Theoretical Description of R–X⋯NH3 Halogen Bond Complexes: Effect of the R Group on the Complex Stability and Sigma-Hole Electron Depletion


 ,
,
Abstract
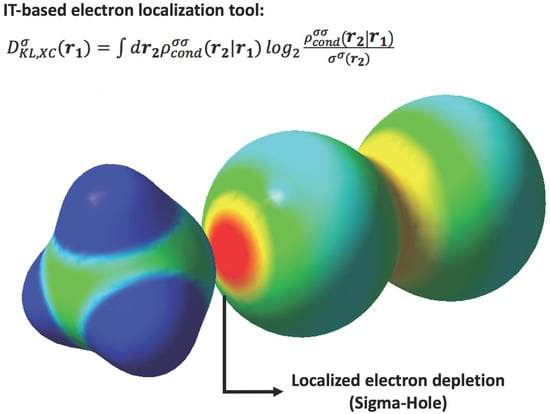
:
1. Introduction
2. Results and Discussion
2.1. Relative Stability of the R–X⋯NH3 Systems
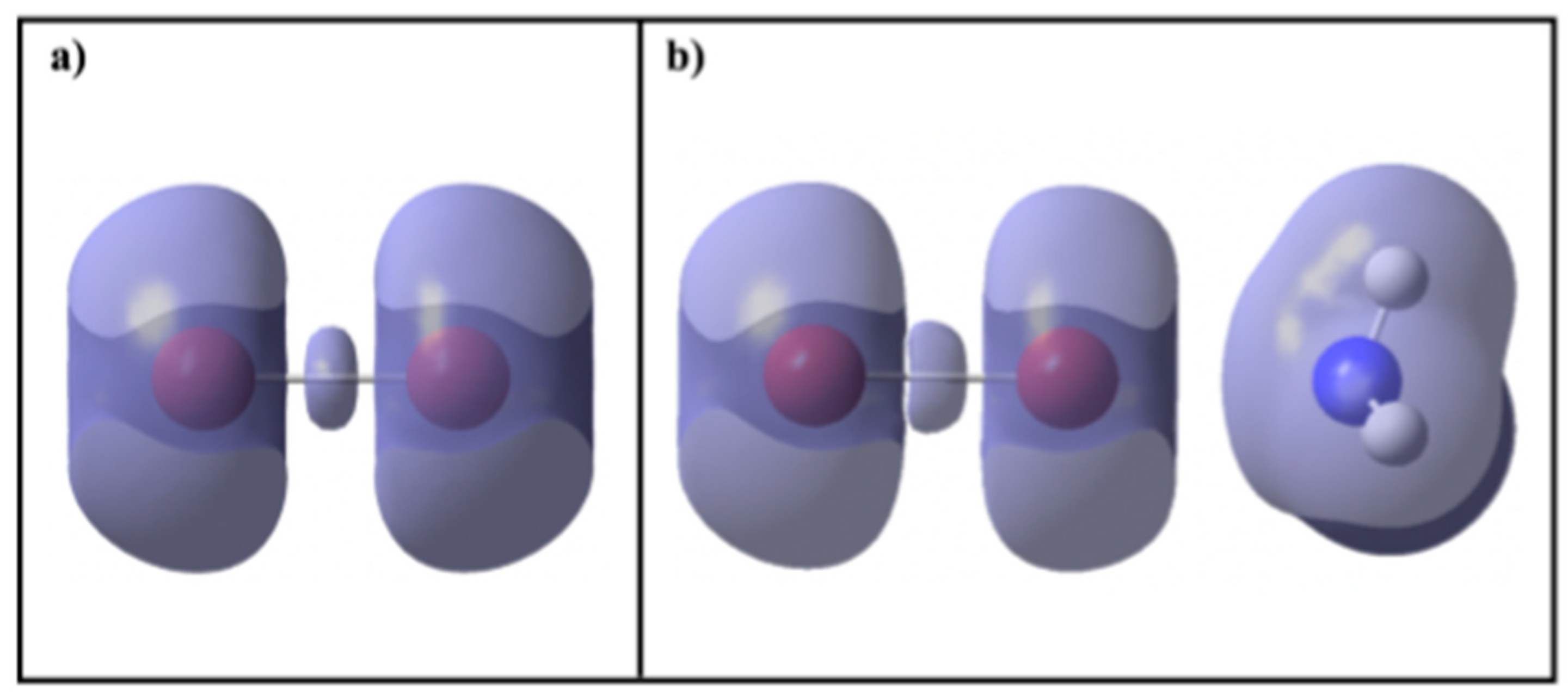
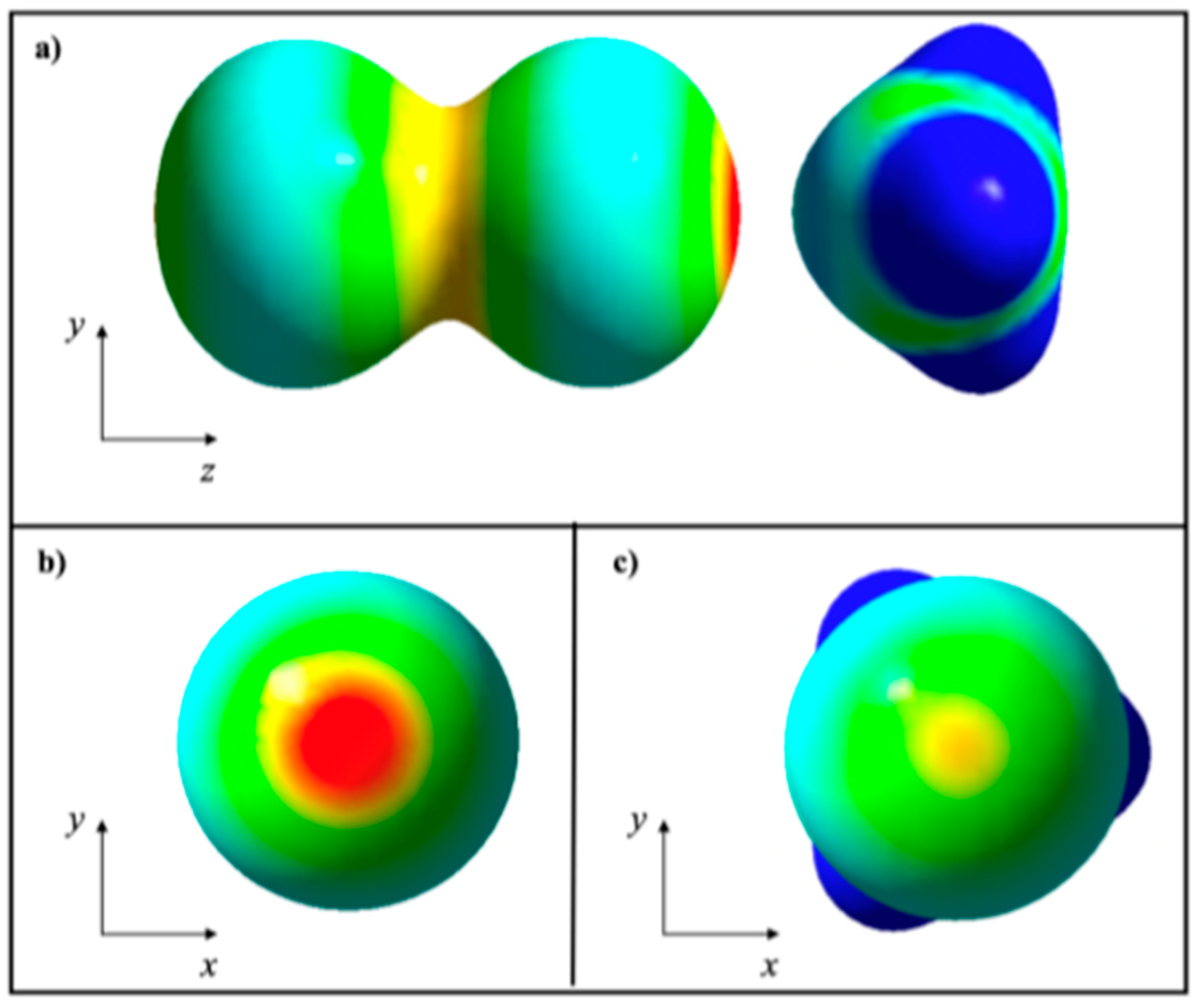
2.2. KLD Analysis of R-Br⋯NH3 Systems
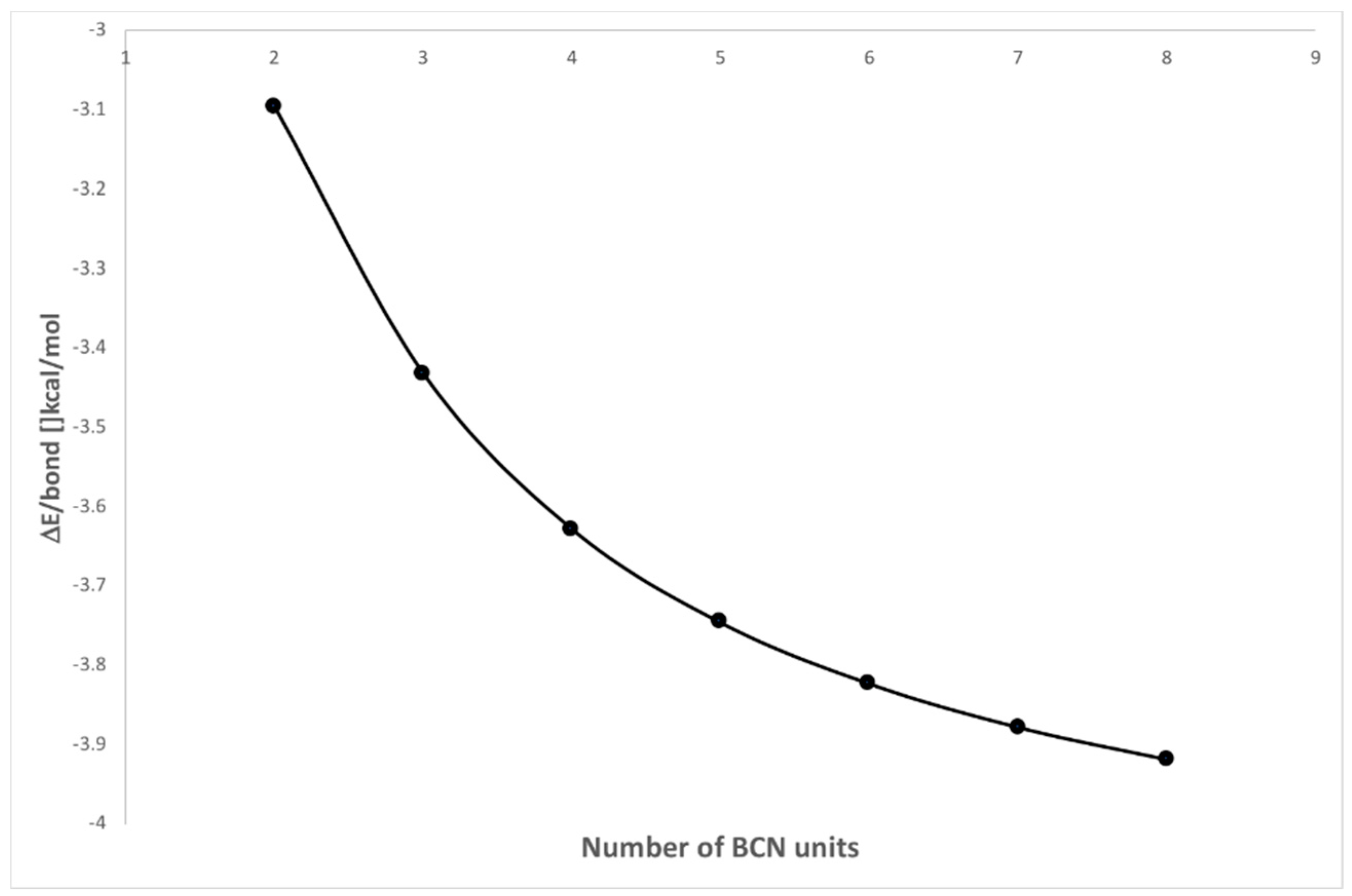
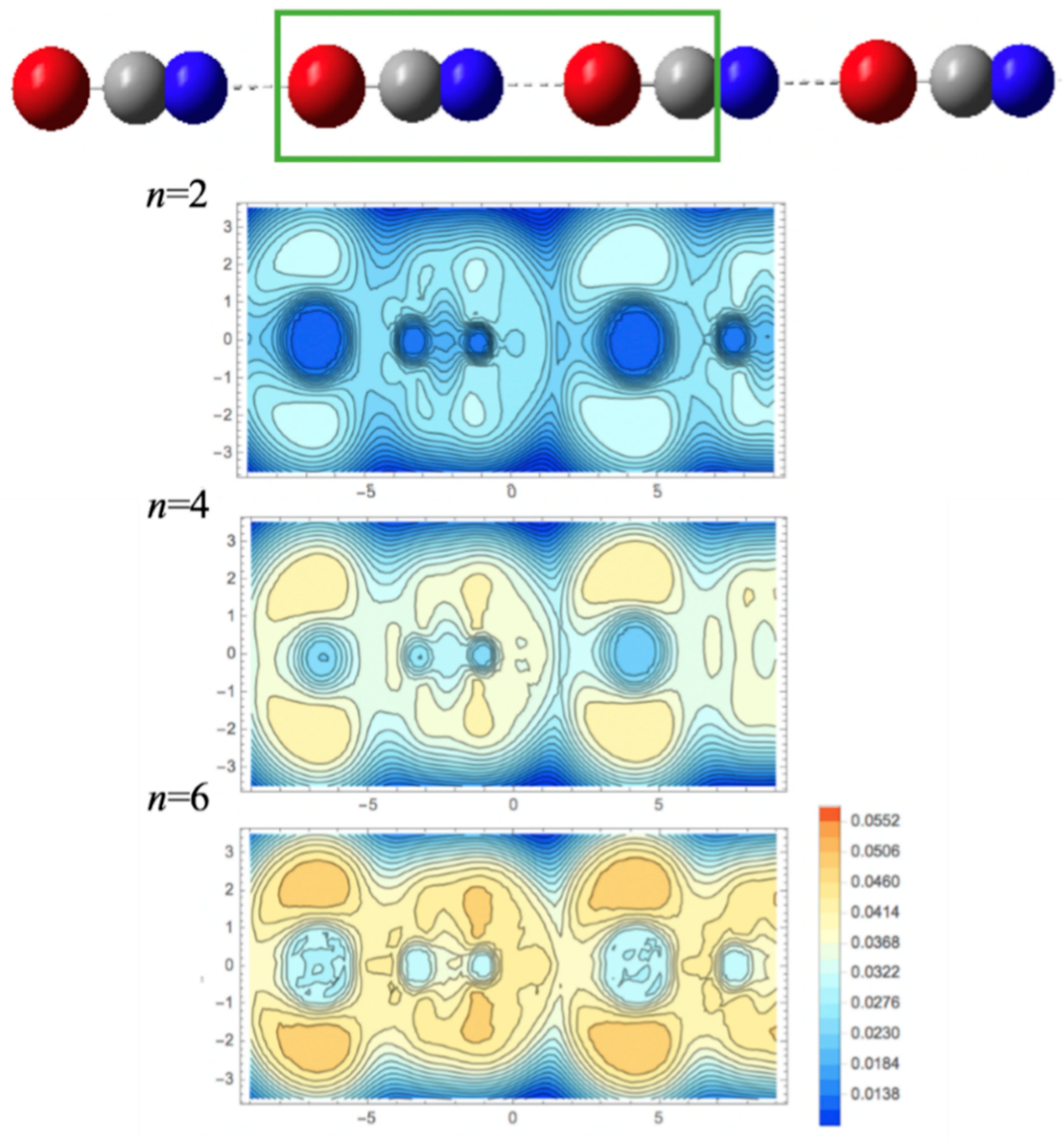
2.3. KLD Analysis of [BrCN]n Chains
3. Models and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Murray, J.S. σ -Hole Interactions: Perspectives and Misconceptions. Crystals 2017, 7, 212. [Google Scholar] [CrossRef] [Green Version]
- Cavallo, G.; Murray, J.S.; Politzer, P.; Pilati, T.; Ursini, M.; Resnati, G. Halogen bonding in hypervalent iodine and bromine derivatives: Halonium salts. Chem. Cryst. Eng. 2017, 4, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Santos, L.A.; Cunha, E.F.F.; Ramalho, T.C. Toward the Classical Description of Halogen Bonds: A Quantum Based Generalized Empirical Potential for Fluorine, Chlorine, and Bromine. J. Phys. Chem. A 2017, 121, 2442–2451. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, W.; Jin, W.J.; Bond, H. σ ‑ Hole Bond vs π ‑ Hole Bond: A Comparison Based on Halogen Bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef] [PubMed]
- Kola, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ ‑ Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, I.; Barrière, O.; Jeannin, O.; Fourmigué, M. Sequential Halogen Bonding with Ditopic Donors: σ‑Hole Evolutions upon Halogen Bond Formation. Cryst. Growth. Des. 2016, 16, 2963–2971. [Google Scholar] [CrossRef]
