
Abstract
The T-box4 (TBX4) gene (OMIM *601719) belongs to the T-box family of transcription regulators that share a conserved homology domain and are expressed at specific sites during various stages of embryonic development. Tbx4 has been found to be a crucial transcriptional regulator in embryonic hindlimb development in animal models. Monoallelic variants in the TBX4 gene are reported to be associated with skeletal defects of the pelvis and lower limbs. We report here a fetus with a novel multiple malformation syndrome associated with sacrococcygeal agenesis, bilateral lower limb aplasia, hypoplastic left heart, bilateral lung hypoplasia, hydroureteronephrosis, and nonimmune fetal hydrops, found to have a homozygous nonsense variant in the TBX4 gene. We propose that biallelic variants in the TBX4 gene are associated with a severe syndromic phenotype of sacrococcygeal agenesis and lower limb reduction defects.
Similar content being viewed by others


Introduction
The T-box family of genes, which share a conserved homology domain, codes for DNA-binding transcription factors that are expressed at specific sites in various stages of embryonic development and plays an important role in the formation of the limbs and other vertebrate organs [1]. Of these, the TBX4 gene (OMIM *601719) is expressed specifically in the embryonic hindlimbs and has been shown to be crucial for the development of hindlimb skeletal elements in multiple vertebrate models [2,3,4,5]. In humans, heterozygous variants in the TBX4 gene have been found to cause the ischiocoxopodopatellar syndrome (OMIM #147891), which is associated with patellar hypoplasia or aplasia with variable involvement of the femur, pelvis, and foot bones, with or without craniofacial anomalies [6]. Till date there have been no reports of any phenotype associated with biallelic variants in the gene. This report describes a novel multiple malformation syndrome comprising caudal regression and bilateral lower limb reduction defects, detected antenatally in the third pregnancy of a consanguineous couple with the two previous similarly affected pregnancies, which was found to be associated with a homozygous novel nonsense variant in the TBX4 gene.
Subject and methods
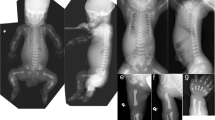
A 21-year-old third gravida, married consanguineously, presented at 17 weeks of gestation with antenatal ultrasonographic findings of nonvisualization of the sacrum and both lower limbs in the fetus (Supplementary Fig. 1a) along with left heart hypoplasia and bilateral renal pyelectasis (Supplementary Fig. 1b). There was no history of maternal diabetes or other teratogenic exposures. Previous two pregnancies of the couple had been terminated in the 5th month of gestation with the same pattern of antenatally detected fetal anomalies; anorectal atresia was additionally documented in the second affected fetus (Fig. 2). The couple decided to opt for termination of this pregnancy also, in view of the multiple major malformations documented in the antenatal scan. Prior to termination, amniocentesis was done and the amniocyte DNA was extracted and preserved. Following termination of this (third) pregnancy, the fetus was brought for autopsy evaluation. Whole body radiographs (anteroposterior and lateral) revealed complete absence of the sacral bones and bilateral lower limb bones (femora, tibiae, fibulae, and foot bones) (Fig. 1a). The occipitofrontal circumference, chest circumference, and abdominal circumference measurements of the fetus corresponded to around 50th centile for 17–18 weeks gestation and crown-rump length corresponded to 50th centile for 16 weeks gestation. Crown-heel length and foot length could not be measured in view of the lower limb reduction defects. Detailed external dysmorphology examination revealed craniofacial dysmorphism in the form of a tall forehead, puffy eyelids, prominent infraorbital creases, broad nose with hypoplastic alae nasi, malar hypoplasia, bilaterally low set ears, and retrognathia (Fig. 1b). Scalp edema, edema over the nape of the neck, and generalized subcutaneous edema suggestive of fetal hydrops were present (Supplementary Fig. 1f). The pelvis appeared triangular in shape with two small nubbin-like structures in the regions corresponding to the lower limbs bilaterally, without any resemblance to normal lower limb anatomy (Fig. 1b, Supplementary Fig. 1g). The labioscrotal folds were unfused and the phallus was prominent and edematous. The anal opening was patent but positioned anteriorly (Supplementary Fig. 1g). Both the upper limbs and the trunk were grossly normal (Fig. 1b). Internal dissection revealed a normal oral cavity and normal neck structures without any tracheal or esophageal anomalies. Thoracic dissection revealed mild pleural effusion, bilateral hypoplastic lungs, and hypoplastic left heart (with hypoplasia of the left ventricle and aorta) without any septal defects, with normal alignment of the great vessels (Supplementary Fig. 1k). There was mild ascites in the abdominal cavity with normal intraperitoneal structures (Supplementary Fig. 1h). Both kidneys appeared enlarged with distended tortuous ureters bilaterally with dilatation of the pelvicalyceal system in the cut section suggestive of bilateral hydroureteronephrosis (Supplementary Fig. 1i, j). The urinary bladder and urethra were normal in appearance. Normal female internal genital structures were present. Intracranial dissection revealed mild enlargement of the lateral ventricles (atrial diameter measuring 10 mm) and normal posterior fossa structures. Histopathology of the rudimentary nubbin-like lower limb structures showed significant growth plate abnormality with unorganized cellular elements and hyaline cartilage (Supplementary Fig. 1l–n).

