 and TGF-β1 also phosphorylated pERK and p38MAPK (data not shown). In all experiments conducted so far no difference was noted between control and IBD cells, and therefore all subsequent experiments were performed using only control HIMF. We next evaluated the specific role of MyD88 and p38MAPK in FN secretion using a RNA interference (RNAi) approach. After determining the optimal knockdown conditions, 10 nM and 24 h were selected as optimal for MyD88 blockade (Supplementary Fig. 4a), and we also verified that MyD88 knockdown persisted throughout the full duration of the experiments (data not shown). Compared to non-transfected and scrambled siRNA-transfected cells, MyD88 siRNA returned FLA-ST-enhanced FN secretion to baseline levels (Fig. 3c), and the same occurred when HIMF were co-cultured with WT S. typhimurium. Of note, TGF-β1-induced FN production remained unaffected by deletion of MyD88. We also investigated the involvement of p38MAPK by using SB203580, a chemical inhibitor which completely inhibited the FLA-ST-induced FN secretion and partly suppressed its TGF-β1-induced secretion (Fig. 3d).

a Co-immunoprecipitation performed using an IRAK1 antibody and levels of MyD88, IRAK1, and TRAF6 determined via immunoblotting. Image representative of n = 4 control HIMF. b Immunoblot performed using specific antibodies to the phosphorylated (P) and total signaling molecule as indicated. Image representative of n = 4 control and 4 IBD HIMF, respectively. c Effect of MyD88 deletion on FLA-ST-, WT S. typhimurium-, and flagellin-negative mutant ΔfliC/fljB-induced FN secretion by HIMF. MyD88 deletion was accomplished by siRNA transfection as described in Materials and methods and followed by exposure to FLA-ST for 72 h. TGF-β1 was used as a negative control for MyD88 siRNA. FN was determined via ELISA. N = 6 control HIMF; *p < 0.01 for FLA-ST siRNA compared to scrambled siRNA. Significant differences between bacterial ligand- and TGF-β1-treated and untreated HIMF are the same as in Fig. 2. d Effect of p38MAPK blockade on FN secretion by HIMF induced by FLA-ST, alone or in combination with other bacterial ligands, or TGF-β1. p38MAPK was blocked by adding SB203580 (10 µM) to HIMF, which were subsequently exposed to the ligands or TGF-β1 for 72 h. FN was determined via ELISA. N = 5 control HIMF. *p < 0.01 for SB203580 treated to untreated HIMF. Significant differences between bacterial ligand- and TGF-β1-treated and untreated HIMF are the same as in Fig. 2.
Post-transcriptional upregulation of flagellin-induced FN secretion
We next investigated the mechanisms involved in increasing flagellin-induced FN secretion and evaluated the HIMF mRNA expression levels of FN, COL1A1, and COL1A2 after incubation with FLA-ST using TGF-β1as a positive control. FLA-ST was used in the same concentration that optimally increased FN and ColI secretion in more than 30 separate HIMF lines. Surprisingly, no increase in mRNA levels for any of the above ECM components was detected at 24, 48, and 72 h (Fig. 4a), while TGF-β1 upregulated the expression of all three ECM components. There was also no upregulation of FN and ColI at 0.5, 1, 2, and 4 h after incubation with FLA-ST, but gene expression of IL-6 was increased at 0.5, 1, and 4 h (Supplementary Fig. 4b). Based on these results we selected a global approach by evaluating gene expression using RNA sequencing in HIMF with and without exposure to FLA-ST for 1 or 24 h.

a Gene expression levels of FN, COL1A1, and COL1A2 by HIMF exposed to FLA-ST or TGF-β1. HIMF were incubated with the stimuli for 24, 48, and 72 h and gene expression determined via quantitative PCR as described in Materials and methods. N = 3 for control HIMF; *p < 0.05 and **p < 0.01 for treated compared to untreated HIMF. b Differential gene expression of flagellin-treated versus control cells using RNA sequencing after 1 h (left panel) or 24 h (right panel). Red points indicate FDR < 0.05 and dashed gray lines indicate two-fold changes. c Principal components analysis (PCA) and heat maps of FDR < 0.2 genes in flagellin-treated versus control cells after 1 h (left panel) or 24 h (right panel). PCA with (red symbols) or without (black symbols) flagellin. When all assessed genes are considered, samples cluster tightly by patient sample with relatively little of the total variance being captured in the first two components. Values are row-normalized log2 counts per million (CPM). Clustering is by Euclidean distance.
To identify differentially expressed genes in HIMF treated with flagellin or not for 1 or 24 h, we employed different statistical RNA-seq analysis workflows based on reads per kilobase of transcript, per Million mapped reads (RPKM) and Fragments Per Kilobase of transcript, per Million mapped reads (FPKM) values, built from the leading DGE tools such as edgeR, DEseq2 and Limma.18,19,20 None of the tools detected differentially expressed genes between two groups (flagellin or no flagellin), given cutoffs with log2 (fold change) >1 and FDR < 0.05, including using principal component analysis (Fig. 4b, c).