- Bauzá, A.; Quiñonero, D.; Deyà, M.; Frontera, A. Halogen Bonding versus Chalcogen and Pnicogen Bonding: A Combined Cambridge Structural Database and Theoretical Study. Cryst. Eng. Comm. 2013, 15, 3137–3144. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Panikkattu, S.; Chopade, P.D.; Desper, J. Competing hydrogen-bond and halogen-bond donors in crystal engineering. Cryst. Eng. Comm. 2013, 15, 3125–3136. [Google Scholar] [CrossRef] [Green Version]
- Awwadi, F.F.; Willett, R.D.; Peterson, K.A.; Twamley, B. The Nature of Halogen ⋯ Halogen Synthons: Crystallographic and Theoretical Studies. Chem-Eur. J. 2006, 12, 8952–8960. [Google Scholar] [CrossRef]
- Li, Q.; Xu, X.; Liu, T.; Jing, B.; Li, W.; Cheng, J.; Gong, B.; Sun, J. Competition between hydrogen bond and halogen bond in complexes of formaldehyde with hypohalous acids. Phys. Chem. Chem. Phys. 2010, 12, 6837–6843. [Google Scholar] [CrossRef] [PubMed]
- Guru Row, T.N.; Parthasarathy, R. Directional Preferences of Nonbonded Atomic Contacts with Divalent Sulfur in Terms of Its Orbital Orientations. 2. S…S Interactions and Nonspherical Shape of Sulfur in Crystals. J. Am. Chem. Soc. 1981, 103, 477–479. [Google Scholar]
- Nagels, N.; Hauchecorne, D.; Herrebout, W.A. Exploring the C-X…π Halogen Bonding Motif: An Infrared and Raman Study of the Complexes of CF3X (X = Cl, Br and I) with the Aromatic Model Compounds Benzene and Toluene. Molecules 2015, 18, 6830–6851. [Google Scholar] [CrossRef] [PubMed]
- Larsen, D.W.; Allredl, A.L. Halogen Complexes. J. Am. Chem. Soc. 1965, 3, 1216–1219. [Google Scholar] [CrossRef]