a Whole body radiograph (anteroposterior view) showing absence of sacrum, pelvic bones, and lower limb bones. b Autopsy findings suggestive of craniofacial dysmorphism, hydropic features, normal upper limbs, normal chest and abdomen, a triangular pelvis, and bilateral lower limb aplasia.
Chromosomal microarray of the fetal DNA with the Affymetrix Cytoscan 750K GeneChip (Affymetric Inc., Thermo Fisher Scientific, Santa Clara, California, United States) did not show any significant copy number variants; however, long stretches of homozygosity involving ~8.83% of the autosomal chromosome complement were noted as expected with parental consanguinity. Whole exome sequencing of the fetal DNA was performed using the Agilent Sure Select V5 exome capture kit (Agilent Technologies, Santa Clara, California, United States). The libraries were sequenced to mean 80–100× coverage on the Illumina sequencing platform (Illumina Inc, San Diego, California, United States). The sequences obtained were aligned to the human reference genome (GRCh37/hg19) using the Burrows-Wheeler Aligner (BWA) program (http://bio-bwa.sourceforge.net/), analyzed using Picard (https://broadinstitute.github.io/picard/) and the Genome Analysis Toolkit (GATK) version 3.6 (https://software.broadinstitute.org/gatk/), and annotated using ANNOVAR (http://annovar.openbioinformatics.org/en/latest/). The total number of on-target variants that passed the variant quality filters was 87,435. Variant filtering was done based on population databases for polymorphic variants, frequency in the local population using in-house data, and location and function of the variants, as detailed in Supplementary Table 1. The variants that remained after the initial filtering were prioritized based on whether they involved genes with OMIM-listed phenotypes and phenotypic similarity of the case with the reported expressed phenotype of these genes. No previously reported pathogenic or likely pathogenic variants were found, but a novel homozygous nonsense variant c.402G>A; p.(Trp134Ter) was identified in the TBX4 gene (transcript id ENST00000240335.1; NM_018488.2). Sanger sequencing-based targeted variant analysis of this TBX4 gene variant confirmed the presence of the variant in the homozygous form in the fetal DNA and in the heterozygous form in both parents (Fig. 2). This variant has not been reported in the gnomAD (https://gnomad.broadinstitute.org/), ExAC (http://exac.broadinstitute.org/), and 1000 Genomes databases (http://www.internationalgenome.org/) and is not present in the in-house database of 700 exomes from the Indian population. This variant has been submitted in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and has been assigned the accession number SCV000925964. It is predicted to be damaging by the in silico prediction tools MutationTaster (http://www.mutationtaster.org/) and CADD (https://cadd.gs.washington.edu/). It involves the tryptophan residue at amino acid position 134 that is conserved across species and the resultant mRNA containing the premature termination codon is predicted to undergo nonsense-mediated decay. Apart from this TBX4 gene variant, the only other variant found in a limb bud development-related gene was a heterozygous missense variant of uncertain significance (VOUS) c.4A>T; p.(Ile2Phe) in the BMP4 gene (transcript id ENST00000245451.4/3; ClinVar accession number SCV000998998). However, as the fetal phenotype did not match the phenotype reported to be associated with the BMP4 gene (OMIM# 607932- Autosomal dominant Syndromic Microphthalmia 6), this VOUS was not considered likely to be the causal variant. Supplementary Table 2 lists the other candidate gene variants that remained after initial filtering of variants.

