
Abnormal Cannabidiol Affects Production of Pro-Inflammatory Mediators and Astrocyte Wound Closure in Primary Astrocytic-Microglial Cocultures


Abstract
:1. Introduction
2. Results
2.1. LPS Stimulates the Production of NO, TNFα and IL-6 in Astrocytic-Microglial Cocultures and Isolated Astrocytes
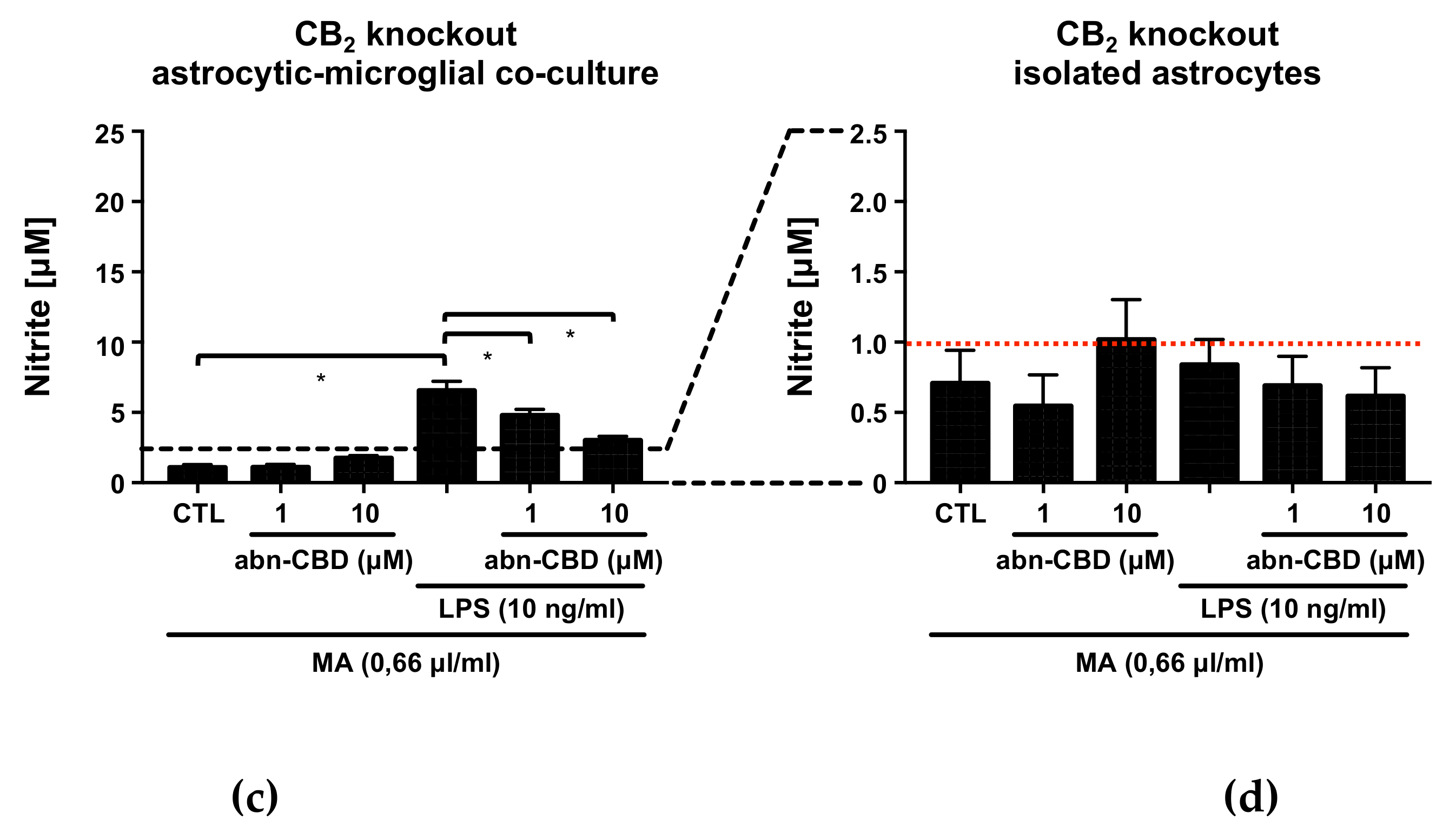
2.2. Abn-CBD Reduces LPS-Induced Production of NO in Astrocytic-Microglial Cocultures
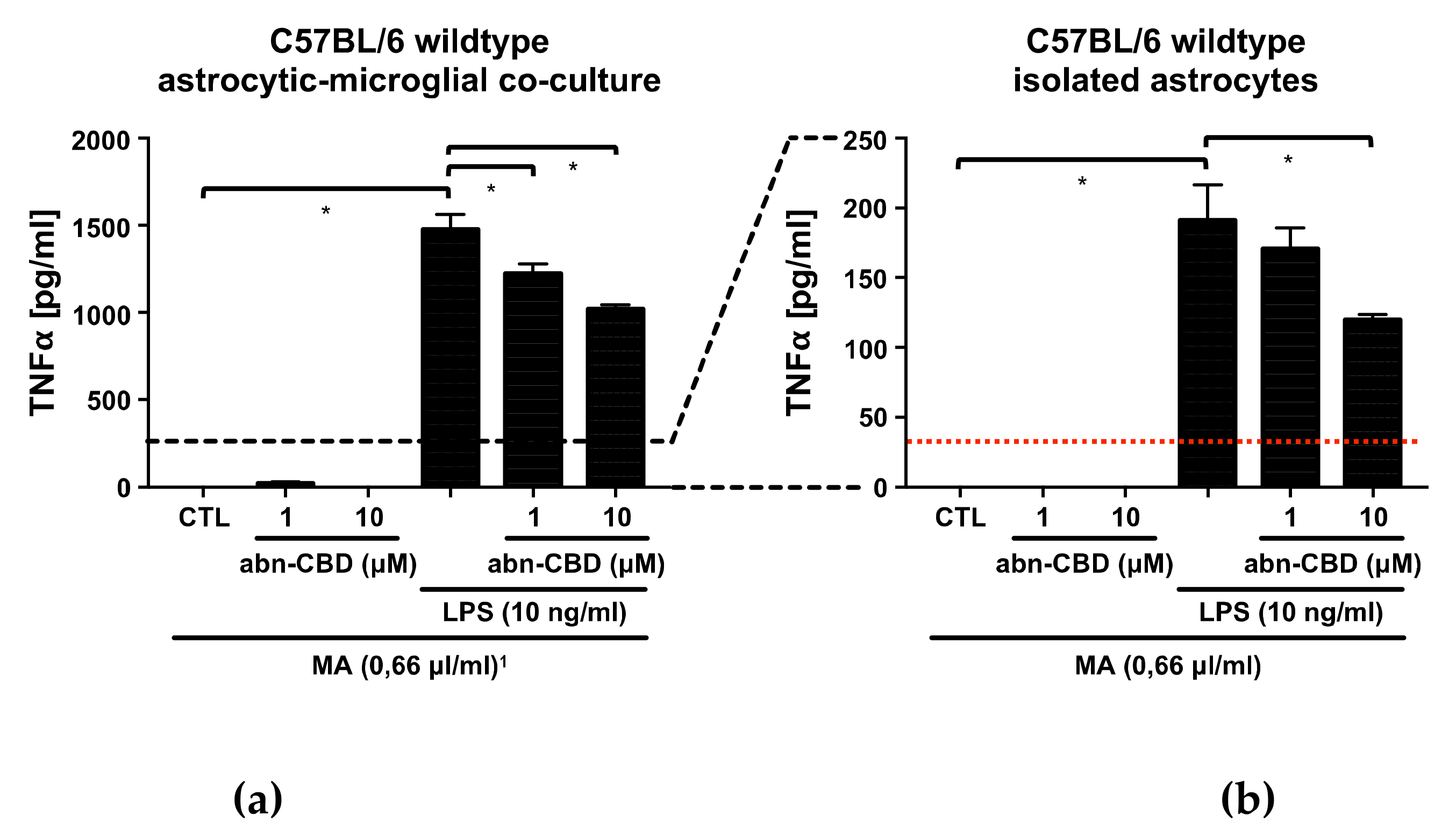
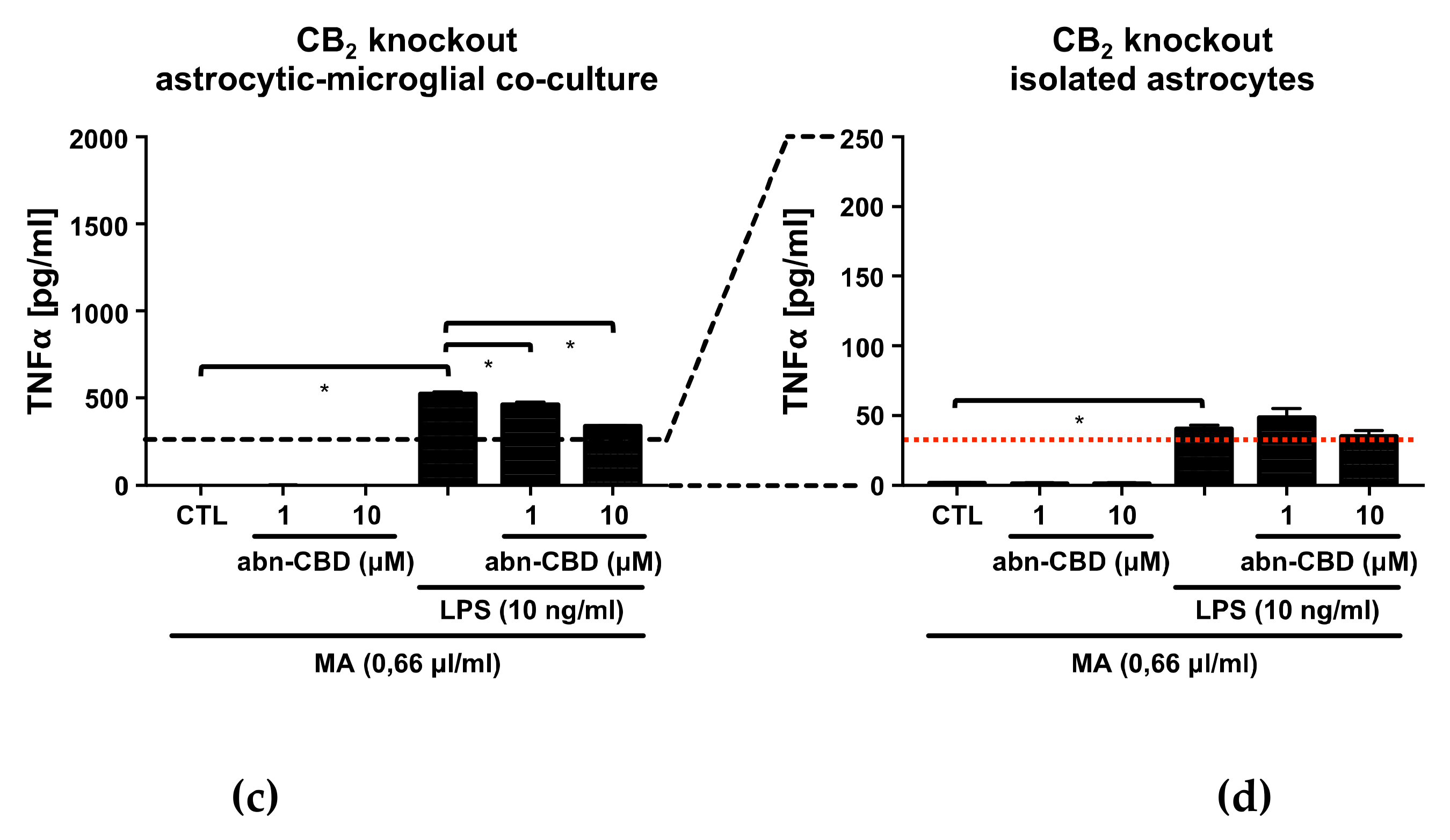
2.3. Abn-CBD Reduces LPS-Induced Production of TNFα in Astrocytic-Microglial Cocultures and Isolated Astrocytic Cultures
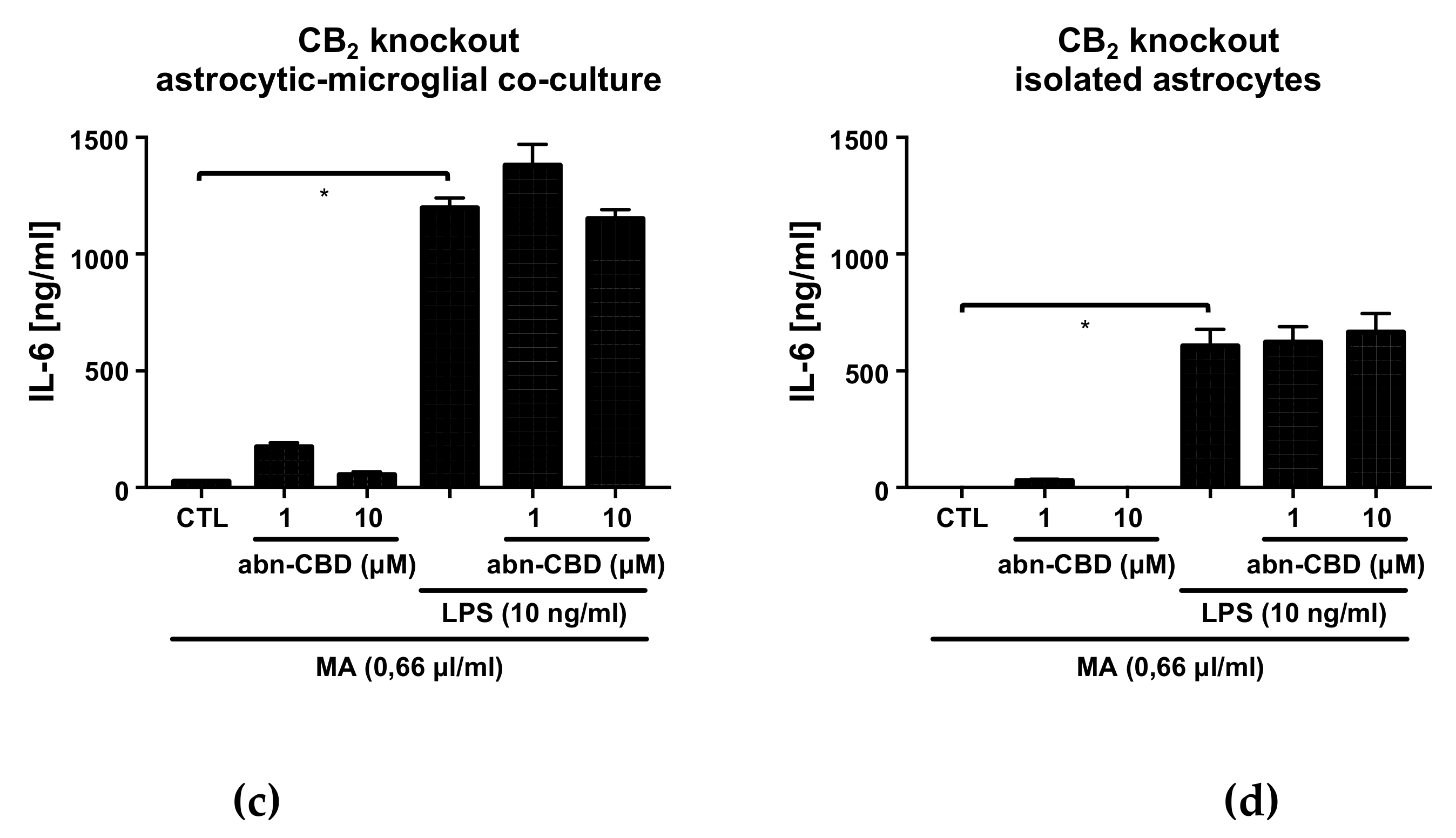
2.4. Abn-CBD has No Effect on LPS-Induced IL-6 Production in Astrocytic-Microglial Cocultures and Isolated Astrocytic Cultures
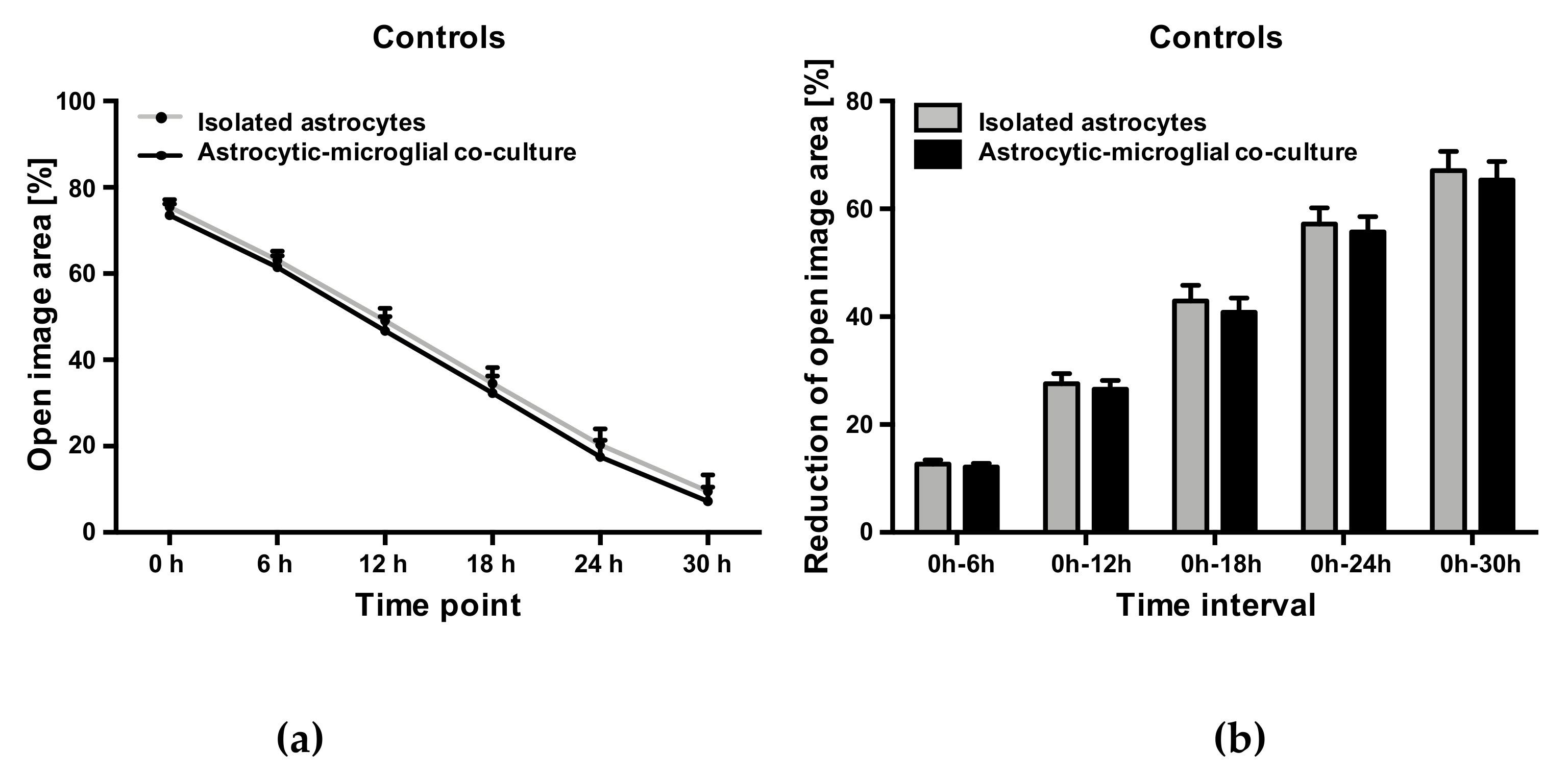
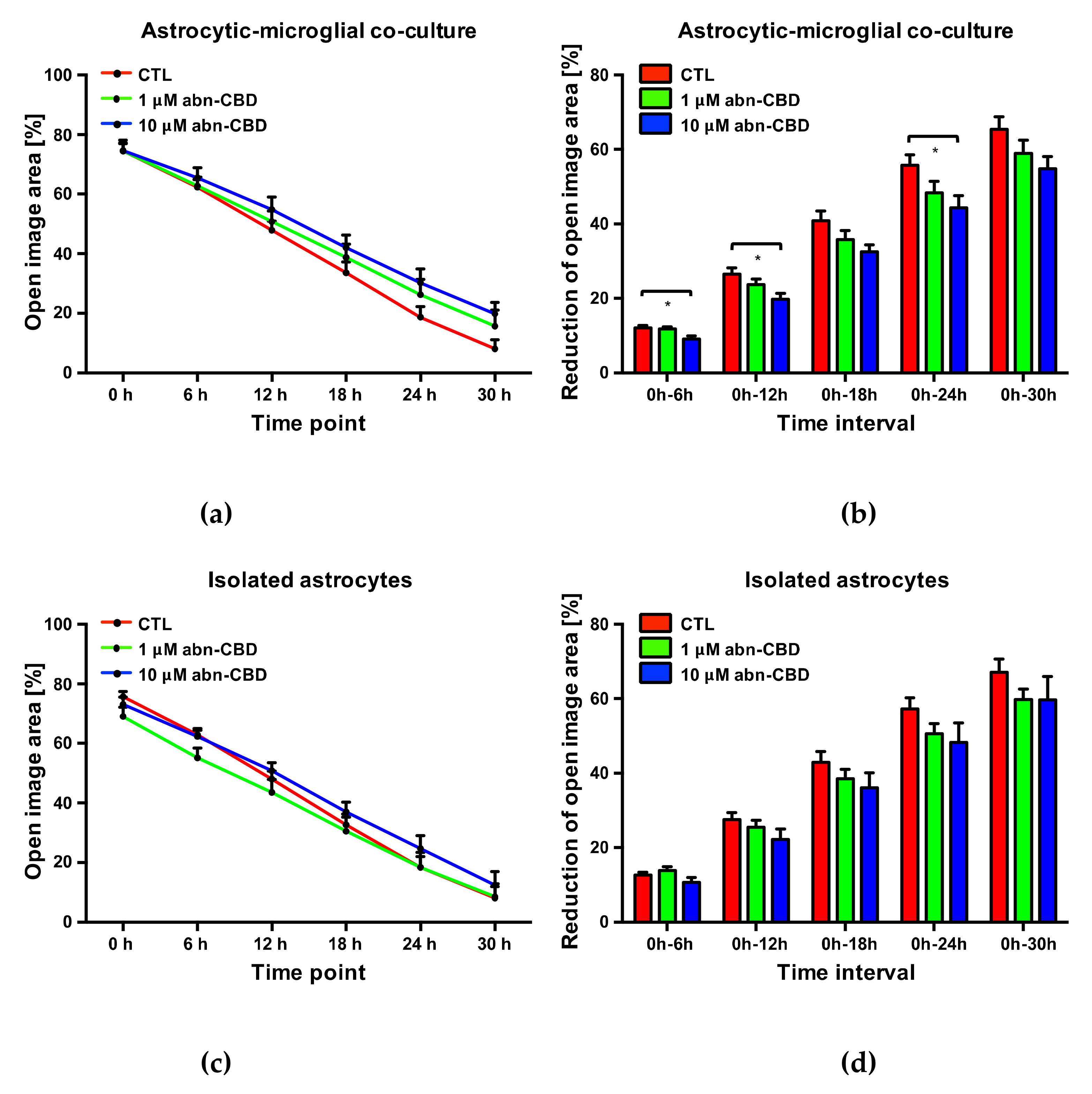
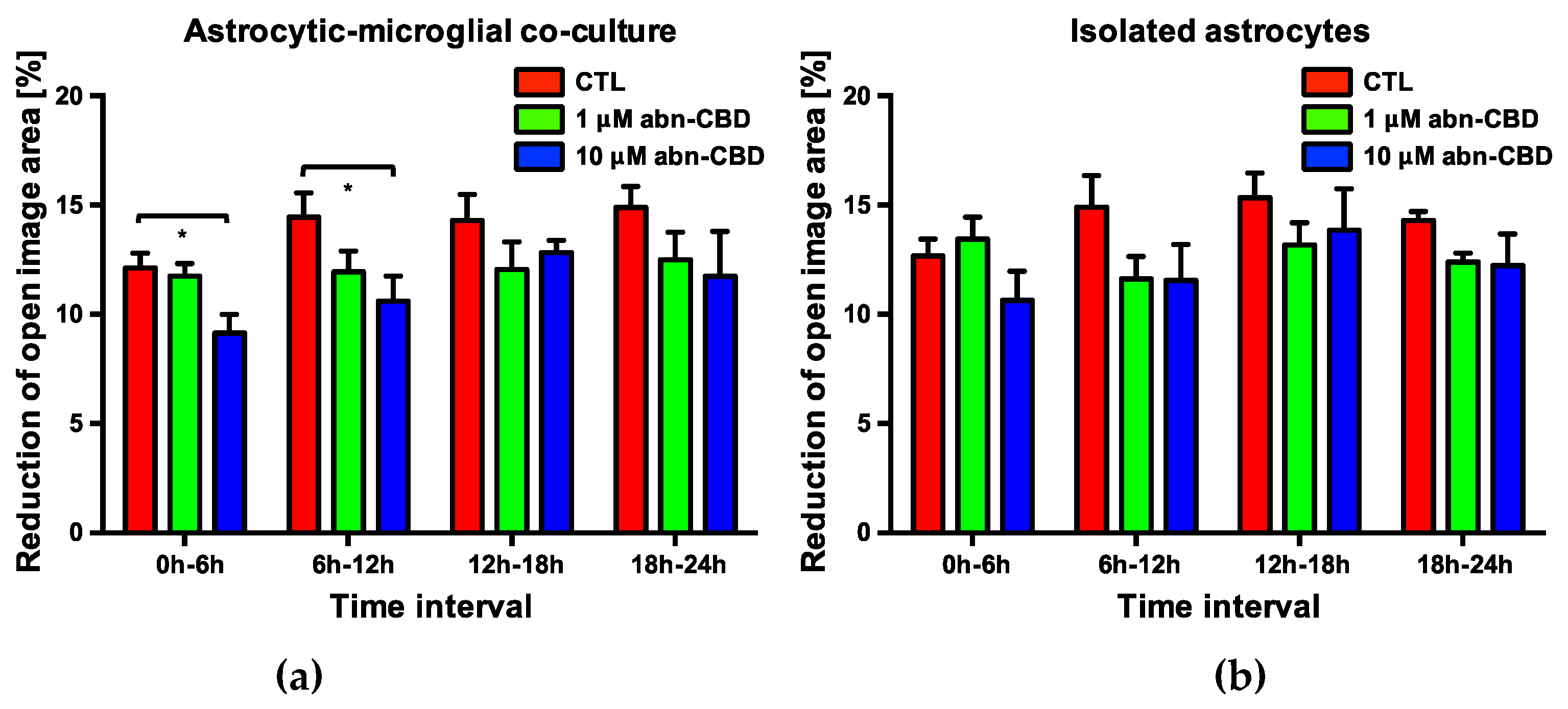
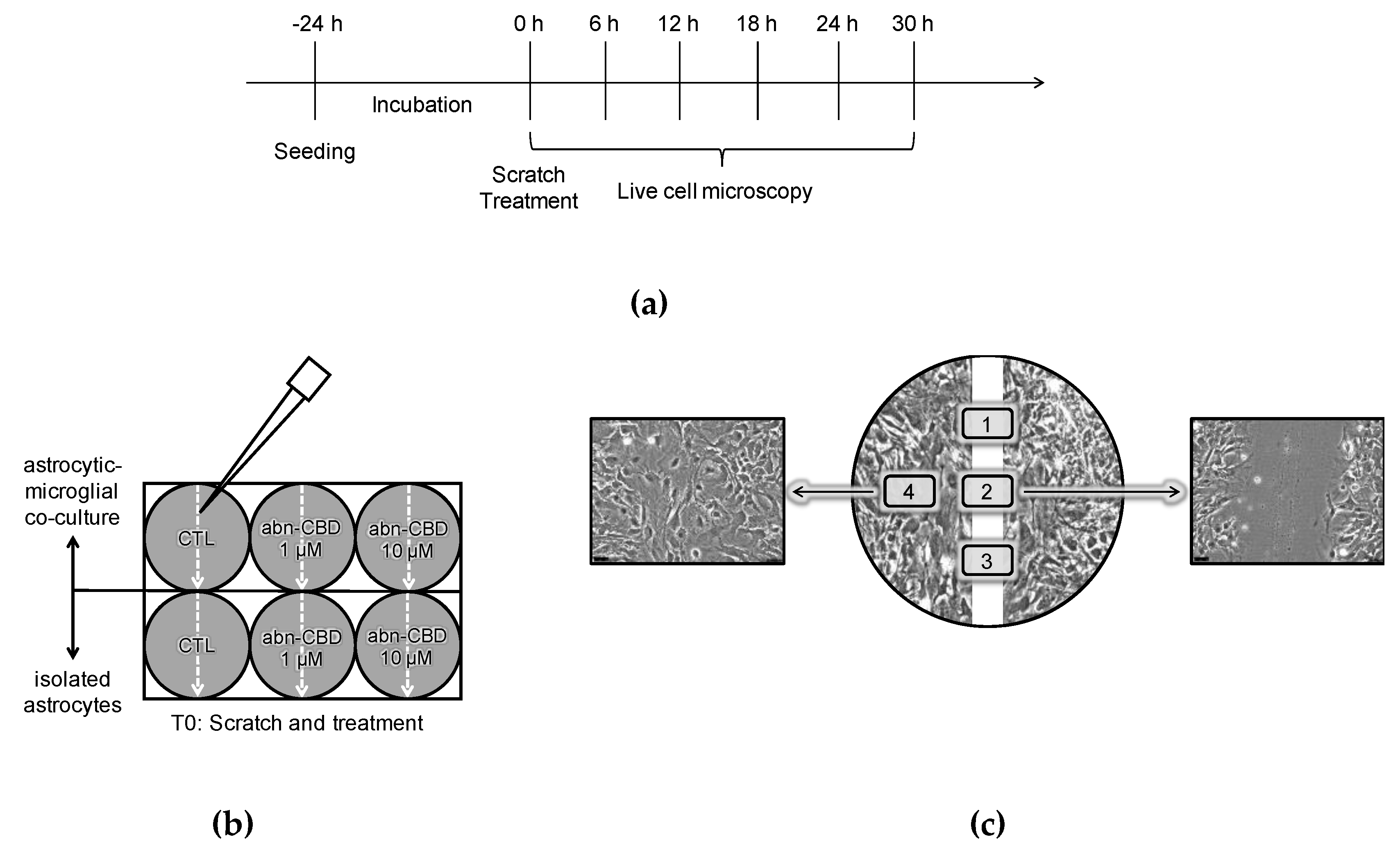
2.5. Abn-CBD Delays Astrocyte Wound Closure in a Microglia-Dependent Manner
3. Discussion
4. Materials and Methods
4.1. Preparation and Generation of Astrocytic-Microglial Cocultures and Isolated Astrocytic Cultures
4.2. Cytokine and Nitrite Measurement
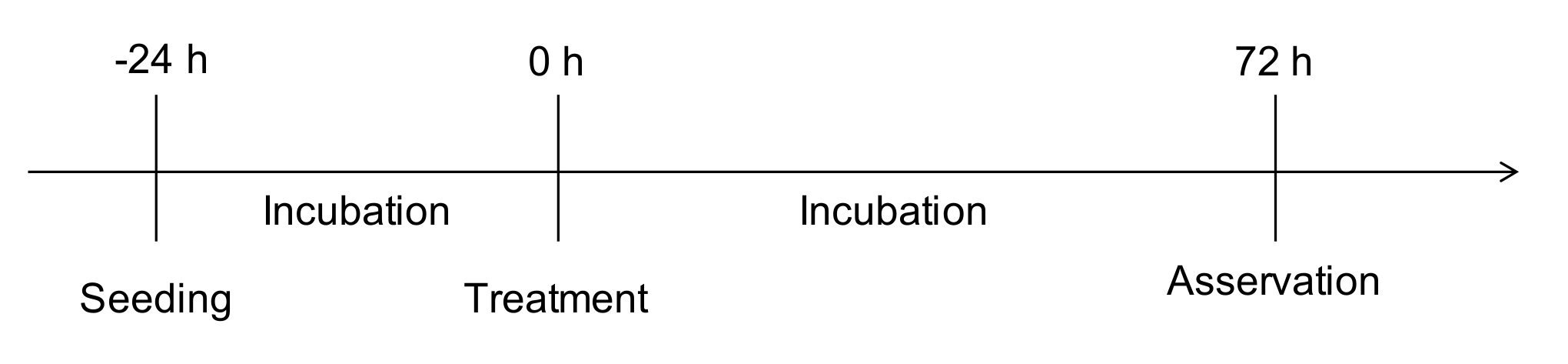
4.3. Scratch-Wound Assay
4.4. Quantification of the Results and Statistical Analysis
4.5. Fluorescence Immunocytochemistry and Confocal Laser Scanning Micoscropy
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dutton, R.P.; McCunn, M. Traumatic brain injury. Curr. Opin. Crit. Care 2003, 9, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, D.; Patil, C.G. Epidemiology and the Global Burden of Stroke. World Neurosurg. 2011, 76, S85–S90. [Google Scholar] [CrossRef] [PubMed]
- McGarry, L.J.; Thompson, D.; Millham, F.H.; Cowell, L.; Snyder, P.J.; Lenderking, W.R.; Weinstein, M.C. Outcomes and costs of acute treatment of traumatic brain injury. J. Trauma 2002, 53, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Coronado, V.G.; Xu, L.; Basavaraju, S.V.; McGuire, L.C.; Wald, M.M.; Faul, M.D.; Guzman, B.R.; Hemphill, J.D. Surveillance for traumatic brain injury-related deaths—United States, 1997–2007. Morb. Mortal. Wkly. Rep. 2011, 60, 1–36. [Google Scholar]
- Prabhakaran, S.; Ruff, I.; Bernstein, R.A. Acute Stroke Intervention: A systematic review. JAMA 2015, 313, 1451–1462. [Google Scholar] [CrossRef]
- Kunz, A.; Dirnagl, U.; Mergenthaler, P. Acute pathophysiological processes after ischaemic and traumatic brain injury. Best Pract. Res. Clin. Anaesthesiol. 2010, 24, 495–509. [Google Scholar] [CrossRef]
- Dirnagl, U. Pathobiology of injury after stroke: The neurovascular unit and beyond. Ann. N. Y. Acad. Sci. 2012, 1268, 21–25. [Google Scholar] [CrossRef]
- Arundine, M.; Tymianski, M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell. Mol. Life Sci. 2004, 61, 657–668. [Google Scholar] [CrossRef]
- Werner, C.; Engelhard, K. Pathophysiology of traumatic brain injury. Br. J. Anaesth. 2007, 99, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Pekny, M.; Pekna, M. Astrocyte Reactivity and Reactive Astrogliosis: Costs and Benefits. Physiol. Rev. 2014, 94, 1077–1098. [Google Scholar] [CrossRef]
