Herpes Simplex Virus Type-2 Paralyzes the Function of Monocyte-Derived Dendritic Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Monocyte-Derived Dendritic Cells (DCs)
2.2. Viruses
2.3. HSV-1 and HSV-2 Infection of iDCs and mDCs
2.4. Construction of Expression Plasmids
2.5. Transient Transfection and HSV-1 or HSV-2 Infection of HEK293T Cells
2.6. Mg/EGTA Treatment of mDCs
2.7. Flow Cytometric Analyses and Antibodies Used
2.8. WesternBlot Analyses and Antibodies Used
2.9. Fibronectin Adhesion Assay
2.10. ICAM Adhesion Assay
2.11. Transwell Migration Assay
2.12. Immunofluorescence Confocal Microscopy
2.13. Statistical Analyses
2.14. Approvals and Legal Requirements
3. Results
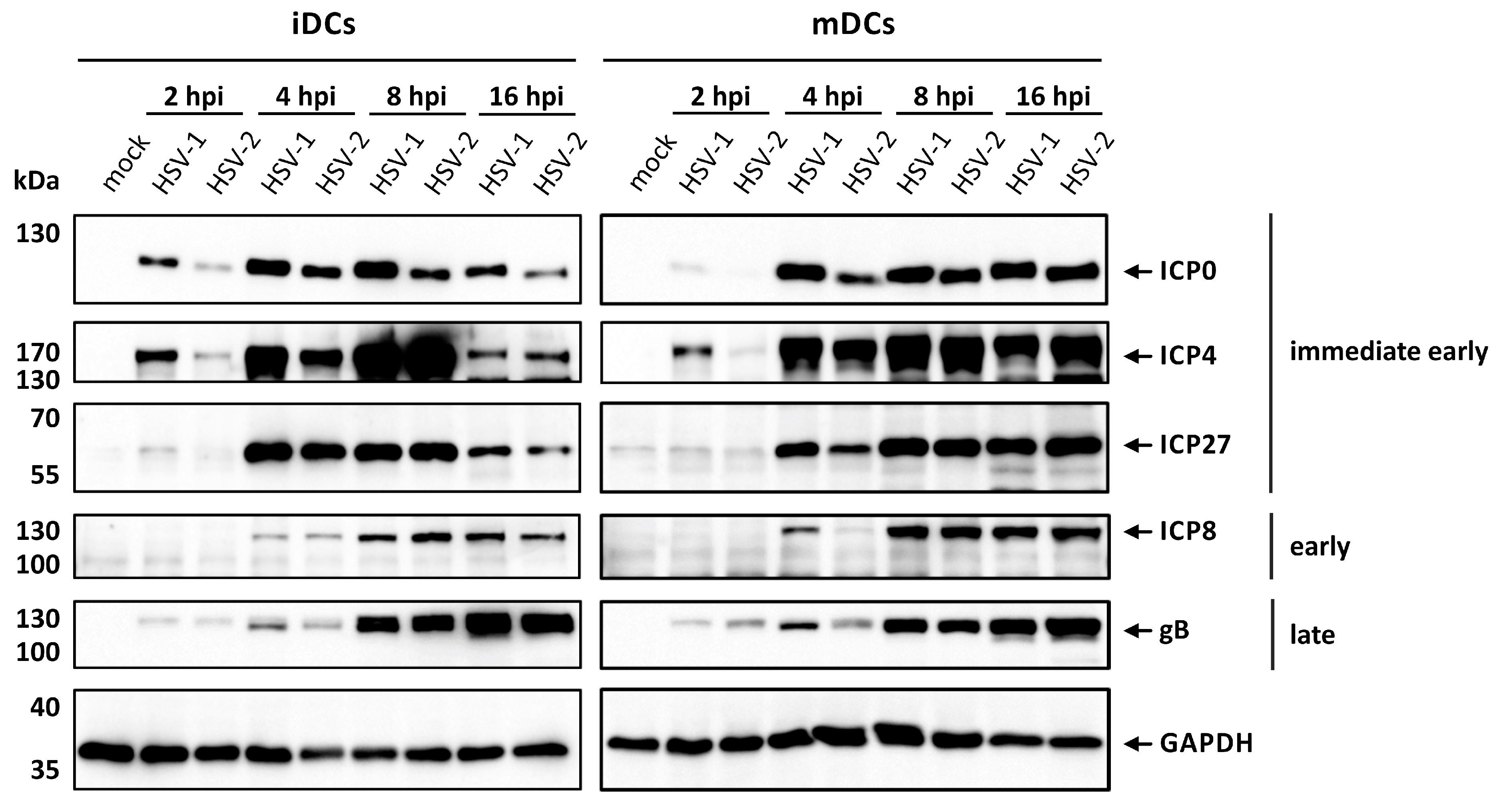
3.1. HSV-2 Successfully Establishes Viral Protein Expression in iDCs and mDCs
3.2. HSV-2 Inhibits DC Maturation When Infecting iDCs
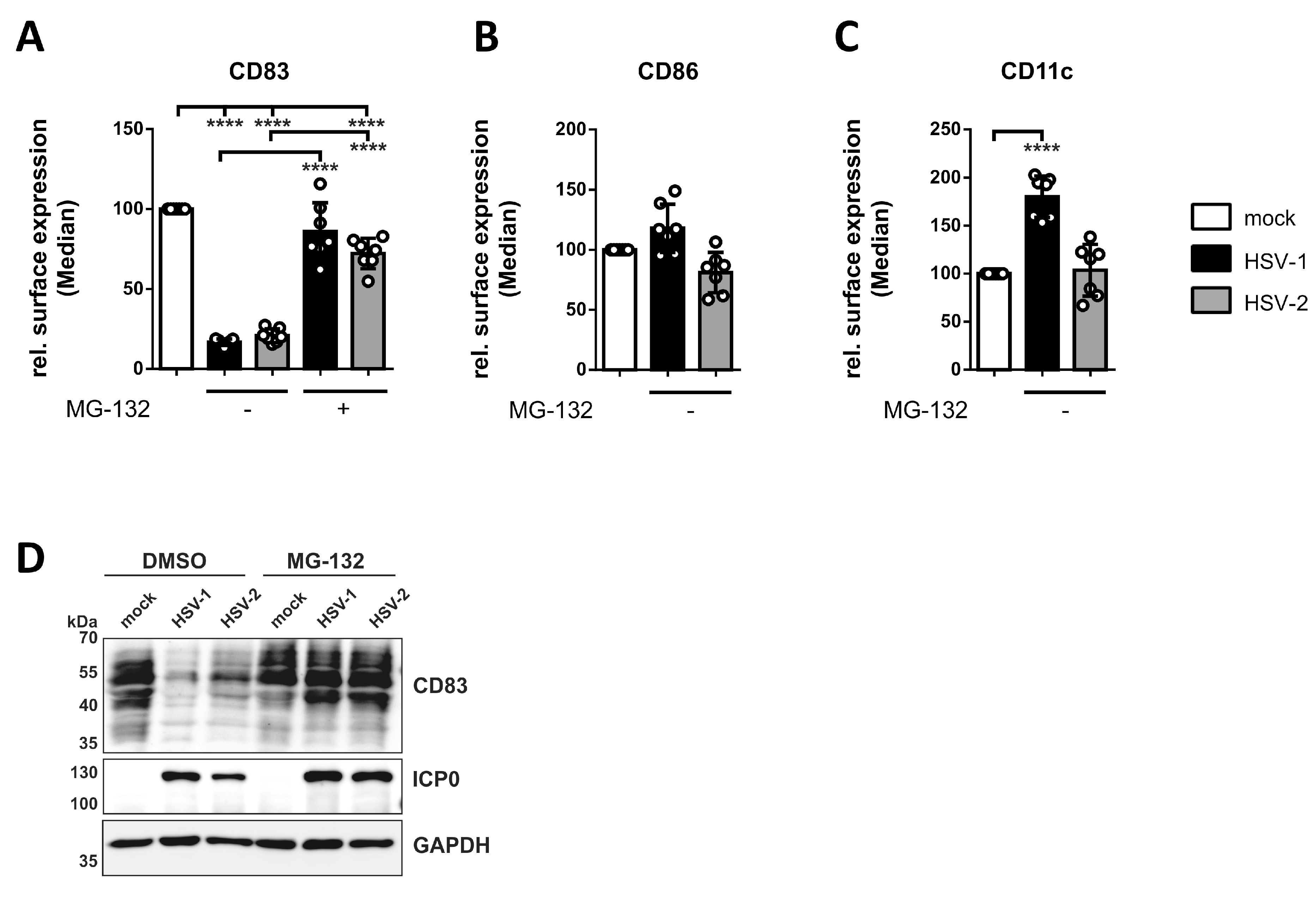
3.3. HSV-2 Induces the Proteasome-Dependent Degradation of CD83 in mDCs
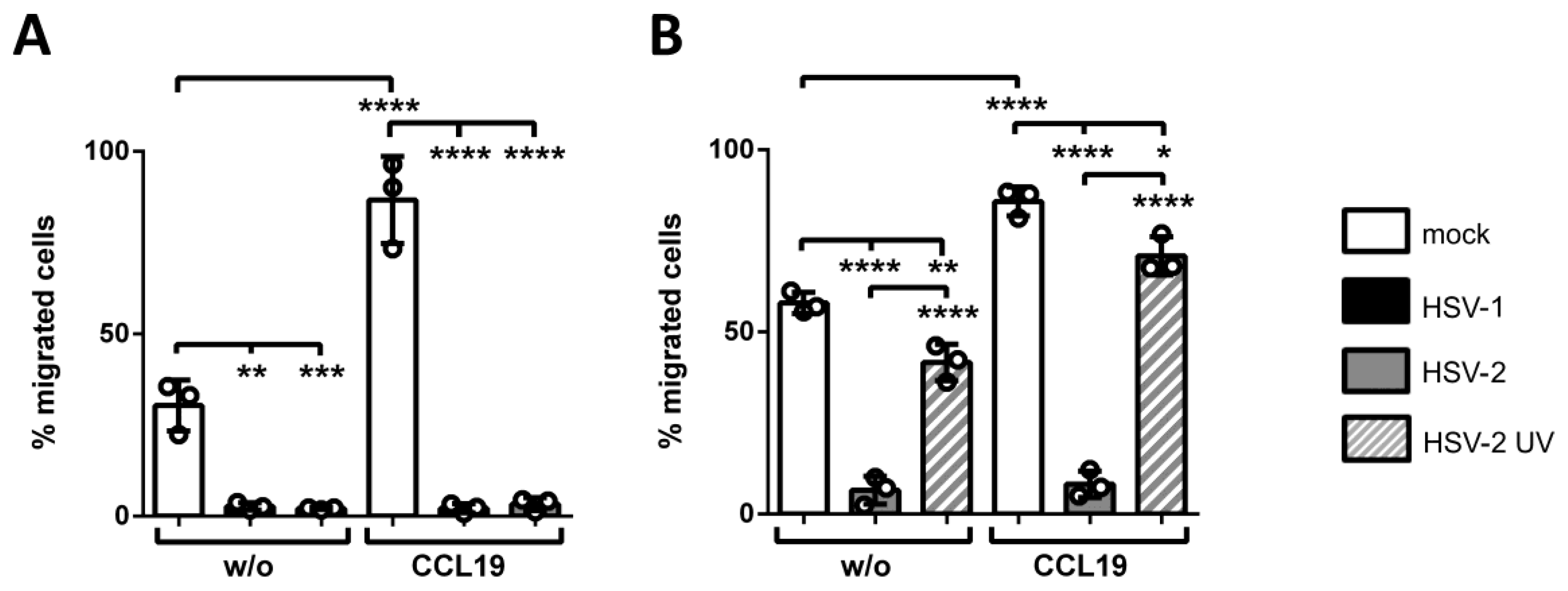
3.4. HSV-2 Inhibits CCL19-Directed Transwell Migration of mDCs Early upon Infection
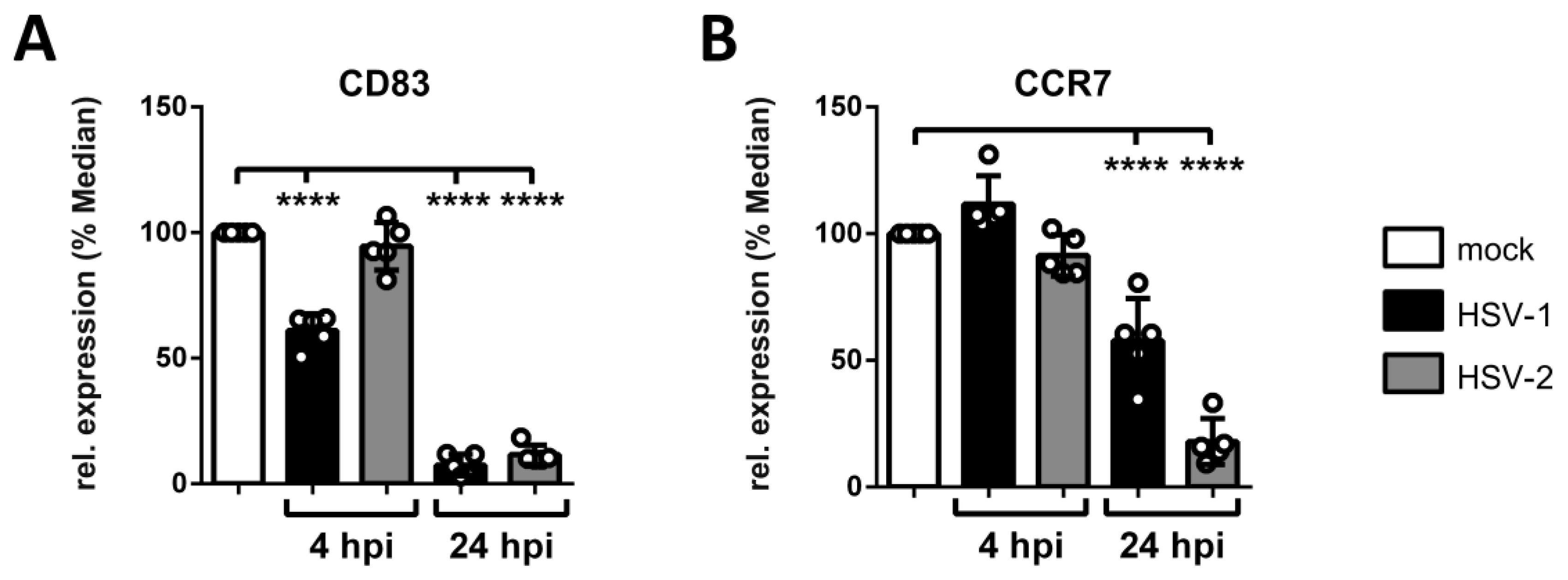
3.5. HSV-2 Mediates the Reduction of CCR7 Surface Expression Late during mDC Infection
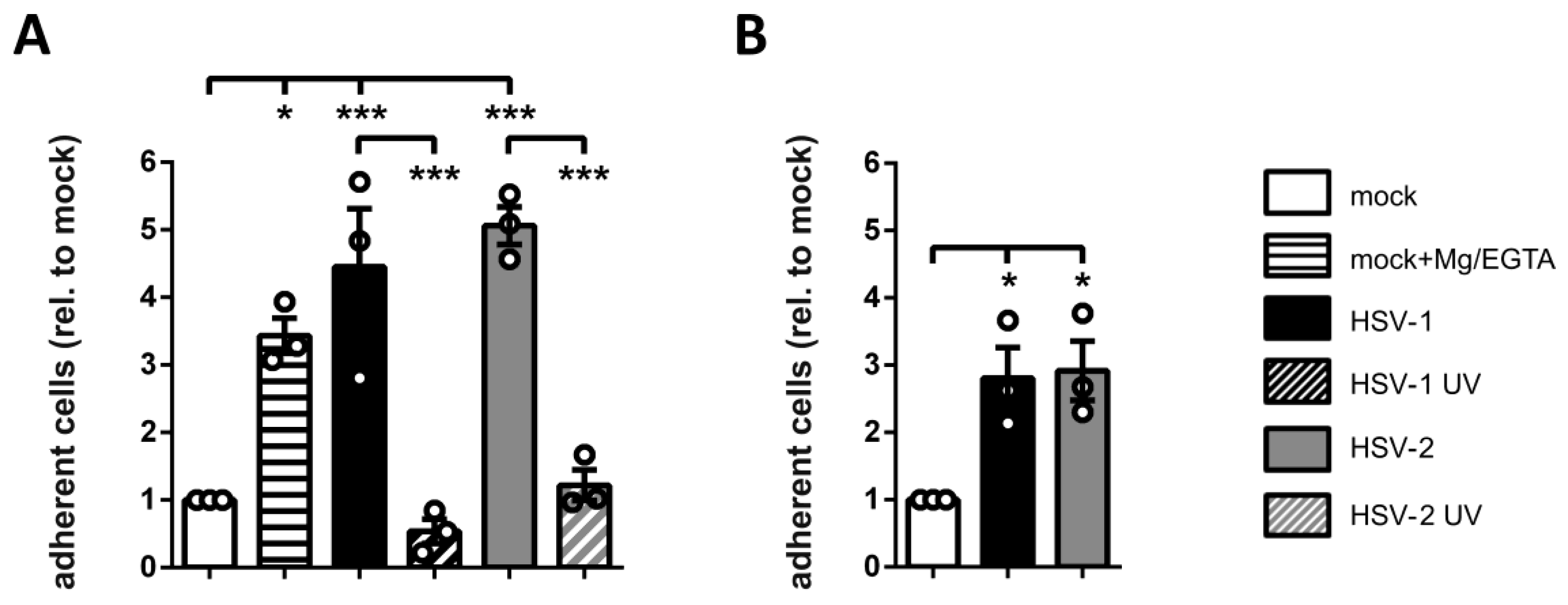
3.6. HSV-2-Infected mDCs Show Increased Adhesion
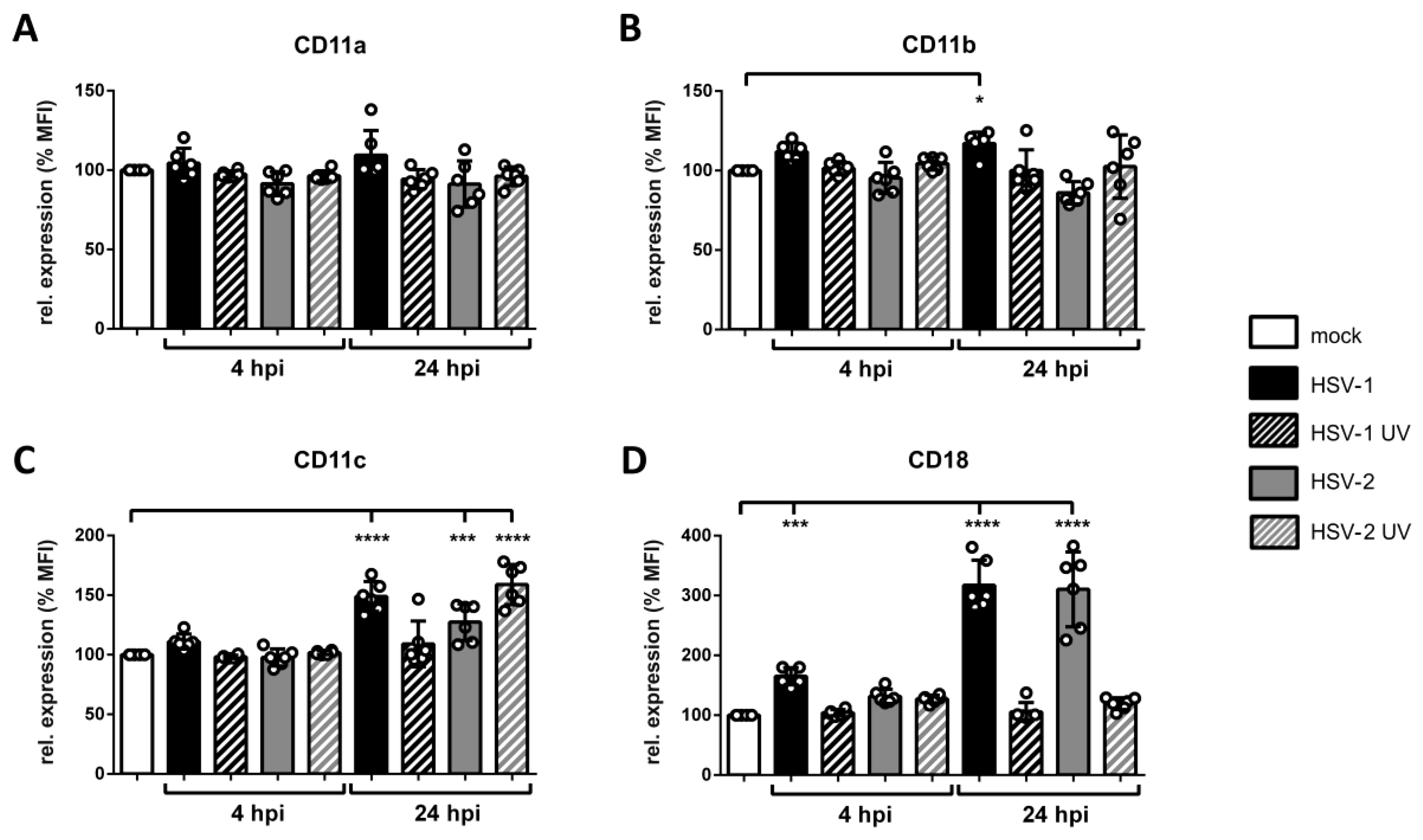
3.7. Surface Expression of β2 Integrin Subunits is Differentially Regulated on HSV-2-Infected mDCs
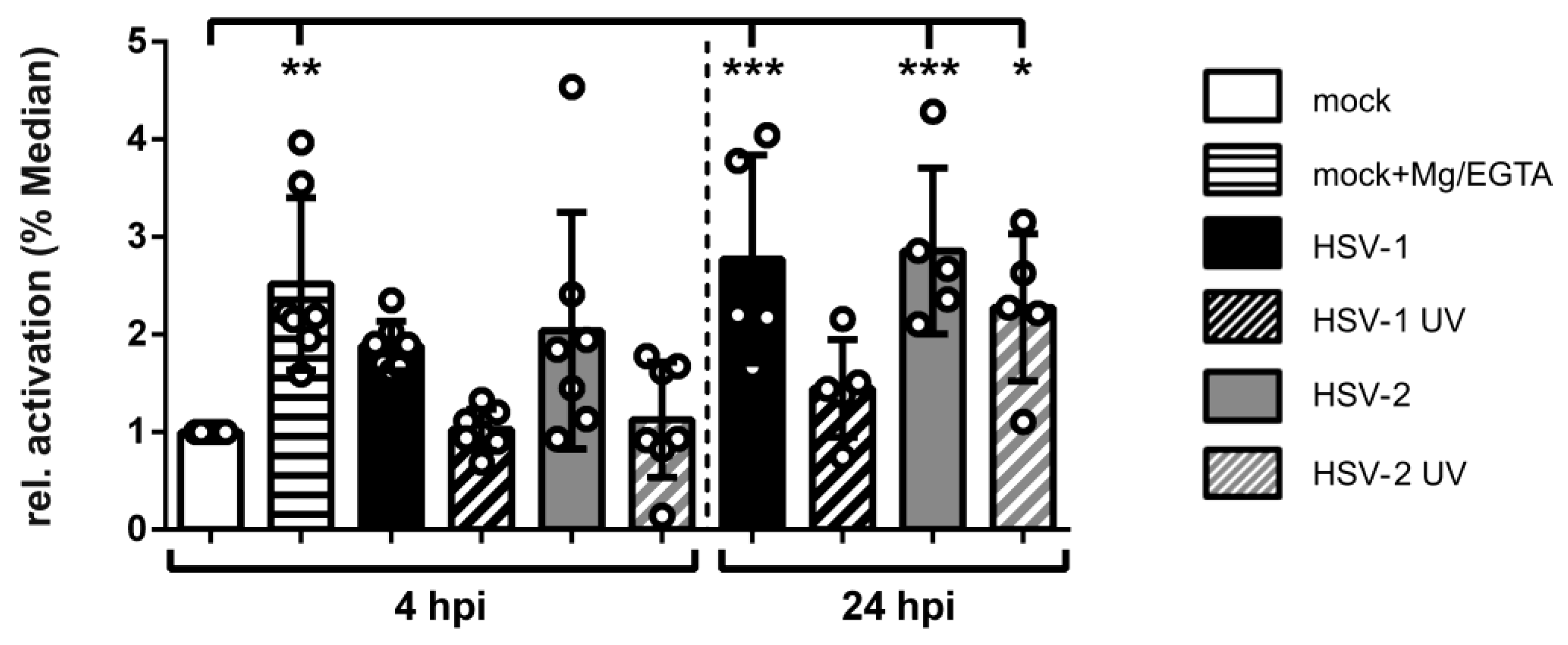
3.8. HSV-2-Infected mDCs Display Increased β2 Integrin Activity
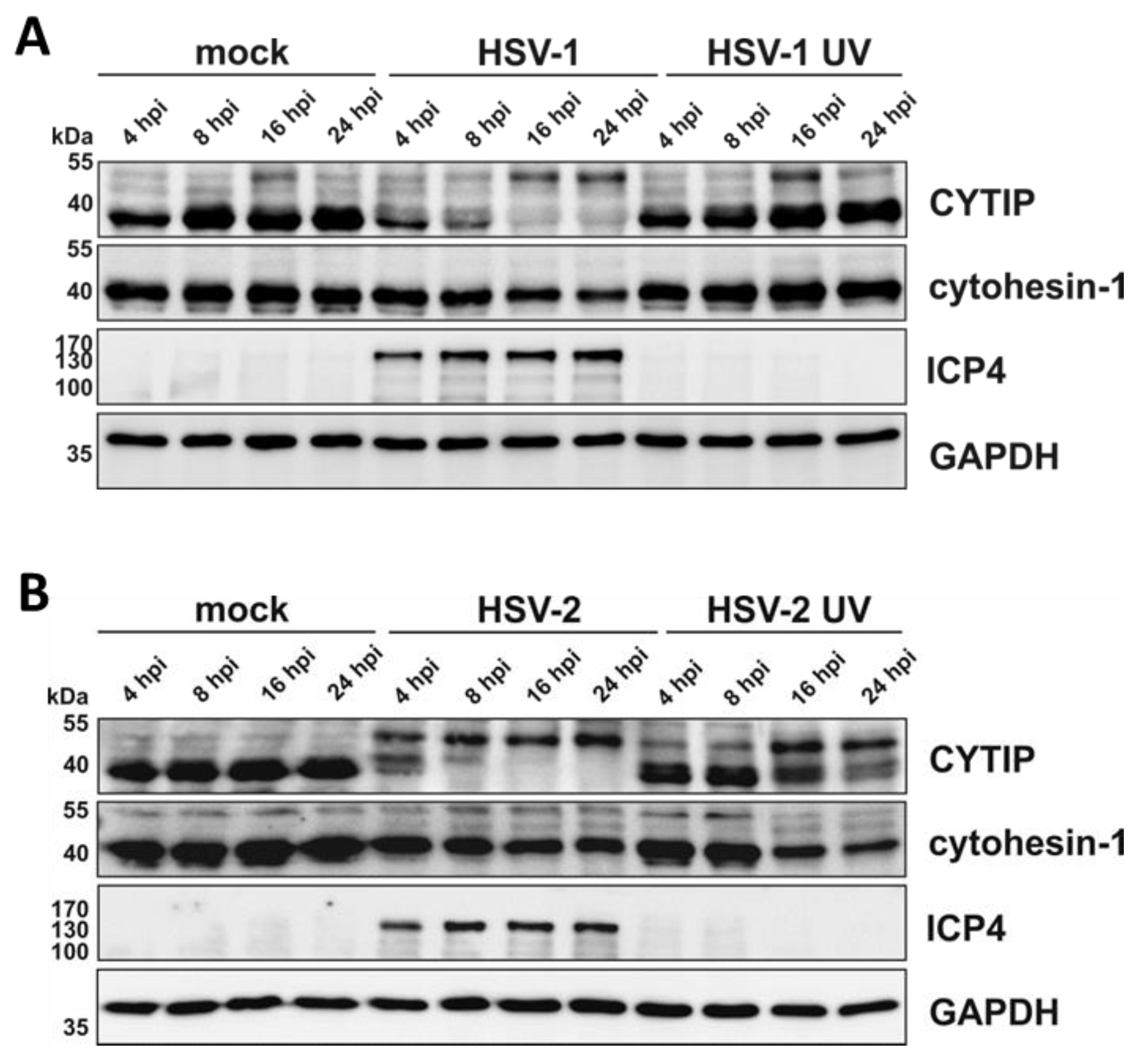
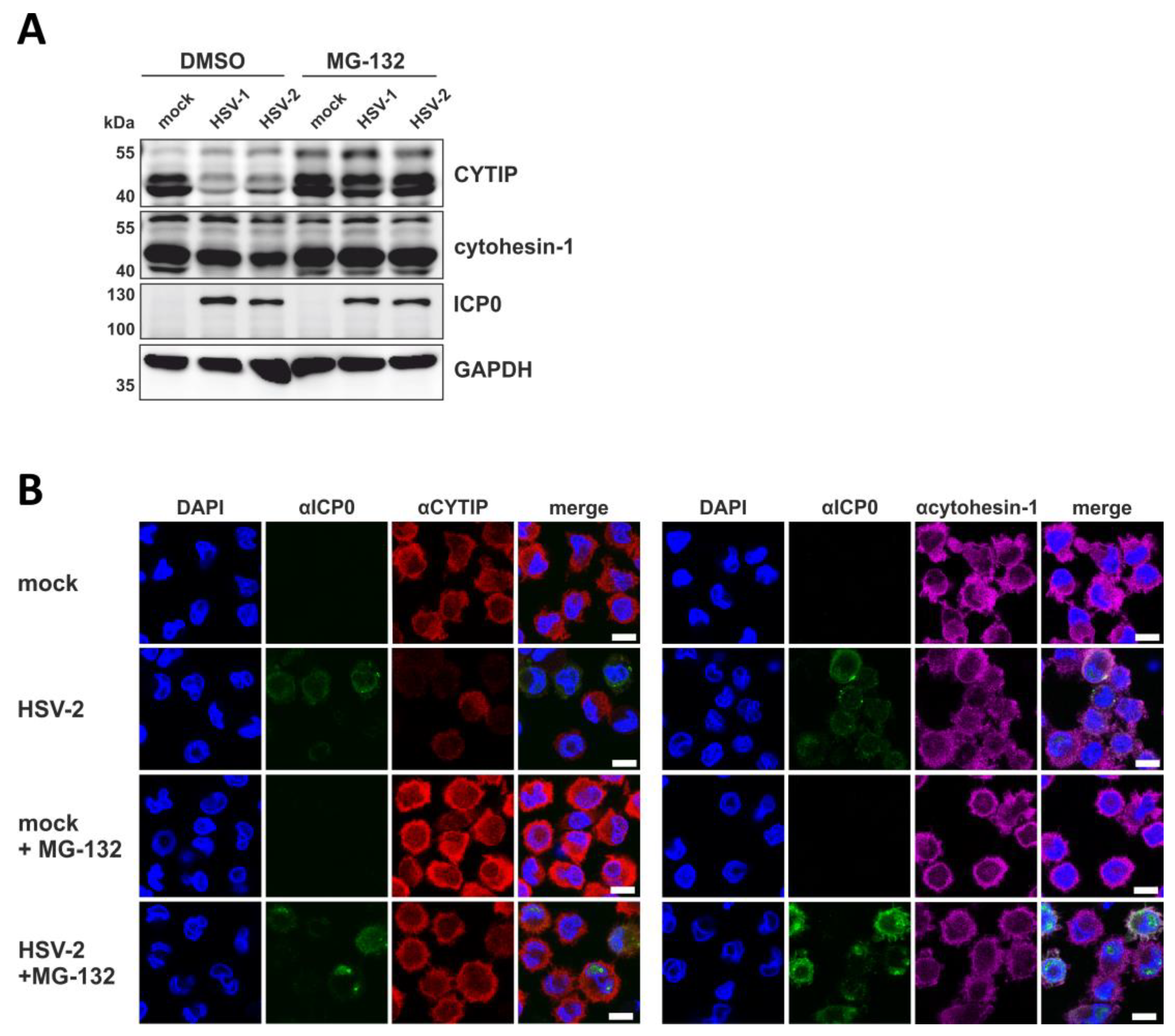
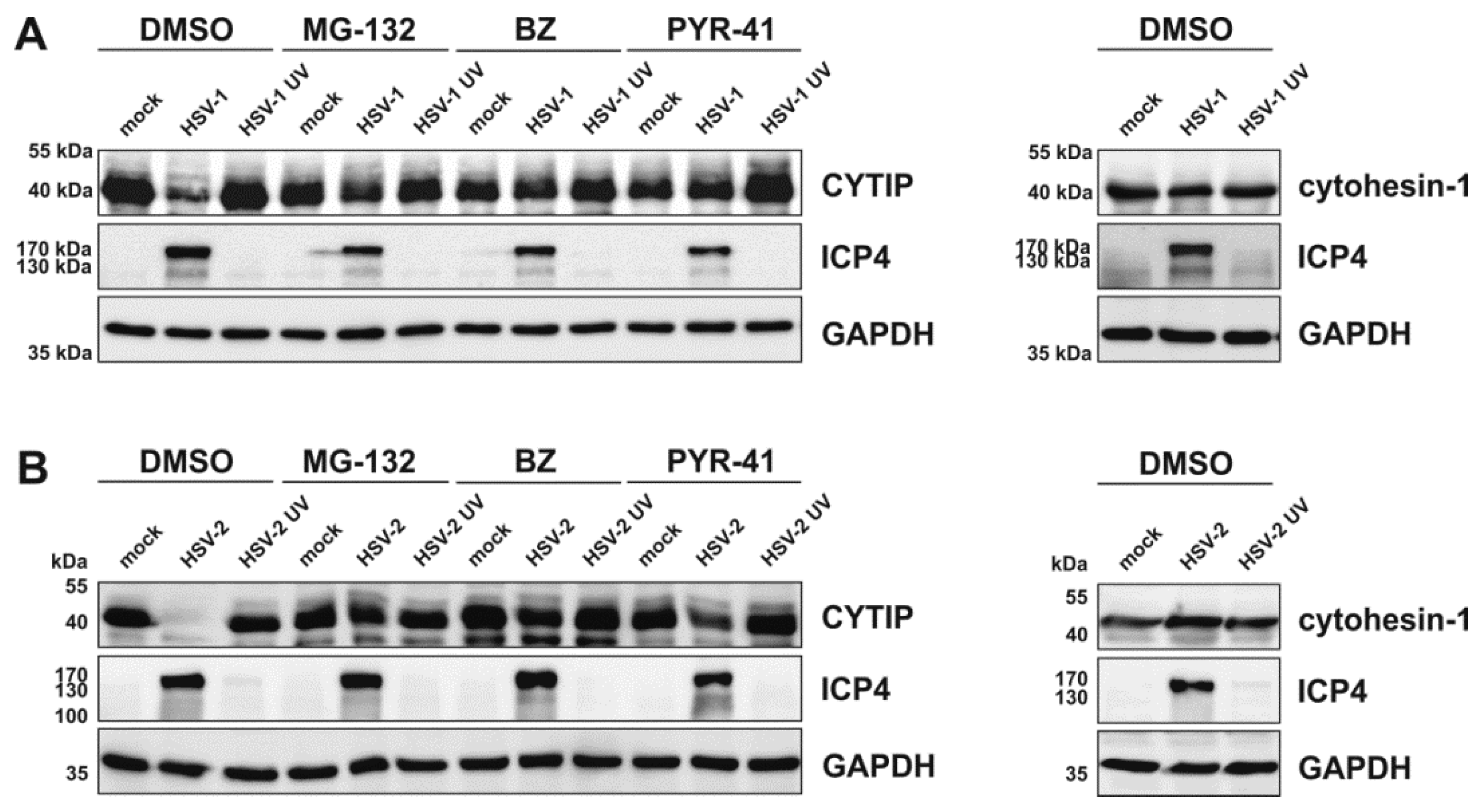
3.9. HSV-2 Mediates Rapid Downmodulation of CYTIP Protein Expression
3.10. HSV-2 Induces a Proteasome and Ubiquitin-Dependent Degradation of CYTIP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef] [PubMed]
- Lanzavecchia, A.; Sallusto, F. The instructive role of dendritic cells on T cell responses: Lineages, plasticity and kinetics. Curr. Opin. Immunol. 2001, 13, 291–298. [Google Scholar] [CrossRef]
- Villadangos, J.A.; Heath, W.R. Life cycle, migration and antigen presenting functions of spleen and lymph node dendritic cells: Limitations of the Langerhans cells paradigm. Semin. Immunol. 2005, 17, 262–272. [Google Scholar] [CrossRef]
- Steinman, R.M. Dendritic cells: Understanding immunogenicity. Eur. J. Immunol. 2007, 37 (Suppl. 1), S53–S60. [Google Scholar] [CrossRef]
- Inaba, K.; Turley, S.; Iyoda, T.; Yamaide, F.; Shimoyama, S.; Reis e Sousa, C.; Germain, R.N.; Mellman, I.; Steinman, R.M. The formation of immunogenic major histocompatibility complex class II-peptide ligands in lysosomal compartments of dendritic cells is regulated by inflammatory stimuli. J. Exp. Med. 2000, 191, 927–936. [Google Scholar] [CrossRef]