We next downloaded the Molecular Signatures Database (MsigDB) H (Hallmark) and C2 (Curated) collection of gene sets provided by the Broad Institute, Cambridge, MA,21 in July 2018.22 Hallmark gene sets summarize and represent specific well-defined biological states or processes and display coherent expression, while C2 collection contains pathway information curated from biomedical literature, knowledge of domain experts and multiple online databases including Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome, and BioCarta.23,24 Gene Set Enrichment Analysis showed that 9 out of 50 hallmark gene sets are upregulated in HIMF in response to flagellin treatment while none of them are significantly enriched at FDR < 0.25 or nominal p value < 0.05; 41 genes were downregulated, of which seven sets were significantly enriched at FDR < 0.25 and p value < 0.05 (Supplementary Table 4A, B). Out of 3493 curated gene 915 sets were upregulated in response to flagellin treatment while only one was significantly enriched at FDR < 0.25 and 12 were significant at p value < 0.05; among 2578 downregulated genes 16 gene sets were significantly enriched at FDR < 0.25 and 246 were significant at p value < 0.05 (Supplementary Table 4C, D). These results confirm the findings of the individual gene analysis and suggest minimal changes at the transcriptional level in HIMF exposed to flagellin compared to unexposed cells. As a control experiment, we exposed the same HIMF to the NOD1 ligand DAP for 1 and 24 h and submitted them to RNA-seq analysis as described above. Exposure to DAP led to significant changes in global transcription and activated pathways, which excludes technical issues for lack of transcriptional changes induced by flagellin (The NOD1 data are part of a separate investigation to be reported elsewhere). We additionally confirmed by immunoblotting the upregulation of FN at the protein level in the same HIMF in response to flagellin (data not shown).
Mechanisms of post-transcriptional ECM upregulation induced by flagellin
Post-transcriptional regulation of ECM production has been reported in liver and skin fibrosis,25,26,27 and determining whether flagellin-induced ECM production is post-transcriptionally regulated could be extremely relevant because intervening in this process may identify novel targets for anti-fibrotic therapies. We therefore explored mechanisms of post-transcriptional regulation by first assessing mRNA stability. Stability of FN mRNA was reduced by incubation with FLA-ST after 24 h, but not at later time points, while that of COL1A1 or COL1A2 mRNA was unaffected. TGF-β1 did not influence the matrix mRNAs stability compared to that of control or FLA-ST-incubated HIMF (Fig. 5a). As next regulatory mechanism, we investigated the initiation of translational activation of the FN, COL1A1, and COL1A2 genes after incubation with and without FLA-ST using a polysome profiling assay.28 When comparing FLA-ST-activated to untreated HIMF a relative increase in mRNA loading for FN, COL1A1, and COL1A2 was detected in the most actively translated fractions (fractions 6–12) with a marked increase in the fraction with the highest translational efficiency (fraction 12). The increase in mRNA induced by TGF-β1 was also translationally regulated (Fig. 5b, c). Statistical analysis of translationally active and inactive fractions revealed significant differences for FN, COL1A1, and COL1A2 in both FLA-ST- and TGF-β1-stimulated HIMF (Supplementary Table 2).

a RNA stability in HIMF exposed to FLA-ST or TGF-β1. HIMF were exposed to actinomycin D (100 ng/ml) for 30 min before the incubation with FLA-ST or TGF-β1 (5 ng/ml) for 24, 48, or 72 h. The levels of mRNA for FN, COL1A1, and COL1A2 as shown as relative to the 0 h baseline. N = 3; *p < 0.05. b UV-absorbance profile of RNP and polysome complexes separated on a sucrose density gradient for HIMF treated with flagellin, TGF-β1, or untreated for 48 h as described in Materials and methods. c Bars indicate the fold change in mRNA expression in flagellin- or TGF-β1-treated HIMF compared to untreated cells. mRNAs from translation-active and translation-inactive fractions were analyzed by quantitative RT-PCR and normalized to GAPDH. Data are representative of five independent experiments for flagellin and four independent experiments for TGF-β1. Error bars represent SEM. d, e Immunoblot performed using specific antibodies to the phosphorylated (P) and total signaling molecule as indicated. HIMF were incubated with LPS, NOD-1L, or flagellin. Image representative of n = 4 HIMF.
To determine which mechanism explained the induction of FN secretion by flagellin, but not TLR4 or NOD1 ligands, we assessed activation of the AKT/mTOR pathway, given its previously described role in initiation of protein translation.29,30 DAP, LPS, or FLA-ST all lead to a phosphorylation of AKT or mTOR (Fig. 5d). We next examined the phosphorylation status of eukaryotic initiation factor 2 alpha (eIF2α) and eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4EBP1). Both molecules are known for their critical involvement in metabolic diseases and are important downstream molecules, broadly recognized for their significant roles in the cap-dependent translation initiation.31 Specifically, eIF2α binds to GTP, Met-tRNA and the 40S ribosome to form the pre-initiation complex. The phosphorylation of eIF2a blocks the formation of the complex and inhibits translation and hence de-phosphorylation of eIF2a in turn enhances translation and protein expression.32 4EBP directly interacts with the eukaryotic translation initiation factor 4E, a limiting component of the recruitment of 40S ribosomal subunits to the 5′ end of mRNAs.33 Interestingly, flagellin, but not DAP or LPS, rapidly and profoundly reduced phosphorylation of eIF2 alpha, the phosphorylation status corresponding to translation initiation. In addition, flagellin increased phosphorylation of 4EBP1 (Fig. 5e). LPS increased 4EPB phosphorylation at a later timepoint and to a lower degree compared to flagellin.