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial Activation in Traumatic Brain Injury. Front. Aging Neurosci. 2017, 9, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, S.A.; Boddeke, H.W.G.M.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S.T. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflammation 2004, 1, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boje, K.M.; Arora, P.K. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992, 587, 250–256. [Google Scholar] [CrossRef]
- Dawson, D.A.; Martin, D.; Hallenbeck, J.M. Inhibition of tumor necrosis factor-alpha reduces focal cerebral ischemic injury in the spontaneously hypertensive rat. Neurosci. Lett. 1996, 218, 41–44. [Google Scholar] [CrossRef]
- Barone, F.C.; Arvin, B.; White, R.F.; Miller, A.; Webb, C.L.; Willette, R.N.; Lysko, P.G.; Feuerstein, G.Z. Tumor necrosis factor-α: A mediator of focal ischemic brain injury. Stroke 1997, 28, 1233–1244. [Google Scholar] [CrossRef]
- Zhao, W.; Xie, W.; Le, W.; Beers, D.R.; He, Y.; Henkel, J.S.; Simpson, E.P.; Yen, A.A.; Xiao, Q.; Appel, S.H. Activated microglia initiate motor neuron injury by a nitric oxide and glutamate-mediated mechanism. J. Neuropathol. Exp. Neurol. 2004, 63, 964–977. [Google Scholar] [CrossRef] [Green Version]
- Erta, M.; Quintana, A.; Hidalgo, J. Interleukin-6, a major cytokine in the central nervous system. Int. J. Biol. Sci. 2012, 8, 1254–1266. [Google Scholar] [CrossRef]
- Abd-El-Basse, E.M. Pro-inflammatory cytokine; tumor-necrosis factor-alpha (TNF-α) inhibits astrocytic support of neuronal survival and neurites outgrowth. Adv. Biosci. Biotechnol. 2013, 4, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M. V Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Eng, L.F.; Ghirnikar, R.S. GFAP and Astrogliosis. In Brain Pathology; Markus, G., Ed.; Wily: New York, NY, USA, 1994; pp. 229–237. [Google Scholar]
- Iannotti, F.A.; Di Marzo, V.; Petrosino, S. Endocannabinoids and endocannabinoid-related mediators: Targets, metabolism and role in neurological disorders. Prog. Lipid Res. 2016, 62, 107–128. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C.; Barth, F.; Bonner, T.I.; Cabral, G.; Casellas, P.; Devane, W.A.; Felder, C.C.; Herkenham, M.; Mackie, K.; Martin, B.R.; et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Felder, C.C.; Joyce, K.E.; Briley, E.M.; Mansouri, J.; Mackie, K.; Blond, O.; Lai, Y.; Ma, A.L.; Mitchell, R.L. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol. Pharmacol. 1995, 48, 443–450. [Google Scholar]
- Maccarrone, M.; Bab, I.; Bíró, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef] [Green Version]
- Stephen, P.H. The Endocannabinoidome: The World of Endocannabinoids and Related Mediators; Di Marzo, V., Wang, J.W., Eds.; Elsevier: London, UK, 2014; pp. 153–175. [Google Scholar]
- Chiurchiù, V. Endocannabinoids and Immunity. Cannabis Cannabinoid Res. 2016, 1, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Chiurchiu, V.; Battistini, L.; Maccarrone, M.; Chiurchiù, V.; Battistini, L.; Maccarrone, M. Endocannabinoid signalling in innate and adaptive immunity. Immunology 2015, 144, 352–364. [Google Scholar] [CrossRef]