- Messina, M.T.; Metrangolo, P.; Navarrini, W.; Radice, S.; Resnati, G.; Zerbi, G. Infrared and Raman analyses of the halogen-bonded non-covalent adducts formed by a, v -diiodoperfluoroalkanes with DABCO and other electron donors. J. Mol. Struct. 2000, 524, 87–94. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G.; Pilati, T.; Liantonio, R.; Meyer, F. Engineering Functional Materials by Halogen Bonding. J. Pol. Sci. A. 2007, 45, 1–15. [Google Scholar] [CrossRef]
- Bolton, O.; Lee, K.; Kim, H.; Lin, K.Y.; Kim, J. organic materials by crystal design. Nat. Chem. 2011, 3, 205–210. [Google Scholar] [CrossRef]
- Christopherson, J.; Topic, F.; Barret, C.J.; Friscic, T. Halogen-Bonded Cocrystals as Optical Materials: Next- generation Control over Light-Matter Interactions. Cryst. Growth. Des. 2018, 18, 1245–1259. [Google Scholar] [CrossRef]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Resnati, G. Halogen Bonding in Supramolecular Chemistry. Minireviews 2008, 47, 6114–6127. [Google Scholar] [CrossRef]
- Lu, Y.; Shi, T.; Wang, Y.; Yang, H.; Yan, X.; Luo, X.; Jiang, H.; Zhu, W. Halogen Bonding—A Novel Interaction for Rational Drug Design? J. Med. Chem. 2009, 52, 2854–2862. [Google Scholar] [CrossRef]
- Parker, A.J.; Stewart, J.; Donald, K.J.; Parish, C.A. Halogen Bonding in DNA Base Pairs. J. Am. Chem. Soc. 2012, 134, 5165–5172. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.; Heidrich, J.; Zimmermann, M.O.; Exner, T.; Boeckler, F.M. Pharmaceutical Modeling Scaffold Effects on Halogen Bonding Strength. J. Chem. Inf. Model. 2019, 59, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Mendez, L.; Henriquez, G.; Sirimulla, S.; Narayan, M. Looking Back, Looking Forward at Halogen Bonding in Drug Discovery. Molecules 2017, 22, 1397. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. σ -Hole Bonding: A Physical Interpretation. Top. Curr. Chem. 2015, 358, 19–42. [Google Scholar] [PubMed]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ -hole. J. Mol. Model 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Bulat, F.A.; Toro-labbé, A.; Brinck, T.; Murray, J.S.; Politzer, P. Quantitative analysis of molecular surfaces: Areas, volumes, electrostatic potentials and average local ionization energies. J. Mol. Model 2010, 16, 1679–1691. [Google Scholar] [CrossRef]
- Brinck, T.; Murray, J.S.; Politzer, P. Surface Electrostatic Potentials of Halogenated Methanes as Indicators of Directional Intermolecular Interactions. Int. J. Quantum Chem. 1992, 64, 57–64. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Clark, T.; Riley, K.E.; Politzer, P. σ -Holes, π -holes and electrostatically-driven interactions. J. Mol. Model 2012, 18, 541–548. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.A.T.; Politzer, P. A Predicted New Type of Directional Noncovalent Interaction. Int. J. Quantum Chem. 2007, 107, 2286–2292. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Lane, P.A.T. σ-Hole Bonding and Hydrogen Bonding: Competitive Interactions. Int. J. Quantum Chem. 2007, 107, 3046–3052. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Halogen Bonding: An Interim Discussion. Chem. Phys. Chem. 2013, 14, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Riley, K.E.; Bulat, F.A.; Murray, J.S. Perspectives on halogen bonding and other sigma-hole interactions: Lex parsimoniae (Occam ’ s Razor). Comp. Theor. Chem. 2012, 998, 2–8. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. Feature article The fundamental nature and role of the electrostatic potential in atoms and molecules. Theor. Chem. Acc. 2002, 108, 134–142. [Google Scholar] [CrossRef]