- Turley, S.J.; Inaba, K.; Garrett, W.S.; Ebersold, M.; Unternaehrer, J.; Steinman, R.M.; Mellman, I. Transport of peptide-MHC class II complexes in developing dendritic cells. Science 2000, 288, 522–527. [Google Scholar] [CrossRef]
- Berchtold, S.; Jones, T.; Mühl-Zürbes, P.; Sheer, D.; Schuler, G.; Steinkasserer, A. The human dendritic cell marker CD83 maps to chromosome 6p23. Ann. Hum. Genet. 1999, 63, 181–183. [Google Scholar] [CrossRef] [Green Version]
- Lechmann, M.; Berchtold, S.; Hauber, J.; Steinkasserer, A. CD83 on dendritic cells: More than just a marker for maturation. Trends Immunol. 2002, 23, 273–275. [Google Scholar] [CrossRef]
- Prechtel, A.T.; Steinkasserer, A. CD83: An update on functions and prospects of the maturation marker of dendritic cells. Arch. Dermatol. Res. 2007, 299, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.J.; Schwarting, R.; Smith, H.M.; Tedder, T.F. A novel cell-surface molecule expressed by human interdigitating reticulum cells, Langerhans cells, and activated lymphocytes is a new member of the Ig superfamily. J. Immunol. 1992, 149, 735–742. [Google Scholar] [PubMed]
- Zhou, L.J.; Tedder, T.F. Human blood dendritic cells selectively express CD83, a member of the immunoglobulin superfamily. J. Immunol. 1995, 154, 3821–3835. [Google Scholar] [PubMed]
- Christensen, J.E.; Thomsen, A.R. Co-ordinating innate and adaptive immunity to viral infection: Mobility is the key. APMIS 2009, 117, 338–355. [Google Scholar] [CrossRef] [PubMed]
- Sozzani, S.; Allavena, P.; D’Amico, G.; Luini, W.; Bianchi, G.; Kataura, M.; Imai, T.; Yoshie, O.; Bonecchi, R.; Mantovani, A. Differential regulation of chemokine receptors during dendritic cell maturation: A model for their trafficking properties. J. Immunol. 1998, 161, 1083–1086. [Google Scholar] [PubMed]
- Förster, R.; Schubel, A.; Breitfeld, D.; Kremmer, E.; Renner-Muller, I.; Wolf, E.; Lipp, M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell 1999, 99, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Ohl, L.; Mohaupt, M.; Czeloth, N.; Hintzen, G.; Kiafard, Z.; Zwirner, J.; Blankenstein, T.; Henning, G.; Förster, R. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity 2004, 21, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Parlato, S.; Santini, S.M.; Lapenta, C.; Di, P.T.; Logozzi, M.; Spada, M.; Giammarioli, A.M.; Malorni, W.; Fais, S.; Belardelli, F. Expression of CCR-7, MIP-3beta, and Th-1 chemokines in type I IFN-induced monocyte-derived dendritic cells: Importance for the rapid acquisition of potent migratory and functional activities. Blood 2001, 98, 3022–3029. [Google Scholar] [CrossRef] [Green Version]
- Randolph, G.J.; Angeli, V.; Swartz, M.A. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat. Rev. Immunol. 2005, 5, 617–628. [Google Scholar] [CrossRef]