- Oláh, A.; Szekanecz, Z.; Bíró, T. Targeting cannabinoid signaling in the immune system: “High“-ly exciting questions, possibilities, and challenges. Front. Immunol. 2017, 8, 1487. [Google Scholar] [CrossRef] [Green Version]
- Panikashvili, D.; Simeonidou, C.; Ben-Shabat, S.; Hanuš, L.; Breuer, A.; Mechoulam, R.; Shohami, E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 2001, 413, 527–531. [Google Scholar] [CrossRef]
- Marsicano, G.; Goodenough, S.; Monory, K.; Hermann, H.; Eder, M.; Cannich, A.; Azad, S.C.; Cascio, M.G.; Ortega-Gutiérrez, S.; Van der Stelt, M.; et al. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 2003, 302, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Kallendrusch, S.; Hobusch, C.; Ehrlich, A.; Nowicki, M.; Ziebell, S.; Bechmann, I.; Geisslinger, G.; Koch, M.; Dehghani, F. Intrinsic up-regulation of 2-AG favors an area specific neuronal survival in different in vitro models of neuronal damage. PLoS ONE 2012, 7, e51208. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.I.; Nicoll, R.A. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 2001, 410, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Schlicker, E.; Kathmann, M. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol. Sci. 2001, 22, 565–572. [Google Scholar] [CrossRef]
- Katona, I.; Urbán, G.M.; Wallace, M.; Ledent, C.; Jung, K.M.; Piomelli, D.; Mackie, K.; Freund, T.F. Molecular Composition of the Endocannabinoid System at Glutamatergic Synapses. J. Neurosci. 2006, 26, 5628–5637. [Google Scholar] [CrossRef] [PubMed]
- Zarruk, J.G.; Fernández-López, D.; García-Yébenes, I.; García-Gutiérrez, M.S.; Vivancos, J.; Nombela, F.; Torres, M.; Burguete, M.C.; Manzanares, J.; Lizasoain, I.; et al. Cannabinoid type 2 receptor activation downregulates stroke-induced classic and alternative brain macrophage/microglial activation concomitant to neuroprotection. Stroke 2012, 43, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Amenta, P.S.; Jallo, J.I.; Tuma, R.F.; Craig Hooper, D.; Elliott, M.B. Cannabinoid receptor type-2 stimulation, blockade, and deletion alter the vascular inflammatory responses to traumatic brain injury. J. Neuroinflammation 2014, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.A.; Varga, K.; Jaárai, Z.; Kunos, G.; Jarai, Z.; Kunos, G. Mesenteric Vasodilation Mediated by Endothelial Anandamide Receptors. Hypertension 1999, 33, 429–434. [Google Scholar] [CrossRef] [Green Version]
- Járai, Z.; Wagner, J.A.; Varga, K.; Lake, K.D.; Compton, D.R.; Martin, B.R.; Zimmer, A.M.; Bonner, T.I.; Buckley, N.E.; Mezey, E.; et al. Cannabinoid-induced mesenteric vasodilation through an endothelial site distinct from CB1 or CB2 receptors. Proc. Natl. Acad. Sci. USA 1999, 96, 14136–14141. [Google Scholar] [CrossRef] [Green Version]
- Begg, M.; Pacher, P.; Bátkai, S.; Osei-Hyiaman, D.; Offertáler, L.; Fong, M.M.; Liu, J.; Kunos, G. Evidence for novel cannabinoid receptors. Pharmacol. Ther. 2005, 106, 133–145. [Google Scholar] [CrossRef]