To assess for functional relevance of the above findings we used specific inhibitors to AKT (GSK690693) and mTOR (Torin 1) as well as a stabilizer of eIF2α phosphorylation (inhibition of eIF2α phosphorylation). Inhibition of mTOR and eIF2α signaling decreased spontaneous FN production by HIMF. Flagellin-induced FN production was reduced by AKT, mTOR, and eIF2α inhibition (Fig. 6a). We next wondered if AKT is necessary for mTOR activation and downstream signaling. Interestingly, we found that blockade of AKT with GSK690693 inhibited mTOR and 4EBP1 phosphorylation as well as eIF2α de-phosphorylation (Fig. 6b). Blockade of mTOR also ameliorated the phosphorylation of 4EBP1 and the de-phosphorylation of eIF2α (Fig. 6c).

a Immunoblot performed for fibronectin using specific antibodies. HIMF were untreated or exposed to flagellin and inhibitors to AKT (GSK690693), mTOR (Torin 1), or eIF2α (Salubrinal) were added as indicated. Image representative of n = 4 control HIMF. *p < 0.05, **p < 0.01, ns not significant. b, c Immunoblot performed using specific antibodies to the phosphorylated (P) and total signaling molecule as indicated. HIMF were incubated with flagellin, with or without the AKT inhibitor GSK690693 (b) or the mTOR inhibitor Torin 1 (c) for 60 min. Image representative of n = 3 HIMF.
Effect of specific deletion of MyD88 in α-SMA-positive cells after induction of intestinal inflammation
We finally tested whether specific deletion of MyD88 in α-SMA-positive cells after induction of experimental inflammation (e.g., after the first cycle of DSS) could ameliorate intestinal fibrosis. Clinical activity score: As before, DSS-fed mice developed clinical signs of colitis as evidenced by a clinical score >0.5 as early as day 2 (Supplementary Fig. 5). There was no clinically meaningful difference in clinical score throughout the experiment when comparing DSS WT and DSS MyD88 deletion mice. There was no difference between WT and MyD88 deleted mice when no DSS was administered and no DSS animals failed to developed clinical signs of colitis. At the end of the experiment at day 38 there was no difference in clinical signs of colitis between the DSS and the no DSS groups. We assessed each of the individual parameters of the clinical score separately (weight loss, rectal bleeding, and stool consistency) and no differences were noted between DSS WT and DSS MyD88 deletion or no DSS WT and no DSS MyD88 deletion. Colon length: The colon in the DSS WT mice was significantly shorter compared to the colon in the no DSS WT mice. There was no difference between the WT and MyD88 deletion groups, irrespective of DSS or no DSS. Inflammation and fibrosis scores: After two cycles of DSS (day 38) there was a significant increase in histologic score in DSS WT mice compared to no DSS animals. There was no difference in the inflammation score, the fibrosis location score, the fibrosis grade, or the type of collagen when comparing the WT with the MyD88 deletion groups (Supplementary Fig. 5). Finally, there was no difference between the DSS WT and DSS MyD88 deletion group in automatic fibrosis determination on Sirius Red and when measuring the wall thickness as described above.
Discussion
The gut microbiota plays a central role in all aspects of health and disease, including wound tissue healing and fibrosis.34 Microbes colonize all epithelial surfaces but, when these are injured, inflamed, or ulcerated, microbes may exert beneficial effects promoting repair or detrimental effects fostering further damage, inflammation, and fibrosis.34 In this context, the gastrointestinal tract is unique because it constantly undergoes epithelial wounding and repair in the presence of a massive luminal microbiota. Moreover, when mucosa integrity is compromised, as in IBD, all PRR-bearing cells will be exposed and respond to PAMPs, including the gut mesenchymal cells. This also holds true for murine intestinal DSS induced inflammation as illustrated by epithelial damage (Fig. 1c) and prior publications indicating increased bacterial translocation.35,36 To understand this response, we conducted experiments using first an α-SMA mesenchymal cell-specific mouse model and second a primary human intestinal mesenchymal cell system and obtained evidence for a dominant role of flagellin in transforming quiescent mesenchymal cells into ECM-producing effector cells. We additionally found evidence that this effect is post-transcriptionally regulated via eIF2α and 4EBP1.