- Weber, M.; Hauschild, R.; Schwarz, J.; Moussion, C.; de Vries, I.; Legler, D.F.; Luther, S.A.; Bollenbach, T.; Sixt, M. Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science 2013, 339, 328–332. [Google Scholar] [CrossRef] [Green Version]
- Nourshargh, S.; Alon, R. Leukocyte migration into inflamed tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cougoule, C.; Lastrucci, C.; Guiet, R.; Mascarau, R.; Meunier, E.; Lugo-Villarino, G.; Neyrolles, O.; Poincloux, R.; Maridonneau-Parini, I. Podosomes, But Not the Maturation Status, Determine the Protease-Dependent 3D Migration in Human Dendritic Cells. Front. Immunol. 2018, 9, 846. [Google Scholar] [CrossRef] [Green Version]
- Renkawitz, J.; Sixt, M. Mechanisms of force generation and force transmission during interstitial leukocyte migration. EMBO Rep. 2010, 11, 744–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Springer, T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell 1994, 76, 301–314. [Google Scholar] [CrossRef]
- Friedl, P.; Borgmann, S.; Brocker, E.B. Amoeboid leukocyte crawling through extracellular matrix: Lessons from the Dictyostelium paradigm of cell movement. J. Leukoc. Biol. 2001, 70, 491–509. [Google Scholar] [PubMed]
- Friedl, P.; Brocker, E.B. Reconstructing leukocyte migration in 3D extracellular matrix by time-lapse videomicroscopy and computer-assisted tracking. Methods Mol. Biol. 2004, 239, 77–90. [Google Scholar] [CrossRef]
- Lämmermann, T.; Bader, B.L.; Monkley, S.J.; Worbs, T.; Wedlich-Soldner, R.; Hirsch, K.; Keller, M.; Förster, R.; Critchley, D.R.; Fassler, R.; et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature 2008, 453, 51–55. [Google Scholar] [CrossRef]
- Lämmermann, T.; Sixt, M. Mechanical modes of ‘amoeboid’ cell migration. Curr. Opin. Cell Biol. 2009, 21, 636–644. [Google Scholar] [CrossRef]
- Schmidt, S.; Friedl, P. Interstitial cell migration: Integrin-dependent and alternative adhesion mechanisms. Cell Tissue Res. 2010, 339, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renkawitz, J.; Schumann, K.; Weber, M.; Lämmermann, T.; Pflicke, H.; Piel, M.; Polleux, J.; Spatz, J.P.; Sixt, M. Adaptive force transmission in amoeboid cell migration. Nat. Cell Biol. 2009, 11, 1438–1443. [Google Scholar] [CrossRef]
- Friedl, P.; Weigelin, B. Interstitial leukocyte migration and immune function. Nat. Immunol. 2008, 9, 960–969. [Google Scholar] [CrossRef]
- Huttenlocher, A.; Ginsberg, M.H.; Horwitz, A.F. Modulation of cell migration by integrin-mediated cytoskeletal linkages and ligand-binding affinity. J. Cell Biol. 1996, 134, 1551–1562. [Google Scholar] [CrossRef] [PubMed]