Sanger sequence chromatograms of the TBX4 gene showing the c.402G>A variant (highlighted by the rectangle) in the homozygous form in the third affected fetus (the index case) and in the heterozygous form in both carrier parents.
As the parents were heterozygous for the TBX4 gene variant and the phenotype of ischiocoxopodopatellar syndrome has previously been described in heterozygotes, clinical re-evaluation of the couple was planned. The mother had been noted to have proximally placed fourth and fifth toes bilaterally in the initial visit. However, as she did not come for follow-up visits after termination of the pregnancy, her skeletal radiographs could not be taken. The father was also found to have proximally placed 4th and 5th toes bilaterally on clinical examination. His skeletal radiographs (of the feet, knee, pelvis, and hip joints) taken in the follow-up visit after completion of molecular genetic testing showed patellar hypoplasia, lesser femoral trochanter hypoplasia, and short fourth and fifth metatarsals bilaterally, which are consistent with the diagnosis of ischiocoxopodopatellar syndrome (Fig. 3).

a Close-up of the father’s feet showing the proximally placed fourth and fifth toes bilaterally. b Anteroposterior radiograph of the right foot showing the short fourth and fifth metatarsals. c Anteroposterior radiograph of the hip joints and femora showing the hypoplastic lesser trochanter of the femur bilaterally (marked with red arrows). d Anteroposterior radiograph of the knee joints showing the hypoplastic patellae bilaterally (marked with red arrows). e Lateral radiographs of the knee joints showing the hypoplastic patellae bilaterally (color figure online).
Discussion
The TbX4 gene has an evolutionarily conserved role in hindlimb initiation and development [7]. It has been found to be specifically expressed only in the hindlimb buds and not in the forelimb buds during embryonic development in many vertebrate models including zebrafish, chick, and mouse [2,3,4,5]. However, gene-swap experiments of Tbx4 with the closely related forelimb-specific gene Tbx5 in the mouse forelimb and similar experiments in the chick and other vertebrate models have suggested that Tbx4 does not confer hindlimb specificity and is not the main determinant of hindlimb morphology, both of which are most likely regulated by the Pitx1 gene [8]. Though direct evidence for hindlimb-specific expression has not been shown in humans, monoallelic variants in the TBX4 gene are reported to cause the ischiocoxopodopatellar syndrome or small patella syndrome, which is associated with abnormalities of the pelvis, knee, and foot [6, 9]. Copy number variations of chromosome 17q23.1q23.2 region especially microduplications that include the TBX4 gene have been found to be associated with isolated congenital talipes equinovarus/club foot deformity [10]. In this reported family, the heterozygous carrier parents had mild phenotypic manifestations. Features of small patella syndrome, as reported in other individuals with monoallelic variants in the TBX4 gene, could be clearly documented clinically and radiographically in the father. The bilateral lower limb aplasia and sacrococcygeal agenesis seen in the homozygous offspring of this heterozygous carrier couple most likely represent the severe end of the phenotypic spectrum associated with TBX4 gene variants.
Don et al. identified variants in the nuclear localization signal (NLS) of Tbx4 in the naturally occurring pelvic finless model of zebrafish, which are postulated to impair the nuclear localisation of the protein and thereby alter the gene expression patterns and lead to complete failure of development of the pelvic fins, which are homologous to tetrapod hindlimbs. Using a TALEN-induced Tbx4-knockout allele they could confirm that variants within the Tbx4 NLS can disrupt pelvic fin development [11]. Till date, no phenotype associated with biallelic loss-of-function variants in the TBX4 gene has been reported in humans. The pattern of caudal regression and bilateral lower limb aplasia in this reported fetus with the homozygous nonsense variant in the TBX4 gene is consistent with the phenotype noted in Tbx4-knockout vertebrate models.