Several lines of evidence suggest a connection between the microbiota and intestinal fibrosis: Ligands to TLR2 or TLR4 induce cytokine and chemokine secretion from cultured intestinal myofibroblasts.13,14 In animal models, microbes may initiate or perpetuate gut inflammation and fibrosis.12 Deletion of the bacterial signaling adaptor molecule MyD88 has been examined in the Salmonella-induced colitis model, but deletion was not restricted to mesenchymal cells and the timing was not controllable by an inducible promoter.37 MyD88 deletion has been reported in ColI-expressing cells, but it was not used in chronic fibrosis models and again no examination of temporal deletion in an inducible system was performed.38 Two recent reports further support the connection between microbes and intestinal fibrosis.15,16 Imai et al.15 found that microbiota driven intestinal fibrosis may be mediated by induction of the IL-33 receptor ST2 on epithelial cells,15 and Jacob et al.16 reported that the pro-fibrotic action of tumor necrosis factor-like cytokine 1 A (TL1A) is abrogated in the absence of a resident microbiota.16 In humans, gene variants located in or near genes involved in bacterial recognition are associated with IBD.39 CD patients carrying variants of the NOD2 gene, an intracellular pattern recognition receptor that binds muramyl dipeptide, a cell wall component of Gram-positive and -negative bacteria, are at increased risk for stricture formation and surgery. Furthermore, antibodies against microbial products are common in patients with IBD and are predictive of a complicated fibrostenotic phenotype.40
We found that deletion of MyD88 specifically in α-SMA-positive cells prior to induction of experimental fibrosis selectively reduced the grade and distribution of fibrosis in the gut wall without affecting clinical signs of colitis or the degree of inflammation. Interestingly, the degree of ECM deposition in the submucosa did not correlate with the thickness of the submucosa, which is consistent with the findings in human UC.5 HIMF express a wide range of microbial-sensing receptors making them fully capable of binding bacterial components. Surprisingly, among several PRRs, only flagellin was able to induce a pro-fibrogenic response in HIMF, as demonstrated by an increased secretion of FN and Col1. This selectivity was unexpected but highly relevant, considering that flagellin-expressing bacteria are abundant in the human intestine.41 Flagellin promoted ECM secretion without inducing proliferation, migration, or enhancing α-SMA expression by HIMF. Moreover, this response was similar among control, CD, or UC HIMF, pointing to an intrinsic property of flagellin rather than a response conditioned by cell differentiation or inflammation, as would be the case for IBD-derived HIMF that are chronically exposed to an inflammatory milieu. We can, however, not exclude that HIMFs loose a specific phenotype after isolation from the intestinal tissues. Any protocol using HIMF requires several passages in vitro in an “artificial” environment to be able to obtain enough cells for the experiments. When the pro-fibrogenic action of flagellin was compared to that of classical mediators of fibrogenesis, it induced comparable or higher secretion of ECM compared to all mediators, except for TGF-β1, reinforcing the inherent and powerful nature of the pro-fibrogenic signal imparted by this bacterial component. Interestingly, deletion of MyD88 specifically in α-SMA-positive cells after induction of experimental intestinal inflammation fibrosis was not reduced. One could speculate that in active inflammation, the contribution of the pro-fibrogenic bacterial signature may be overridden by the presence of a strong TGF-β signal or that already established tissue damage becomes self-perpetuating.42 Nonetheless, the TLR5-dependent signature could be especially important in early stages of the fibrogenic response, by amplifying weak TGF-β signals. In addition to TLRs, MyD88 also receives signals from the IL-1 receptor10 and we cannot exclude an effect of this pathway on the in vivo results. We however found that IL-1β did not significantly increase FN production in vitro in HIMF, strengthening the hypothesis that the responses on fibrosis are TLR5 mediated.
Multiple cells produce active TGF-β1, including mesenchymal cells, and strategies to target HIMF-derived TGF-β1 for anti-fibrotic therapy make sense in spite of the danger of concomitantly blocking its vital immunoregulatory properties.43 Nevertheless, the pro-fibrotic effect of flagellin was completely independent of TGF-β1 and additive to the pro-fibrotic action TGF-β1, similarly to what has been reported in the lung.44 This is notable because the steady presence of microbiota in the gut represents a constant pro-fibrotic signal instead of the ever-changing types and levels of other pro-fibrotic signals found in an inflamed intestine.8
We investigated whether flagellin, in addition to its intrinsic pro-fibrogenic activity, could also promote inflammation. As measured by HIMF production of IL-6, this response was weak and separate from that induced by other TLRs and NLR ligands, all of which were weak inducers of FN production unless combined with flagellin. These results illustrate a clear functional compartmentalization of various PRRs in regard to their effect on mesenchymal cells, and suggest that shifts in microbiota composition, as those occurring in IBD,45 could preferentially promote pro-fibrotic or proinflammatory activities in the gut. Of note, in vivo deletion of MyD88 in α-SMA-positive cells prior to induction of colitis not only reduced fibrosis but also the degree of inflammation, suggesting that bacterial sensing of myofibroblasts and smooth muscle cells is an active contributor to chronic inflammation.
Since the fibrogenic mechanisms of flagellin have not been previously investigated, we explored its underlying signaling pathways in HIMF. These mechanisms involved canonical TLR5 pathways similar to those of immune cells,46 including MyD88, IRAK1, p38MAPK, and NFκB, and blockade of MyD88 and p38MAPK lessened production of FN. Despite a robust flagellin-induced increase in FN and Col1 protein detected by immunoblot, ELISA, RIA, and immunocytochemistry in all HIMF lines, this response was not associated with a correspondent increase in mRNA, suggesting post-transcriptional regulation of gene expression. This was confirmed by RNA sequencing. Flagellin did not affect RNA stability of FN or Col1, but upregulated the translational activity of both genes as shown by the polysome profiling assays. This regulatory step in ECM production has been described in response to growth factors such as insulin growth factor-1 and TGF-β1,47,48 but its induction by a bacterial component is a novel finding that further highlights the influence of the microbiota on fibrogenic processes.
It has been reported that MyD88 is essential to link TLR5 engagement to PI3K in colonic epithelial cells.49 Our finding of increased phosphorylation of AKT and mTOR suggests that flagellin regulates the ECM production through the PI3K/AKT/mTOR signaling pathway in HIMF. Eukaryotic initiation factor—eIF2 alpha and eukaryotic initiation factor 4E-binding protein—4EBP1 are two important downstream molecules, which are broadly recognized for their significant roles in the cap-dependent translation initiation.31,50 The decreased phosphorylation of eIF2 alpha and the increased phosphorylation of 4EBP1 following flagellin treatment as well as the decreased flagellin-induced FN production by AKT, mTOR, and p-eIF2 alpha inhibition suggests the post-transcriptional regulation of ECM production in HIMF. We further assessed the phosphorylation of eIF2 alpha in response to AKT inhibition and mTOR inhibition, respectively. AKT inhibition but not mTOR inhibition caused the elevated phosphorylation of eIF2 alpha, which indicates AKT may directly determine the ECM production through the eIF2 alpha phosphorylation pathway.