- Offertáler, L.; Mo, F.M.; Bátkai, S.; Liu, J.; Begg, M.; Razdan, R.K.; Martin, B.R.; Bukoski, R.D.; Kunos, G. Selective ligands and cellular effectors of a G protein-coupled endothelial cannabinoid receptor. Mol. Pharmacol. 2003, 63, 699–705. [Google Scholar] [CrossRef] [Green Version]
- Mackie, K.; Stella, N. Cannabinoid Receptors and Endocannabinoids: Evidence for New Players. AAPS J. 2006, 8, E298–E306. [Google Scholar] [CrossRef] [PubMed]
- Walter, L.; Franklin, A.; Witting, A.; Wade, C.; Xie, Y.; Kunos, G.; Mackie, K.; Stella, N. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J. Neurosci. 2003, 23, 1398–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, A.; Stella, N. Arachidonylcyclopropylamide increases microglial cell migration through cannabinoid CB2 and abnormal-cannabidiol-sensitive receptors. Eur. J. Pharmacol. 2003, 474, 195–198. [Google Scholar] [CrossRef]
- Baker, D.; Pryce, G.; Davies, W.L.; Hiley, C.R. In silico patent searching reveals a new cannabinoid receptor. Trends Pharmacol. Sci. 2006, 27, 1–4. [Google Scholar] [CrossRef]
- Lauckner, J.E.; Jensen, J.B.; Chen, H.-Y.; Lu, H.-C.; Hille, B.; Mackie, K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHugh, D.; Hu, S.S.; Rimmerman, N.; Juknat, A.; Vogel, Z.; Walker, J.M.; Bradshaw, H.B. N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci. 2010, 11, 44. [Google Scholar] [CrossRef] [Green Version]
- McHugh, D. GPR18 in microglia: Implications for the CNS and endocannabinoid system signalling. Br. J. Pharmacol. 2012, 167, 1575–1582. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Chu, A.; Li, W.; Wang, B.; Shelton, F.; Otero, F.; Nguyen, D.G.; Caldwell, J.S.; Chen, Y.A. Lipid G Protein-coupled Receptor Ligand Identification Using β-Arrestin PathHunterTM Assay. J. Biol. Chem. 2009, 284, 12328–12338. [Google Scholar] [CrossRef] [Green Version]
- Finlay, D.B.; Joseph, W.R.; Grimsey, N.L.; Glass, M. GPR18 undergoes a high degree of constitutive trafficking but is unresponsive to N-Arachidonoyl Glycine. PeerJ. 2016, 4, e1835. [Google Scholar] [CrossRef] [Green Version]
- Console-Bram, L.; Brailoiu, E.; Brailoiu, G.C.; Sharir, H.; Abood, M.E. Activation of GPR18 by cannabinoid compounds: A tale of biased agonism. Br. J. Pharmacol. 2014, 171, 3908–3917. [Google Scholar] [CrossRef] [Green Version]
- Kreutz, S.; Koch, M.; Böttger, C.; Ghadban, C.; Korf, H.W.; Dehghani, F. 2-Arachidonoylglycerol elicits neuroprotective effects on excitotoxically lesioned dentate gyrus granule cells via abnormal-cannabidiol-sensitive receptors on microglial cells. Glia 2009, 57, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Kreutz, S.; Koch, M.; Ghadban, C.; Korf, H.W.; Dehghani, F. Cannabinoids and neuronal damage: Differential effects of THC, AEA and 2-AG on activated microglial cells and degenerating neurons in excitotoxically lesioned rat organotypic hippocampal slice cultures. Exp. Neurol. 2007, 203, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Mishima, K.; Hayakawa, K.; Abe, K.; Ikeda, T.; Egashira, N.; Iwasaki, K.; Fujiwara, M. Cannabidiol Prevents Cerebral Infarction Via a Serotonergic 5-Hydroxytryptamine1A Receptor-Dependent Mechanism. Stroke 2005, 36, 1071–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, L.C.; Wagner, D.A.; Glogowski, J.; Skipper, P.L.; Wishnok, J.S.; Tannenbaum, S.R. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal. Biochem. 1982, 126, 131–138. [Google Scholar] [CrossRef]