Monoallelic variants in the TBX4 gene have also been implicated in causation of acinar dysplasia and lung hypoplasia [12]. The reported fetus had significant bilateral lung hypoplasia in addition to left heart hypoplasia. The hydropic changes noted in the fetus are likely to have resulted from congestive heart failure secondary to the left heart hypoplasia. The other anomalies present in this case namely sacral agenesis/caudal regression, bilateral hydroureteronephrosis, and mild ventriculomegaly have not been previously reported in association with the TBX4 gene in either human subjects or in vertebrate models.
This is the first report of a human phenotype associated with biallelic variants in the TBX4 gene. The in silico prediction for the identified nonsense TBX4 gene variant, its Mendelian segregation pattern in the family and absence of any other significant gene variants or chromosomal copy number variants, along with phenotypic similarities with knockout vertebrate models for this gene, suggests that the pattern of anomalies noted in this family could represent a new syndrome associated with biallelic pathogenic variants in the TBX4 gene.
References
Naiche LA, Harrelson Z, Kelly RG, et al. T-box genes in vertebrate development. Annu Rev Genet. 2005;39:219–39.
Gibson-Brown JJ, Agulnik SI, Chapman DL, Alexiou M, Garvey N, Silver LM, et al. Evidence of a role for T-box genes in the evolution of limb morphogenesis and the specification of forelimb/hindlimb identity. Mech Dev. 1996;56:93–101.
Naiche LA, Papaioannou VE. Loss of Tbx4 blocks hindlimb development and affects vascularization and fusion of the allantois. Development. 2003;130:2681–93.
Tamura K, Yonei-Tamura S, Izpisúa Belmonte JC. Differential expression of Tbx4 and Tbx5 in Zebrafish fin buds. Mech Dev. 1999;87:181–4.
Horton AC, Mahadevan NR, Minguillon C, Osoegawa K, Rokhsar DS, Ruvinsky I, et al. Conservation of linkage and evolution of developmental function within the Tbx2/3/4/5 subfamily of T-box genes: implications for the origin of vertebrate limbs. Dev Genes Evol. 2008;218:613–28.
Bongers EM, Duijf PH, van Beersum SE, Schoots J, Van Kampen A, Burckhardt A, et al. Mutations in the human TBX4 gene cause small patella syndrome. Am J Hum Genet. 2004;74:1239–48.
Sheeba CJ, Logan MP. The roles of T-Box genes in vertebrate limb development. Curr Top Dev Biol. 2017;122:355–81.
Minguillon C, Del Buono J, Logan MP. Tbx5 and Tbx4 are not sufficient to determine limb-specific morphologies but have common roles in initiating limb outgrowth. Dev Cell. 2005;8:75–84.
Vanlerberghe C, Jourdain AS, Dieux A, Toutain A, Callewaert B, Dupuis-Girod S, et al. Small patella syndrome: New clinical and molecular insights into a consistent phenotype. Clin Genet. 2017;92:676–8.
Alvarado DM, Aferol H, McCall K, Huang JB, Techy M, Buchan J, et al. Familial isolated clubfoot is associated with recurrent chromosome 17q23.1q23.2 microduplications containing TBX4. Am J Hum Genet. 2010;87:154–60.
Don EK, de Jong-Curtain TA, Doggett K, Hall TE, Heng B, Badrock AP, et al. Genetic basis of hindlimb loss in a naturally occurring vertebrate model. Biol Open. 2016;5:359–66.
Karolak JA, Vincent M, Deutsch G, Gambin T, Cogné B, Pichon O, et al. Complex compound inheritance of lethal lung developmental disorders due to disruption of the TBX-FGF pathway. Am J Hum Genet. 2019;104:213–28.
Acknowledgements
Molecular genetic testing of this family was done with the help of funding support obtained from DST—SERB (Department of Science and Technology—Science and Engineering Research Board) project number EMR/2016/003887.
Funding
Molecular genetic testing of this family was done with the help of the funding support obtained from DST-SERB (Department of Science and Technology—Science and Engineering Research Board) project number EMR/2016/003887.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ranganath, P., Perala, S., Nair, L. et al. A newly recognized multiple malformation syndrome with caudal regression associated with a biallelic c.402G>A variant in TBX4. Eur J Hum Genet 28, 669–673 (2020). https://doi.org/10.1038/s41431-020-0572-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-0572-5

{kind=link}