- Mulliken, S. Molecular Compounds and their Spectra, 111. J. Am. Chem. Soc. 1952, 74, 811–824. [Google Scholar] [CrossRef]
- McDowell, S.A.C. Sigma-hole cooperativity in anionic [FX•••CH3]- (X, Y = Cl, Br) complexes. Chem. Phys. Lett. 2014, 598, 1–4. [Google Scholar] [CrossRef]
- Lohr, H.; Engel, A.; Josel, H.; Vogtle, F.; Schuh, W. Three-Dimensional Linkage by Electron Donor-Acceptor Interactions: Complexes of Organic Ammonium Halides with Triiodomethane. J. Org. Chem. 1984, 49, 1621–1627. [Google Scholar] [CrossRef]
- Pinter, B.; Nagels, N.; Herrebout, W.A.; Proft, F. De Halogen Bonding from a Hard and Soft Acids and Bases Perspective: Investigation by Using Density Functional Theory Reactivity Indices. Chem-Eur. J. 2013, 19, 519–530. [Google Scholar] [CrossRef]
- Lucia, S.; Hobza, P. CCSD[T] Describes Noncovalent Interactions Better than the CCSD(T), CCSD(TQ), and CCSDT Methods. J. Chem. Theory Comput. 2013, 9, 364–369. [Google Scholar]
- Wolters, L.P.; Schyman, P.; Pavan, M.J.; William, L.; Bickelhaupt, F.M.; Kozuch, S. The many faces of halogen bonding: A review of theoretical models and methods. WIREs Comput. Mol. Sci. 2014, 4, 523–540. [Google Scholar] [CrossRef]
- Quiñonero, D. Sigma-Hole Carbon-Bonding Interactions in Carbon-Carbon Double Bonds: An Unnoticed Contact. Phys. Chem. Chem. Phys. 2017, 19, 15530–15540. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Hobza, P.; Piton, M. Scaled MP3 Non-Covalent Interaction Energies Agree Closely with Accurate CCSD (T) Benchmark Data. Chem. Phys. Chem. 2009, 10, 282–289. [Google Scholar]
- Kozuch, S.; Martin, J.M.L. Halogen Bonds: Benchmarks and Theoretical Analysis. J. Chem. Theory Comput. 2013, 9, 1918–1931. [Google Scholar] [CrossRef] [PubMed]