- Kelm, M. Nitric oxide metabolism and breakdown. Biochim. Biophys. Acta Bioenerg. 1999, 1411, 273–289. [Google Scholar] [CrossRef] [Green Version]
- Mikkelsen, R.B.; Wardman, P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene 2003, 22, 5734–5754. [Google Scholar] [CrossRef] [Green Version]
- Shohami, E.; Cohen-Yeshurun, A.; Magid, L.; Algali, M.; Mechoulam, R. Endocannabinoids and traumatic brain injury. Br. J. Pharmacol 2011, 163, 1402–1410. [Google Scholar] [CrossRef] [Green Version]
- Puffenbarger, R.A.; Boothe, A.C.; Cabral, G.A. Cannabinoids inhibit LPS-inducible cytokine mRNA expression in rat microglial cells. Glia 2000, 29, 58–69. [Google Scholar] [CrossRef]
- Facchinetti, F.; Del Giudice, E.; Furegato, S.; Passarotto, M.; Leon, A. Cannabinoids ablate release of TNFα in rat microglial cells stimulated with lypopolysaccharide. Glia 2003, 41, 161–168. [Google Scholar] [CrossRef]
- Eljaschewitsch, E.; Witting, A.; Mawrin, C.; Lee, T.; Schmidt, P.M.; Wolf, S.; Hoertnagl, H.; Raine, C.S.; Schneider-Stock, R.; Nitsch, R.; et al. The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron 2006, 49, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Martin-Moreno, A.M.; Reigada, D.; Ramirez, B.G.; Mechoulam, R.; Innamorato, N.; Cuadrado, A.; de Ceballos, M.L. Cannabidiol and Other Cannabinoids Reduce Microglial Activation In Vitro and In Vivo: Relevance to Alzheimer’s Disease. Mol. Pharmacol. 2011, 79, 964–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janefjord, E.; Mååg, J.L.V.; Harvey, B.S.; Smid, S.D. Cannabinoid effects on β amyloid fibril and aggregate formation, neuronal and microglial-activated neurotoxicity in vitro. Cell. Mol. Neurobiol. 2014, 34, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Bsibsi, M.; Ravid, R.; Gveric, D.; van Noort, J.M. Broad expression of Toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 1013–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettenmann, H.; Hanisch, U.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Olson, J.K.; Miller, S.D. Microglia Initiate Central Nervous System Innate and Adaptive Immune Responses through Multiple TLRs. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef] [Green Version]
- Pålsson-McDermott, E.M.; O’Neill, L.A.J. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology 2004, 113, 153–162. [Google Scholar] [CrossRef]
- Rosenberger, K.; Dembny, P.; Derkow, K.; Engel, O.; Krüger, C.; Wolf, S.A.; Kettenmann, H.; Schott, E.; Meisel, A.; Lehnardt, S. Intrathecal heat shock protein 60 mediates neurodegeneration and demyelination in the CNS through a TLR4- and MyD88-dependent pathway. Mol. Neurodegener. 2015, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Lehnardt, S. Innate immunity and neuroinflammation in the CNS: The role of microglia in toll-like receptor-mediated neuronal injury. Glia 2010, 58, 253–263. [Google Scholar] [CrossRef]