- Berrier, A.L.; Yamada, K.M. Cell-matrix adhesion. J. Cell. Physiol. 2007, 213, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Hogg, N.; Henderson, R.; Leitinger, B.; McDowall, A.; Porter, J.; Stanley, P. Mechanisms contributing to the activity of integrins on leukocytes. Immunol. Rev. 2002, 186, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Kinashi, T. Intracellular signalling controlling integrin activation in lymphocytes. Nat. Rev. Immunol. 2005, 5, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Hyun, Y.M.; Lefort, C.T.; Kim, M. Leukocyte integrins and their ligand interactions. Immunol. Res. 2009, 45, 195–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schittenhelm, L.; Hilkens, C.M.; Morrison, V.L. Beta2 Integrins As Regulators of Dendritic Cell, Monocyte, and Macrophage Function. Front. Immunol. 2017, 8, 1866. [Google Scholar] [CrossRef]
- Kishimoto, T.K.; O’Connor, K.; Lee, A.; Roberts, T.M.; Springer, T.A. Cloning of the beta subunit of the leukocyte adhesion proteins: Homology to an extracellular matrix receptor defines a novel supergene family. Cell 1987, 48, 681–690. [Google Scholar] [CrossRef]
- Anceriz, N.; Vandal, K.; Tessier, P.A. S100A9 mediates neutrophil adhesion to fibronectin through activation of beta2 integrins. Biochem. Biophys. Res. Commun. 2007, 354, 84–89. [Google Scholar] [CrossRef] [Green Version]
- Gahmberg, C.G.; Tolvanen, M.; Kotovuori, P. Leukocyte adhesion--structure and function of human leukocyte beta2-integrins and their cellular ligands. Eur. J. Biochem. 1997, 245, 215–232. [Google Scholar] [CrossRef]
- Fagerholm, S.C.; Guenther, C.; Llort, A.M.; Savinko, T.; Uotila, L.M. Beta2-Integrins and Interacting Proteins in Leukocyte Trafficking, Immune Suppression, and Immunodeficiency Disease. Front. Immunol. 2019, 10, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, S.; Etzioni, A. Leukocyte adhesion deficiencies. Ann. N. Y. Acad. Sci. 2012, 1250, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.S.; Weyrich, A.S.; Zimmerman, G.A. Lessons from rare maladies: Leukocyte adhesion deficiency syndromes. Curr. Opin. Hematol. 2013, 20, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharffetter-Kochanek, K.; Lu, H.; Norman, K.; van Nood, N.; Munoz, F.; Grabbe, S.; McArthur, M.; Lorenzo, I.; Kaplan, S.; Ley, K.; et al. Spontaneous skin ulceration and defective T cell function in CD18 null mice. J. Exp. Med. 1998, 188, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogg, N.; Patzak, I.; Willenbrock, F. The insider’s guide to leukocyte integrin signalling and function. Nat. Rev. Immunol. 2011, 11, 416–426. [Google Scholar] [CrossRef]
- Shattil, S.J.; Kim, C.; Ginsberg, M.H. The final steps of integrin activation: The end game. Nat. Rev. Mol. Cell Biol. 2010, 11, 288–300. [Google Scholar] [CrossRef] [Green Version]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef]
- Schwartz, M.A.; Ginsberg, M.H. Networks and crosstalk: Integrin signalling spreads. Nat. Cell Biol. 2002, 4, E65–E68. [Google Scholar] [CrossRef]
- Dib, K. BETA 2 integrin signaling in leukocytes. Front. Biosci. 2000, 5, D438–D451. [Google Scholar] [CrossRef] [Green Version]