The stimulatory action of flagellin on ECM production adds a whole new dimension to the dynamic and complex process of intestinal fibrosis.8 The relevance of the TLR5 pathway in driving fibrosis supports the existing evidence from experimental liver and cardiac fibrosis, where deletion of TLR5 ameliorated ECM accumulation.51,52 Ongoing studies will explore the specific role of TLR5 in experimental intestinal fibrosis in vivo. Flagellin clearly induced FN secretion in HIMF, but this response was lower than the response of HIMF to TGF-β1, which suggests that flagellin increases the non-TGF-β1-dependent portion of FN production.
In summary, by describing novel and distinct mechanisms of flagellin-induced increase in ECM production by intestinal mesenchymal cells, this study lends support to the notion of microbially induced intestinal fibrogenesis.1 Intestinal fibrosis and stricture formation are induced by multiple mediators53 and involve not only ECM production but also activation of myofibroblasts, increased autocrine cytokine production as well as cellular hyperplasia and hypertrophy in the muscular propria.2 Flagellin showed a selective effect on ECM production in our system and could contribute to the development or progression of inflammation-independent progression of stricture formation. In this regard, it is intriguing that bacterial flagellin is a dominant antigen in CD54 and that some TLR5 gene variants can protect against CD,55 events not observed in UC or control subjects. Therefore, selective modulation of flagellated bacteria or blockade of signals downstream of TLR5 could become novel pathophysiology-based strategies to prevent or treat intestinal fibrosis. Examples for compounds that modulate this pathway have been tested in experimental systems including the anti-microbial peptide TP359 or TLR5 antibodies,56,57 but to date this strategy has not been employed in humans. Systemically targeting bacterial responses, by, e.g., inhibiting MyD88 signaling, may carry a risk of enhancing bacterial replication inside the body. While α-SMA cell-selective strategies are not yet available, mesenchymal cell targeted approaches have been tested in experimental liver fibrosis using the platelet-derived growth factor beta receptor or fibroblast growth factor-inducible 14 (Fn14).58,59 With the development of new endpoints for clinical trials in fibrostenosing CD the implementation of this strategy may become reality.60
Materials and methods
Experimental DSS fibrosis
We used an approach of α-SMA-specific deletion of MyD88 in vivo. We controlled the timing of deletion by selectively activating α-SMA-linked Cre-recombinase with intraperitoneal tamoxifen administration. This was accomplished by crossing α-SMA CreERT2 mice (kindly provided by Dr. Wang61) with “floxed” MyD88 (Jackson laboratories, Bar Harbor, ME) counterparts (Supplementary Fig. 1a). All mice were on a C57/BL6 background and were bred, genotyped, and maintained in the Biomedical Research Unit at the Cleveland Clinic using standard protocols. Genotyping was performed by ear notching. Chronic DSS colitis was induced as previously described.62,63,64,65 All animal experiments were approved by the local IACUC committee (Protocol 2013-0990). Two distinct approaches were used: (a) α-SMA-specific MyD88 deletion prior to fibrosis induction (preventive model) and (b) after establishing inflammation (resolution model). A complete description of the two approaches can be found in Supplementary Information, Materials and Methods
Isolation of human intestinal fibroblasts
HIMF were obtained as explants of surgically resected intestinal mucosa, grown to subconfluence in Dulbecco’s minimal essential medium supplemented with 10% fetal bovine serum (FBS) and antibiotics, and established as long-term cultures fed twice weekly and subcultured at confluence. A complete description of the isolation of HIMF can be found in Supplementary Information, Materials and Methods.
Cell cultures
HIMF monolayers were cultured in 24-well plates (Corning Inc., Corning, NY) at 40,000 cells/well and exposed to individual PRR ligands or cytokines alone or in combination for 72 h unless otherwise stated. For immunoblot and RNA sequencing experiments HIMF were cultured in six-well plates (Corning) at a concentration of 100,000 cells/well. In all experiments HIMF were serum-deprived 24 h prior to addition of stimuli, and all experiments were performed under serum-free conditions to allow the measurement of ECM components.