- Yang, Q.W.; Xiang, J.; Zhou, Y.; Zhong, Q.; Li, J.C. Targeting HMGB1/TLR4 signaling as a novel approach to treatment of cerebral ischemia. Front. Biosci. 2010, 2, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Solà, C.; Casal, C.; Tusell, J.M.; Serratosa, J. Astrocytes enhance lipopolysaccharide-induced nitric oxide production by microglial cells. Eur. J. Neurosci. 2002, 16, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Ledeboer, A.; Brevé, J.J.P.; Poole, S.; Tilders, F.J.H.; Van Dam, A.M. Interleukin-10, interleukin-4, and transforming growth factor-β differentially regulate lipopolysaccharide-induced production of pro-inflammatory cytokines and nitric oxide in co-cultures of rat astroglial and microglial cells. Glia 2000, 30, 134–142. [Google Scholar] [CrossRef]
- Saura, J. Microglial cells in astroglial cultures: A cautionary note. J. Neuroinflammation 2007, 4, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehghani, F.; Conrad, A.; Kohl, A.; Korf, H.W.; Hailer, N.P. Clodronate inhibits the secretion of proinflammatory cytokines and NO by isolated microglial cells and reduces the number of proliferating glial cells in excitotoxically injured organotypic hippocampal slice cultures. Exp. Neurol. 2004, 189, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Showalter, V.M.; Compton, D.R.; Martin, B.R.; Abood, M.E. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): Identification of cannabinoid receptor subtype selective ligands. J. Pharmacol. Exp. Ther. 1996, 278, 969–999. [Google Scholar]
- Begg, M.; Mo, F.M.; Offertaler, L.; Bátkai, S.; Pacher, P.; Razdan, R.K.; Lovinger, D.M.; Kunos, G. G protein-coupled endothelial receptor for atypical cannabinoid ligands modulates a Ca2+-dependent K+ current. J. Biol. Chem. 2003, 278, 46188–46194. [Google Scholar] [CrossRef] [Green Version]
- Chao, C.C.; Hu, S.; Molitor, T.W.; Shaskan, E.G.; Peterson, P.K. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J. Immunol. 1992, 149, 2736–2741. [Google Scholar]
- Brown, G.C. Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem. Soc. Trans. 2007, 35, 1119–1121. [Google Scholar] [CrossRef] [Green Version]
- Sawada, M.; Suzumura, A.; Marunouchi, T. TNFα induces IL-6 production by astrocytes but not by microglia. Brain Res. 1992, 583, 296–299. [Google Scholar] [CrossRef]
- Neumann, H.; Schweigreiter, R.; Yamashita, T.; Rosenkranz, K.; Wekerle, H.; Barde, Y.A. Tumor necrosis factor inhibits neurite outgrowth and branching of hippocampal neurons by a rho-dependent mechanism. J. Neurosci. 2002, 22, 854–862. [Google Scholar] [CrossRef]
- Medina, S.; Martínez, M.; Hernanz, A. Antioxidants Inhibit the Human Cortical Neuron Apoptosis Induced by Hydrogen Peroxide, Tumor Necrosis Factor Alpha, Dopamine and Beta-amyloid Peptide 1-42. Free Radic. Res. 2002, 36, 1179–1184. [Google Scholar] [CrossRef] [PubMed]