- Hynes, R.O. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell 1992, 69, 11–25. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Carman, C.V.; Springer, T.A. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science 2003, 301, 1720–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiger, C.; Nagel, W.; Boehm, T.; van Kooyk, Y.; Figdor, C.G.; Kremmer, E.; Hogg, N.; Zeitlmann, L.; Dierks, H.; Weber, K.S.; et al. Cytohesin-1 regulates beta-2 integrin-mediated adhesion through both ARF-GEF function and interaction with LFA-1. EMBO J. 2000, 19, 2525–2536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolanus, W.; Nagel, W.; Schiller, B.; Zeitlmann, L.; Godar, S.; Stockinger, H.; Seed, B. Alpha L beta 2 integrin/LFA-1 binding to ICAM-1 induced by cytohesin-1, a cytoplasmic regulatory molecule. Cell 1996, 86, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Weber, K.S.; Weber, C.; Ostermann, G.; Dierks, H.; Nagel, W.; Kolanus, W. Cytohesin-1 is a dynamic regulator of distinct LFA-1 functions in leukocyte arrest and transmigration triggered by chemokines. Curr. Biol. 2001, 11, 1969–1974. [Google Scholar] [CrossRef] [Green Version]
- Boehm, T.; Hofer, S.; Winklehner, P.; Kellersch, B.; Geiger, C.; Trockenbacher, A.; Neyer, S.; Fiegl, H.; Ebner, S.; Ivarsson, L.; et al. Attenuation of cell adhesion in lymphocytes is regulated by CYTIP, a protein which mediates signal complex sequestration. EMBO J. 2003, 22, 1014–1024. [Google Scholar] [CrossRef] [Green Version]
- Theodoridis, A.A.; Eich, C.; Figdor, C.G.; Steinkasserer, A. Infection of dendritic cells with herpes simplex virus type 1 induces rapid degradation of CYTIP, thereby modulating adhesion and migration. Blood 2011, 118, 107–115. [Google Scholar] [CrossRef]
- Hofer, S.; Pfeil, K.; Niederegger, H.; Ebner, S.; Nguyen, V.A.; Kremmer, E.; Auffinger, M.; Neyer, S.; Furhapter, C.; Heufler, C. Dendritic cells regulate T-cell deattachment through the integrin-interacting protein CYTIP. Blood 2006, 107, 1003–1009. [Google Scholar] [CrossRef] [Green Version]
- Paz-Bailey, G.; Ramaswamy, M.; Hawkes, S.J.; Geretti, A.M. Herpes simplex virus type 2: Epidemiology and management options in developing countries. Postgrad. Med. J. 2008, 84, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Robinson, N.J. Age-specific prevalence of infection with herpes simplex virus types 2 and 1: A global review. J. Infect. Dis. 2002, 186 (Suppl. 1), S3–S28. [Google Scholar] [CrossRef]
- Weiss, H. Epidemiology of herpes simplex virus type 2 infection in the developing world. J. IHMF 2004, 11 (Suppl. 1), 24A–35A. [Google Scholar]
- Whitley, R.J.; Roizman, B. Herpes simplex virus infections. Lancet 2001, 357, 1513–1518. [Google Scholar] [CrossRef]
- Looker, K.J.; Magaret, A.S.; May, M.T.; Turner, K.M.; Vickerman, P.; Gottlieb, S.L.; Newman, L.M. Global and Regional Estimates of Prevalent and Incident Herpes Simplex Virus Type 1 Infections in 2012. PLoS ONE 2015, 10, e0140765. [Google Scholar] [CrossRef] [Green Version]