RNA sequencing
Total RNA was extracted from 700,000 non-IBD human intestinal fibroblast cells that were either untreated or treated by the recombinant flagellin FLA-ST at 10 ng/ml for 24 h using the RNeasy Mini Kit (Qiagen, Germany). RNA quality was assessed using an Agilent bio-analyzer; samples with a RNA quality (RIN) score of >9.0 were used in RNA-seq. RNA-seq libraries were generated using Illumina TruSEQ kits following the manufacturer’s protocol and libraries were sequenced using an Illumina HiSEQ4000 using Illumina reagents and protocols. Single-ended 50 bp reads were trimmed with Trim Galore! (v.0.3.7) and checked for quality with FastQC (v0.11.3) (http://www.bioinformatics.babraham.ac.uk/projects) before alignment to the human genome (GRCh38.p10 using Gencode Release 27 annotations). Reads were aligned using STAR (v.2.5.3a)66 in single- (24 h samples) or two-pass mode (1 h samples) using default parameters and quantMode GeneCounts. For two-pass alignment, splice junctions from the first pass for all samples were combined and filtered to remove chrM and non-standard scaffolds as well as non-canonical junctions and those supported by <10 reads. Overall, an average of 84% of reads aligned uniquely resulting in an average of 26 million reads per sample. Raw counts were loaded into R67 and filtered to exclude transcripts with <0.5 reads per million mapped before performing differential expression (DE) analysis with edgeR.20 Filtering retained 30% (17,365) of transcripts at 1 h and 41.4% (23,414) at 24 h. Given a strong patient effect, the linear model included patient ID as a blocking factor in addition to the treatment term. Voom.,68 +imma.,19 and DESeq2 (ref. 18) were also used and yielded similar results. DEXSeq (v.1.22.0)69 was used to assess differential exon usage at 1 h, again blocking on patient ID. Values generated with the cpm function of edgeR, including library size normalization and log2 conversion, were used in figures. Heat maps were generated using pheatmap.70 Other plots were made using in-house R scripts (available upon request). RNA sequencing data have been deposited in Gene Expression Omnibus (GEO) under accession number GSE128384.
Statistical analysis
Data were analyzed using analysis of variance (ANOVA) for independent groups. Repeated measures for the same experiment were analyzed by using Student’s paired t-test. Values were expressed as mean ± SEM, and statistical significance was set at p < 0.05. For the polysome analysis data are presented as mean (95% CI) and the upper one-sided t-test was used to test whether the means were significantly higher than 1. Statistical significance was set at p < 0.05. All analyses were performed using SAS (version 9.3; The SAS Institute Inc., Cary, NC).
Detailed descriptions of experimental fibrosis endpoints, reagents, measurement of FN, collagen and cytokines, immunofluorescence, immunostaining, immunoblotting, co-immunoprecipitation, quantitative reverse transcriptase polymerase chain reaction, reverse transcription polymerase chain reaction, RNA stability, transfection, proliferation assay, migration assay, and polysome profiling can be found in the Supplementary Information, Materials and Methods.
References
Rieder, F., Brenmoehl, J., Leeb, S., Scholmerich, J. & Rogler, G. Wound healing and fibrosis in intestinal disease. Gut 56, 130–139 (2007).
Rieder, F., Fiocchi, C. & Rogler, G. Mechanisms, management, and treatment of fibrosis in patients with inflammatory bowel diseases. Gastroenterology 152, 340–350 (2017). e346.
Cosnes, J. et al. Impact of the increasing use of immunosuppressants in Crohn’s disease on the need for intestinal surgery. Gut 54, 237–241 (2005).
Gordon, I. O., Agrawal, N., Goldblum, J. R., Fiocchi, C. & Rieder, F. Fibrosis in ulcerative colitis: mechanisms, features, and consequences of a neglected problem. Inflamm. Bowel Dis. 20, 2198–2206 (2014).
Gordon, I. O. et al. Fibrosis in ulcerative colitis is directly linked to severity and chronicity of mucosal inflammation. Aliment. Pharm. Ther. 47, 922–939 (2018).
Armuzzi, A. et al. Results of the 2nd scientific workshop of the ECCO (IV): therapeutic strategies to enhance intestinal healing in inflammatory bowel disease. J. Crohn’s Colitis 6, 492–502 (2012).
Friedman, S. L., Sheppard, D., Duffield, J. S. & Violette, S. Therapy for fibrotic diseases: nearing the starting line. Sci. Transl. Med. 5, 167sr161 (2013).
Rieder, F. The gut microbiome in intestinal fibrosis: environmental protector or provocateur? Sci. Transl. Med. 5, 190ps110 (2013).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010).
Moresco, E. M., LaVine, D. & Beutler, B. Toll-like receptors. Curr. Biol. 21, R488–R493 (2011).
Mow, W. S. et al. Association of antibody responses to microbial antigens and complications of small bowel Crohn’s disease. Gastroenterology 126, 414–424 (2004).
Rieder, F., Kessler, S., Sans, M. & Fiocchi, C. Animal models of intestinal fibrosis: new tools for the understanding of pathogenesis and therapy of human disease. Am. J. Physiol. 303, G786–G801 (2012).
Otte, J. M., Rosenberg, I. M. & Podolsky, D. K. Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology 124, 1866–1878 (2003).
Burke, J. P. et al. Bacterial lipopolysaccharide promotes profibrotic activation of intestinal fibroblasts. Br. J. Surg. 97, 1126–1134 (2010).
Imai, J. et al. Flagellin-mediated activation of IL-33-ST2 signaling by a pathobiont promotes intestinal fibrosis. Mucosal Immunol. 12, 632–643 (2019).
Jacob, N. et al. Inflammation-independent TL1A-mediated intestinal fibrosis is dependent on the gut microbiome. Mucosal Immunol. 11, 1466–1476 (2018).
Vijay-Kumar, M., Aitken, J. D. & Gewirtz, A. T. Toll like receptor-5: protecting the gut from enteric microbes. Semin Immunopathol. 30, 11–21 (2008).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Subramanian, A., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. 102, 15545–15550 (2005).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Vastrik, I. et al. Reactome: a knowledge base of biologic pathways and processes. Genome Biol. 8, R39 (2007).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Stefanovic, B. et al. Posttranscriptional regulation of collagen alpha1(I) mRNA in hepatic stellate cells. Mol. Cell Biol. 17, 5201–5209 (1997).