- Urbina, A.S.; Torres, F.J.; Rincon, L. The electron localization as the information content of the conditional pair density. J. Chem. Phys. 2016, 144, 244104. [Google Scholar] [CrossRef]
- Rincón, L.; Almeida, R.; Contreras, P.L.; Javier Torres, F. The information content of the conditional pair probability. Chem. Phys. Lett. 2015, 635, 116–119. [Google Scholar] [CrossRef]
- Rincon, L.; Torres, F.J.; Becerra, M.; Liu, S.; Fritsch, A.; Rincon, L.; Torres, F.J.; Becerra, M.; Liu, S.; Fritsch, A. On the separation of the information content of the Fermi and Coulomb holes and their influence on the electronic properties of molecular systems. Mol. Phys. 2019, 117, 610–625. [Google Scholar] [CrossRef]
- Torres, F.J.; Rincón, L.; Zambrano, C.; Mora, J.R.; Méndez, M. A review on the information content of the pair density as a tool for the description of the electronic properties in molecular systems. Int. J. Quantum Chem. 2019, 119, e25763. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F.W.; Carroll, M.T.; Cheeseman, J.R.; Chang, C. Properties of Atoms in Molecules: Atomic Volumes. J. Am. Chem. Soc. 1987, 109, 7968–7979. [Google Scholar] [CrossRef]
- Anderson, L.N.; Aquino, F.W.; Raeber, A.E.; Chen, X.; Wong, B.M. Halogen Bonding Interactions: Revised Benchmarks and a New Assessment of Exchange vs. Dispersion. J. Chem. Theory Comput. 2018, 14, 180–190. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Zou, W.; Kraka, E. Strengthening of hydrogen bonding with the push-pull effect. Chem. Phys. Lett. 2017, 685, 251–258. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. B.01 2016; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Rincón, L.; Torres, F.J.; Almeida, R. Is the Pauli exclusion principle the origin of electron localisation? Mol. Phys. 2018, 116, 578–587. [Google Scholar] [CrossRef]