- Milman, G.; Maor, Y.; Abu-Lafi, S.; Horowitz, M.; Gallily, R.; Batkai, S.; Mo, F.-M.; Offertaler, L.; Pacher, P.; Kunos, G.; et al. N-arachidonoyl L-serine, an endocannabinoid-like brain constituent with vasodilatory properties. Proc. Natl. Acad. Sci. USA 2006, 103, 2428–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Núñez, E.; Benito, C.; Pazos, M.R.; Barbachano, A.; Fajardo, O.; González, S.; Tolón, R.M.; Romero, J. Cannabinoid CB 2 receptors are expressed by perivascular microglial cells in the human brain: An immunohistochemical study. Synapse 2004, 53, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Ashton, J.C.; Glass, M. The cannabinoid CB2 receptor as a target for inflammation-dependent neurodegeneration. Curr. Neuropharmacol. 2007, 5, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Carlisle, S.J.; Marciano-Cabral, F.; Staab, A.; Ludwick, C.; Cabral, G.A. Differential expression of the CB2 cannabinoid receptor by rodent macrophages and macrophage-like cells in relation to cell activation. Int. Immunopharmacol. 2002, 2, 69–82. [Google Scholar] [CrossRef]
- Maresz, K.; Carrier, E.J.; Ponomarev, E.D.; Hillard, C.J.; Dittel, B.N. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J. Neurochem. 2005, 95, 437–445. [Google Scholar] [CrossRef]
- Cabral, G.A.; Marciano-Cabral, F. Cannabinoid receptors in microglia of the central nervous system: Immune functional relevance. J. Leukoc. Biol. 2005, 78, 1192–1197. [Google Scholar] [CrossRef]
- Alferink, J.; Specht, S.; Arends, H.; Schumak, B.; Schmidt, K.; Ruland, C.; Lundt, R.; Kemter, A.; Dlugos, A.; Kuepper, J.M.; et al. Cannabinoid receptor 2 modulates susceptibility to experimental cerebral malaria through a CCL17-dependent Mechanism. J. Biol. Chem. 2016, 291, 19517–19531. [Google Scholar] [CrossRef] [Green Version]
- Sawada, M.; Kondo, N.; Suzumura, A.; Marunouchi, T. Production of tumor necrosis factor-alpha by microglia and astrocytes in culture. Brain Res. 1989, 491, 394–397. [Google Scholar] [CrossRef]
- Lin, J.H.C.; Weigel, H.; Cotrina, M.L.; Liu, S.; Bueno, E.; Hansen, A.J.; Hansen, T.W.; Goldman, S.; Nedergaard, M. Gap-junction-mediated propagation and amplification of cell injury. Nat. Neurosci. 1998, 1, 494–500. [Google Scholar] [CrossRef]
- Ebrahimi, F.; Koch, M.; Pieroh, P.; Ghadban, C.; Hobusch, C.; Bechmann, I.; Dehghani, F. Time dependent neuroprotection of mycophenolate mofetil: Effects on temporal dynamics in glial proliferation, apoptosis, and scar formation. J. Neuroinflammation 2012, 9, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinozaki, Y.; Shibata, K.; Yoshida, K.; Shigetomi, E.; Gachet, C.; Ikenaka, K.; Tanaka, K.F.; Koizumi, S. Transformation of Astrocytes to a Neuroprotective Phenotype by Microglia via P2Y1 Receptor Downregulation. Cell Rep. 2017, 19, 1151–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etienne-Manneville, S. In vitro assay of primary astrocyte migration as a tool to study Rho GTPase function in cell polarization. Methods Enzymol. 2006, 406, 565–578. [Google Scholar] [PubMed]
- Etienne-Manneville, S.; Hall, A. Integrin-Mediated Activation of Cdc42 Controls Cell Polarity in Migrating Astrocytes through PKCζ. Cell 2001, 106, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Yao, F.; Li, K.; Zhang, L.; Yin, G.; Du, M.; Wu, B. Fisetin regulates astrocyte migration and proliferation in vitro. Int. J. Mol. Med. 2017, 39, 783–790. [Google Scholar] [CrossRef] [Green Version]
- Adams, M.D.; Earnhardt, J.T.; Martin, B.R.; Harris, L.S.; Dewey, W.L.; Razdan, R.K. A cannabinoid with cardiovascular activity but no overt behavioral effects. Experientia 1977, 33, 1204–1205. [Google Scholar] [CrossRef]