Eckes, B., Mauch, C., Huppe, G. & Krieg, T. Differential regulation of transcription and transcript stability of pro-alpha 1(I) collagen and fibronectin in activated fibroblasts derived from patients with systemic scleroderma. Biochem. J. 315, 549–554 (1996).
Lindquist, J. N., Stefanovic, B. & Brenner, D. A. Regulation of collagen alpha1(I) expression in hepatic stellate cells. J. Gastroenterol. 35, 80–83 (2000).
Yao, P. et al. Coding region polyadenylation generates a truncated tRNA synthetase that counters translation repression. Cell 149, 88–100 (2012).
Mamane, Y., Petroulakis, E., LeBacquer, O. & Sonenberg, N. mTOR, translation initiation and cancer. Oncogene 25, 6416–6422 (2006).
Ma, X. M. & Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318 (2009).
Laplante, M. & Sabatini, D. M. mTOR signaling in growth control and disease. Cell 149, 274–293 (2012).
Kimball, S. R. Eukaryotic initiation factor eIF2. Int. J. Biochem. Cell Biol. 31, 25–29 (1999).
Grewal, S. S. Insulin/TOR signaling in growth and homeostasis: a view from the fly world. Int. J. Biochem. Cell Biol. 41, 1006–1010 (2009).
Scales, B. S. & Huffnagle, G. B. The microbiome in wound repair and tissue fibrosis. J. Pathol. 229, 323–331 (2013).
Panpetch, W. Additional Candida albicans administration enhances the severity of dextran sulfate solution induced colitis mouse model through leaky gut-enhanced systemic inflammation and gut-dysbiosis but attenuated by Lactobacillus rhamnosus L34. Gut Microbes 10, 1–16 (2019).
Andrade, M. E. Pretreatment and treatment with L-arginine attenuate weight loss and bacterial translocation in dextran sulfate sodium colitis. J. Parenter. Enter. Nutr. 40, 1131–1139 (2016).
Mansson, L. E. et al. MyD88 signaling promotes both mucosal homeostatic and fibrotic responses during Salmonella-induced colitis. Am. J. Physiol. 303, G311–G323 (2012).
Beswick, E. J. et al. TLR4 activation enhances the PD-L1-mediated tolerogenic capacity of colonic CD90+ stromal cells. J. Immunol. 193, 2218–2229 (2014).
Franke, A. et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat, Genet. 42, 1118 (2010).
Rieder, F., Lawrance, I. C., Leite, A. & Sans, M. Predictors of fibrostenotic Crohn’s disease. Inflamm. Bowel Dis. 17, 2000–2007 (2011).
Kolmeder, C. A. et al. Comparative metaproteomics and diversity analysis of human intestinal microbiota testifies for its temporal stability and expression of core functions. PLoS ONE 7, e29913 (2012).
Johnson, L. A. et al. Intestinal fibrosis is reduced by early elimination of inflammation in a mouse model of IBD: impact of a “Top-Down” approach to intestinal fibrosis in mice. Inflamm. Bowel Dis. 18, 460–471 (2012).
Diebold, R. J. et al. Early-onset multifocal inflammation in the transforming growth factor beta 1-null mouse is lymphocyte mediated. Proc. Natl Acad. Sci. USA 92, 12215–12219 (1995).
Kondo, Y. et al. Induction of epithelial-mesenchymal transition by flagellin in cultured lung epithelial cells. Am. J. Physiol. 303, L1057–L1069 (2012).
Kaur, N., Chen, C. C., Luther, J. & Kao, J. Y. Intestinal dysbiosis in inflammatory bowel disease. Gut Microbes 2, 211–216 (2011).
Seki, E. & Brenner, D. A. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology 48, 322–335 (2008).
Blackstock, C. D. et al. Insulin-like growth factor-1 increases synthesis of collagen type I via induction of the mRNA-binding protein LARP6 expression and binding to the 5’ stem-loop of COL1a1 and COL1a2 mRNA. J. Biol. Chem. 289, 7264–7274 (2014).
Boudreau, N., Clausell, N., Boyle, J. & Rabinovitch, M. Transforming growth factor-beta regulates increased ductus arteriosus endothelial glycosaminoglycan synthesis and a post-transcriptional mechanism controls increased smooth muscle fibronectin, features associated with intimal proliferation. Lab. Invest. 67, 350–359 (1992).
Rhee, S. H., Kim, H., Moyer, M. P. & Pothoulakis, C. Role of MyD88 in phosphatidylinositol 3-kinase activation by flagellin/toll-like receptor 5 engagement in colonic epithelial cells. J. Biol. Chem. 281, 18560–18568 (2006).
Richter, J. D. & Sonenberg, N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 433, 477–480 (2005).
Shu, M. et al. Inhibition of MAPK and NF-kappaB signaling pathways alleviate carbon tetrachloride (CCl4)-induced liver fibrosis in Toll-like receptor 5 (TLR5) deficiency mice. Biochem. Biophys. Res. Commun. 471, 233–239 (2016).
Liu, Y. et al. Toll-like receptor 5 deficiency attenuates interstitial cardiac fibrosis and dysfunction induced by pressure overload by inhibiting inflammation and the endothelial-mesenchymal transition. Biochim. Biophys. Acta 1852, 2456–2466 (2015).
Rieder, F. & Fiocchi, C. Intestinal fibrosis in IBD—a dynamic, multifactorial process. Nat. Rev. 6, 228–235 (2009).