- Levy, M. Correlation Energy Functionals of One-Matrices and Hartree-Fock Densities. In Density Matrices and Density Functionals; Erdahl, R., Smith, V., Eds.; Springer: Berlin, Germany, 1987; p. 479. [Google Scholar]
- Kirk, D.B.; Hwu, W.W. Programming Massively Parallel Processors. A Hands-on Approach; Elsevier Inc.: Amsterdam, The Netherlands, 2010. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
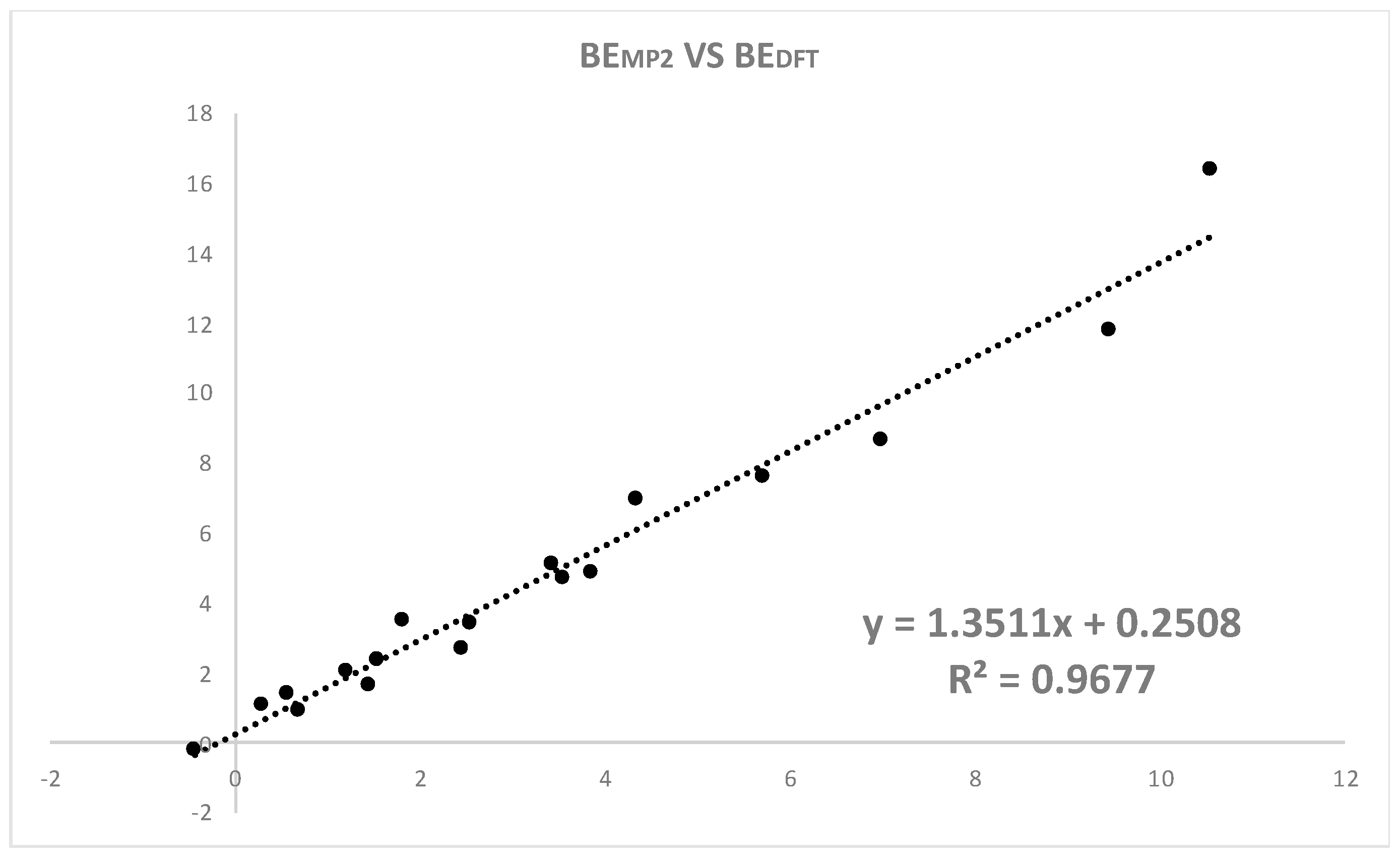
| Halogen Bond Donor | R Substituent | DX⋯N | ΔqN | ΔqX | ΔqxN | BEDFT | BEMP2 | BECCSD(T) |
|---|---|---|---|---|---|---|---|---|
| Cl | Cl | 2.52 (2.58) | 0.04 (0.00) | 0.00 (0.05) | −0.10 (−0.10) | 7.0 (7.9) | 4.4 (6.0) | 3.8 |
| CF3 | 2.97 (3.01) | −0.01 (−0.01) | 0.03 (0.04) | −0.02 (−0.02) | 2.8 (3.2) | 2.5 (3.3) | 2.5 | |
| CHF2 | 3.05 (3.10) | −0.01 (−0.01) | 0.03 (0.04) | −0.02 (−0.02) | 1.7 (2.0) | 1.5 (2.1) | 1.5 | |
| CH2F | 3.24 (3.28) | 0.00 (0.00) | 0.01 (0.01) | −0.01 (−0.01) | 1.0 (1.2) | 0.7 1.3) | 0.7 | |
| CH3 | 3.27 (3.37) | 0.00 (0.00) | 0.02 (0.03) | −0.01 (−0.01) | −0.1 (0.0) | −0.4 (−0.1) | −0.3 | |
| Br | Br | 2.55 (2.50) | 0.04 (0.01) | 0.02 (0.08) | −0.14 (−0.17) | 8.7 (12.4) | 7.0 (11.5) | 5.0 |
| CF3 | 2.88 (2.91) | −0.01 (−0.02) | 0.03 (0.05) | −0.03 (−0.03) | 4.9 (7.1) | 3.9 (6.5) | 3.9 | |
| CHF2 | 2.99 (3.03) | −0.01 (−0.01) | 0.04 (0.05) | −0.03 (−0.03) | 3.5 (5.2) | 2.6 (4.7) | 2.6 | |
| CH2F | 3.05 (3.11) | 0.00 (−0.01) | 0.03 (0.04) | −0.02 (−0.03) | 2.4 (4.0) | 1.6 (3.4) | 1.6 | |
| CH3 | 3.08 (3.17) | 0.00 (0.00) | 0.03 (0.04) | −0.02 (−0.03) | 1.5 (3.0) | 0.6 (2.2) | 0.6 | |
| I | I | 2.58 (2.62) | 0.08 (0.02) | 0.06(0.13) | −0.23 (−0.25) | 16.4 (17.2) | 10.6 (12.7) | 8.7 |