- Looker, K.J.; Magaret, A.S.; Turner, K.M.; Vickerman, P.; Gottlieb, S.L.; Newman, L.M. Global estimates of prevalent and incident herpes simplex virus type 2 infections in 2012. PLoS ONE 2015, 10, e114989. [Google Scholar] [CrossRef] [Green Version]
- Nahmias, A.J.; Keyserling, H.; Lee, F.K. Herpes Simplex Viruses 1 and 2. In Viral Infections of Humans: Epidemiology and Control, 3rd ed.; Evans, A.S., Ed.; Springer: Boston, MA, USA, 1989; pp. 393–418. [Google Scholar] [CrossRef]
- Arduino, P.G.; Porter, S.R. Herpes Simplex Virus Type 1 infection: Overview on relevant clinico-pathological features. J. Oral Pathol. Med. 2008, 37, 107–121. [Google Scholar] [CrossRef]
- Whitley, R.J.; Miller, R.L. Immunologic approach to herpes simplex virus. Viral Immunol. 2001, 14, 111–118. [Google Scholar] [CrossRef]
- Freeman, E.E.; Weiss, H.A.; Glynn, J.R.; Cross, P.L.; Whitworth, J.A.; Hayes, R.J. Herpes simplex virus 2 infection increases HIV acquisition in men and women: Systematic review and meta-analysis of longitudinal studies. AIDS 2006, 20, 73–83. [Google Scholar] [CrossRef]
- Lieveld, M.; Carregosa, A.; Benoy, I.; Redzic, N.; Berth, M.; Vanden Broeck, D. A high resolution melting (HRM) technology-based assay for cost-efficient clinical detection and genotyping of herpes simplex virus (HSV)-1 and HSV-2. J. Virol. Methods 2017, 248, 181–186. [Google Scholar] [CrossRef]
- Wald, A. Genital HSV-1 infections. Sex. Transm. Infect. 2006, 82, 189–190. [Google Scholar] [CrossRef]
- Whitley, R.; Baines, J. Clinical management of herpes simplex virus infections: Past, present, and future. F1000Res. 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Koelle, D.M.; Corey, L. Herpes simplex: Insights on pathogenesis and possible vaccines. Annu. Rev. Med. 2008, 59, 381–395. [Google Scholar] [CrossRef]
- Tan, I.L.; McArthur, J.C.; Venkatesan, A.; Nath, A. Atypical manifestations and poor outcome of herpes simplex encephalitis in the immunocompromised. Neurology 2012, 79, 2125–2132. [Google Scholar] [CrossRef] [Green Version]
- Honess, R.W.; Roizman, B. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 1974, 14, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Diefenbach, R.J.; Miranda-Saksena, M.; Douglas, M.W.; Cunningham, A.L. Transport and egress of herpes simplex virus in neurons. Rev. Med. Virol. 2008, 18, 35–51. [Google Scholar] [CrossRef]
- Enquist, L.W.; Husak, P.J.; Banfield, B.W.; Smith, G.A. Infection and spread of alphaherpesviruses in the nervous system. Adv. Virus Res. 1998, 51, 237–347. [Google Scholar] [CrossRef]