Lodes, M. J. et al. Bacterial flagellin is a dominant antigen in Crohn disease. J. Clin. Invest. 113, 1296–1306 (2004).
Gewirtz, A. T. et al. Dominant-negative TLR5 polymorphism reduces adaptive immune response to flagellin and negatively associates with Crohn’s disease. Am. J. Physiol. 290, G1157–G1163 (2006).
Dosunmu, E. F. et al. The anti-microbial peptide TP359 attenuates inflammation in human lung cells infected with Pseudomonas aeruginosa via TLR5 and MAPK pathways. PLoS ONE 12, e0176640 (2017).
Blohmke, C. J. et al. Innate immunity mediated by TLR5 as a novel antiinflammatory target for cystic fibrosis lung disease. J. Immunol. 180, 7764–7773 (2008).
Bansal, R. et al. Novel engineered targeted interferon-gamma blocks hepatic fibrogenesis in mice. Hepatology 54, 586–596 (2011).
Huang, L. et al. Highly selective targeting of hepatic stellate cells for liver fibrosis treatment using a D-enantiomeric peptide ligand of Fn14 Identified by mirror-image mRNA display. Mol. Pharm. 14, 1742–1753 (2017).
Rieder, F. et al. An expert consensus to standardise definitions, diagnosis and treatment targets for anti-fibrotic stricture therapies in Crohn’s disease. Aliment. Pharm. Ther. 48, 347–357 (2018).
Worthley, D. L. et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 160, 269–284 (2015).
Lawrance, I. C. et al. A murine model of chronic inflammation-induced intestinal fibrosis down-regulated by antisense NF-kappa B. Gastroenterology 125, 1750–1761 (2003).
Fichtner-Feigl, S. et al. Induction of IL-13 triggers TGF-beta1-dependent tissue fibrosis in chronic 2,4,6-trinitrobenzene sulfonic acid colitis. J. Immunol. 178, 5859–5870 (2007).
Rieder, F. et al. Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am. J. Pathol. 179, 2660–2673 (2012).
Rieder, F. et al. The selective phosphodiesterase 4 inhibitor roflumilast and phosphodiesterase 3/4 inhibitor pumafentrine reduce clinical score and TNF expression in experimental colitis in mice. PLoS ONE 8, e56867 (2013).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Team, R. Core. R: A Language and Environment for Statistical Computing [WWW Document]. R Found. Stat. Comput., Vienna, Austria. https://www.r-project.org/. Accessed 4.1.15 (2015).
Liu, R. et al. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res. 43, e97 (2015).
Anders, S., Reyes, A. & Huber, W. Detecting differential usage of exons from RNA-seq data. Genome Res. 22, 2008–2017 (2012).
Kolde, R. pheatmap: Pretty Heatmaps. R package version 1.0. 8 (2015). http://CRAN.R-project.org/package=pheatmap.
Acknowledgements
This work was supported by grants from the National Institutes of Health (DK50984 and DK069854 to C.F.; T32DK083251, P30DK097948 Pilot, K08DK110415 to F.R.), the European Crohn’s and Colitis Organization and Digest Science (to C.F. and F.R.), the Deutsche Forschungsgemeinschaft (RI1735/2-1 to F.R.), and in part by the Crohn’s and Colitis Foundation of America (No.2308 to F.R.) and the National Center for Advancing Translational Sciences of the National Institutes of Health (UL1TR000439). The authors thank Dr. Melanie Rhee for the initial characterization of TLR and NLR expression in human intestinal mesenchymal cells, Dr. Luigi Franchi for critical review of the manuscript (IFM Therapeutics, Boston, MA), and acknowledge the support of the Departments of Colorectal Surgery and Pathology of the Cleveland Clinic. Tissue samples were provided by the Human Tissue Procurement Facility of the Cleveland Clinic. The authors wish to thank the University of Chicago Genomics Facility for RNA sequencing, Dr. Eleni Stylianou for support of the molecular immunology aspects of this work as well as Dr. Madeleine Lemieux for tremendous support with the analysis of sequencing data.
Author information
Authors and Affiliations
Contributions
S.Z., D.D., P.Y., S.E.O., C.F. and F.R. designed research; all authors performed research; P.Y., G.G., P.L.F., S.E.O., C.F. and F.R. contributed materials; S.Z., P.Y., R.L., S.E.O., C.F. and F.R. analyzed data; and all authors wrote the paper.
Corresponding author
Ethics declarations
Competing interests
F.R. is consultant to Allergan, AbbVie, Boehringer-Ingelheim, Celgene, Gilead, Helmsley, Janssen, Morphic, Pliant, Pfizer, Receptos, RedX, Roche, Samsung, Takeda, Thetis, and UCB. All other authors have no conflict of interest to declare.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhao, S., Dejanovic, D., Yao, P. et al. Selective deletion of MyD88 signaling in α-SMA positive cells ameliorates experimental intestinal fibrosis via post-transcriptional regulation. Mucosal Immunol 13, 665–678 (2020). https://doi.org/10.1038/s41385-020-0259-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-020-0259-9
This article is cited by
-
Revisiting fibrosis in inflammatory bowel disease: the gut thickens
Nature Reviews Gastroenterology & Hepatology (2022)
-
Fibroblasts as immune regulators in infection, inflammation and cancer
Nature Reviews Immunology (2021)
-
Fibrosis: from mechanisms to medicines
Nature (2020)