| CF3 | 2.80 (2.84) | 0.03 (−0.01) | 0.01 (0.05) | −0.04 (−0.04) | 11.8 (12.3) | 9.5 (10.7) | 8.9 | |
| CHF2 | 2.94 (3.01) | 0.03 (0.00) | 0.03 (0.06) | −0.04 (−0.04) | 7.7 (8.1) | 5.7 (6.7) | 5.4 | |
| CH2F | 3.07 (3.15) | 0.02 (0.00) | 0.03 (0.06) | −0.04 (−0.04) | 5.1 (5.5) | 3.4 (4.2) | 3.2 | |
| CH3 | 3.14 (3.26) | 0.02 (0.00) | 0.03 (0.05) | −0.04 (−0.04) | 3.5 (3.9) | 1.8 (2.5) | 1.6 |
| Halogen Bond Donor | R Substituent | BEMP2 | Pop (Br) | |
|---|---|---|---|---|
| Br | Br | 7.0 | 0.194 | 5.56 |
| CF3 | 3.9 | 0.259 | 5.64 | |
| CHF2 | 2.6 | 0.269 | 5.69 | |
| CH2F | 1.6 | 0.275 | 5.73 | |
| CH3 | 0.6 | 0.271 | 5.75 | |
| CH3CH2 | 0.3 | 0.278 | 5.78 | |
| CH2CH | 1.2 | 0.270 | 5.77 | |
| CHC | 3.6 | 0.249 | 5.59 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zurita, J.; Rodriguez, V.; Zambrano, C.; Mora, J.R.; Rincón, L.; Torres, F.J. Theoretical Description of R–X⋯NH3 Halogen Bond Complexes: Effect of the R Group on the Complex Stability and Sigma-Hole Electron Depletion. Molecules 2020, 25, 530. https://doi.org/10.3390/molecules25030530
Zurita J, Rodriguez V, Zambrano C, Mora JR, Rincón L, Torres FJ. Theoretical Description of R–X⋯NH3 Halogen Bond Complexes: Effect of the R Group on the Complex Stability and Sigma-Hole Electron Depletion. Molecules. 2020; 25(3):530. https://doi.org/10.3390/molecules25030530
Chicago/Turabian StyleZurita, Juan, Vladimir Rodriguez, Cesar Zambrano, Jose Ramón Mora, Luis Rincón, and F. Javier Torres. 2020. "Theoretical Description of R–X⋯NH3 Halogen Bond Complexes: Effect of the R Group on the Complex Stability and Sigma-Hole Electron Depletion" Molecules 25, no. 3: 530. https://doi.org/10.3390/molecules25030530