- Moutaftsi, M.; Mehl, A.M.; Borysiewicz, L.K.; Tabi, Z. Human cytomegalovirus inhibits maturation and impairs function of monocyte-derived dendritic cells. Blood 2002, 99, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Senechal, B.; Boruchov, A.M.; Reagan, J.L.; Hart, D.N.; Young, J.W. Infection of mature monocyte-derived dendritic cells with human cytomegalovirus inhibits stimulation of T-cell proliferation via the release of soluble CD83. Blood 2004, 103, 4207–4215. [Google Scholar] [CrossRef] [Green Version]
- Heilingloh, C.S.; Grosche, L.; Kummer, M.; Mühl-Zürbes, P.; Kamm, L.; Scherer, M.; Latzko, M.; Stamminger, T.; Steinkasserer, A. The Major Immediate-Early Protein IE2 of Human Cytomegalovirus Is Sufficient to Induce Proteasomal Degradation of CD83 on Mature Dendritic Cells. Front. Microbiol. 2017, 8, 119. [Google Scholar] [CrossRef]
- Grosche, L.; Drassner, C.; Mühl-Zürbes, P.; Kamm, L.; Le-Trilling, V.T.K.; Trilling, M.; Steinkasserer, A.; Heilingloh, C.S. Human Cytomegalovirus-Induced Degradation of CYTIP Modulates Dendritic Cell Adhesion and Migration. Front. Immunol. 2017, 8, 461. [Google Scholar] [CrossRef]
- Pfeiffer, I.A.; Zinser, E.; Strasser, E.; Stein, M.F.; Dörrie, J.; Schaft, N.; Steinkasserer, A.; Knippertz, I. Leukoreduction system chambers are an efficient, valid, and economic source of functional monocyte-derived dendritic cells and lymphocytes. Immunobiology 2013, 218, 1392–1401. [Google Scholar] [CrossRef]
- Düthorn, A.; Turan, A.; Drassner, C.; Mühl-Zürbes, P.; Heilingloh, C.S.; Steinkasserer, A.; Grosche, L. siRNA Electroporation to Modulate Autophagy in Herpes Simplex Virus Type 1-Infected Monocyte-Derived Dendritic Cells. J. Vis. Exp. 2019, e60190. [Google Scholar] [CrossRef] [PubMed]
- Turan, A.; Grosche, L.; Krawczyk, A.; Mühl-Zürbes, P.; Drassner, C.; Düthorn, A.; Kummer, M.; Hasenberg, M.; Voortmann, S.; Jastrow, H.; et al. Autophagic degradation of lamins facilitates the nuclear egress of herpes simplex virus type 1. J. Cell Biol. 2019, 218, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Coffin, R.S.; MacLean, A.R.; Latchman, D.S.; Brown, S.M. Gene delivery to the central and peripheral nervous systems of mice using HSV1 ICP34.5 deletion mutant vectors. Gene Ther. 1996, 3, 886–891. [Google Scholar]
- Coffin, R.S.; Thomas, S.K.; Thomas, N.S.; Lilley, C.E.; Pizzey, A.R.; Griffiths, C.H.; Gibb, B.J.; Wagstaff, M.J.; Inges, S.J.; Binks, M.H.; et al. Pure populations of transduced primary human cells can be produced using GFP expressing herpes virus vectors and flow cytometry. Gene Ther. 1998, 5, 718–722. [Google Scholar] [CrossRef] [Green Version]
- Grosche, L.; Döhner, K.; Düthorn, A.; Hickford-Martinez, A.; Steinkasserer, A.; Sodeik, B. Herpes Simplex Virus Type 1 Propagation, Titration and Single-step Growth Curves. Bio-Protocol 2019, 9, e3441. [Google Scholar] [CrossRef]
- Guan, S.; Cheng, M.; Law, S.K. The integrin alphaL leg region controls the Mg/EGTA mediated activation of LFA-1. Biochem. Biophys. Res. Commun. 2015, 458, 251–255. [Google Scholar] [CrossRef]