- Buckley, N.E.; McCoy, K.L.; Mezey, E.; Bonner, T.; Zimmer, A.; Felder, C.C.; Glass, M.; Zimmer, A. Immunomodulation by cannabinoids is absent in mice deficient for the cannabinoid CB(2) receptor. Eur. J. Pharmacol. 2000, 396, 141–149. [Google Scholar] [CrossRef]
- Buckley, N.E. The peripheral cannabinoid receptor knockout mice: An update. Br. J. Pharmacol. 2008, 153, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Kumamaru, H.; Saiwai, H.; Kobayakawa, K.; Kubota, K.; van Rooijen, N.; Inoue, K.; Iwamoto, Y.; Okada, S. Liposomal clodronate selectively eliminates microglia from primary astrocyte cultures. J. Neuroinflammation 2012, 9, 116. [Google Scholar] [CrossRef] [Green Version]
- Kohl, A.; Dehghani, F.; Korf, H.W.; Hailer, N.P. The bisphosphonate clodronate depletes microglial cells in excitotoxically injured organotypic hippocampal slice cultures. Exp. Neurol. 2003, 181, 1–11. [Google Scholar] [CrossRef]
- Mönkkönen, H.; Rogers, M.J.; Makkonen, N.; Niva, S.; Auriola, S.; Mönkkönen, J. The cellular uptake and metabolism of clodronate in RAW 264 macrophages. Pharm. Res. 2001, 18, 1550–1555. [Google Scholar] [CrossRef] [PubMed]
- Frith, J.C.; Mönkkönen, J.; Auriola, S.; Mönkkönen, H.; Rogers, M.J. The Molecular Mechanism of Action of the Antiresorptive and Antiinflammatory Drug Clodronate: Evidence for the Formation in Vivo of a Metabolite That Inhibits Bone Resorption and Causes Osteoclast and Macrophage Apoptosis. Arthritis Rheum. 2001, 44, 2201–2210. [Google Scholar] [CrossRef]
- Engvall, E.; Perlmann, P. Enzyme-linked immunosorbent assay (ELISA) quantitative assay of immunoglobulin G. Immunochemistry 1971, 8, 871–874. [Google Scholar] [CrossRef]
- Gebäck, T.; Schulz, M.M.P.; Koumoutsakos, P.; Detmar, M. TScratch: A novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques 2009, 46, 265–274. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Substances |
|---|---|
| 1 | CTL 0.66 µL/mL methylacetate (MA) |
| 2 | Abn-CBD 1 µM + 0.6 µL/mL MA |
| 3 | Abn-CBD 10 µM including 0.66 µL/mL MA |
| 4 | LPS 10 ng/mL + 0.66 µL/mL MA |
| 5 | LPS 10 ng/mL + abn-CBD 1 µM + 0.6 µL/mL MA |
| 6 | LPS 10 ng/mL + abn-CBD 10 µM including 0.66 µL/mL MA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardinal von Widdern, J.; Hohmann, T.; Dehghani, F. Abnormal Cannabidiol Affects Production of Pro-Inflammatory Mediators and Astrocyte Wound Closure in Primary Astrocytic-Microglial Cocultures. Molecules 2020, 25, 496. https://doi.org/10.3390/molecules25030496
Cardinal von Widdern J, Hohmann T, Dehghani F. Abnormal Cannabidiol Affects Production of Pro-Inflammatory Mediators and Astrocyte Wound Closure in Primary Astrocytic-Microglial Cocultures. Molecules. 2020; 25(3):496. https://doi.org/10.3390/molecules25030496
Chicago/Turabian StyleCardinal von Widdern, Julian, Tim Hohmann, and Faramarz Dehghani. 2020. "Abnormal Cannabidiol Affects Production of Pro-Inflammatory Mediators and Astrocyte Wound Closure in Primary Astrocytic-Microglial Cocultures" Molecules 25, no. 3: 496. https://doi.org/10.3390/molecules25030496