- Prechtel, A.T.; Turza, N.M.; Kobelt, D.J.; Eisemann, J.I.; Coffin, R.S.; McGrath, Y.; Hacker, C.; Ju, X.; Zenke, M.; Steinkasserer, A. Infection of mature dendritic cells with herpes simplex virus type 1 dramatically reduces lymphoid chemokine-mediated migration. J. Gen. Virol. 2005, 86, 1645–1657. [Google Scholar] [CrossRef]
- Salio, M.; Cella, M.; Suter, M.; Lanzavecchia, A. Inhibition of dendritic cell maturation by herpes simplex virus. Eur. J. Immunol. 1999, 29, 3245–3253. [Google Scholar] [CrossRef]
- Stefanidou, M.; Ramos, I.; Mas, C.V.; Trepanier, J.B.; Rosenbaum, S.; Fernandez-Sesma, A.; Herold, B.C. Herpes simplex virus 2 (HSV-2) prevents dendritic cell maturation, induces apoptosis, and triggers release of proinflammatory cytokines: Potential links to HSV-HIV synergy. J. Virol. 2013, 87, 1443–1453. [Google Scholar] [CrossRef] [Green Version]
- Kummer, M.; Turza, N.M.; Mühl-Zürbes, P.; Lechmann, M.; Boutell, C.; Coffin, R.S.; Everett, R.D.; Steinkasserer, A.; Prechtel, A.T. Herpes simplex virus type 1 induces CD83 degradation in mature dendritic cells with immediate-early kinetics via the cellular proteasome. J. Virol. 2007, 81, 6326–6338. [Google Scholar] [CrossRef] [Green Version]
- Bosnjak, L.; Jones, C.A.; Abendroth, A.; Cunningham, A.L. Dendritic cell biology in herpesvirus infections. Viral Immunol. 2005, 18, 419–433. [Google Scholar] [CrossRef]
- Hogg, N.; Bennett, R.; Cabanas, C.; Dransfield, I. Leukocyte integrin activation. Kidney Int. 1992, 41, 613–616. [Google Scholar] [CrossRef] [Green Version]
- Hogg, N.; Selvendran, Y. An anti-human monocyte/macrophage monoclonal antibody, reacting most strongly with macrophages in lymphoid tissue. Cell Immunol. 1985, 92, 247–253. [Google Scholar] [CrossRef]
- Kamata, T.; Tieu, K.K.; Tarui, T.; Puzon-McLaughlin, W.; Hogg, N.; Takada, Y. The role of the CPNKEKEC sequence in the beta(2) subunit I domain in regulation of integrin alpha(L)beta(2) (LFA-1). J. Immunol. 2002, 168, 2296–2301. [Google Scholar] [CrossRef] [Green Version]
- Salas, A.; Shimaoka, M.; Kogan, A.N.; Harwood, C.; von Andrian, U.H.; Springer, T.A. Rolling adhesion through an extended conformation of integrin alphaLbeta2 and relation to alpha I and beta I-like domain interaction. Immunity 2004, 20, 393–406. [Google Scholar] [CrossRef] [Green Version]
- Cristea, I.M. The Host-Pathogen Ecosystem Viewed Through the Prism of Proteomics. Mol. Cell Proteom. 2017, 16, S1–S4. [Google Scholar] [CrossRef] [Green Version]
- Sironi, M.; Cagliani, R.; Forni, D.; Clerici, M. Evolutionary insights into host-pathogen interactions from mammalian sequence data. Nat. Rev. Genet. 2015, 16, 224–236. [Google Scholar] [CrossRef]
- Berard, A.R.; Coombs, K.M.; Severini, A. Quantification of the host response proteome after herpes simplex virus type 1 infection. J. Proteome Res. 2015, 14, 2121–2142. [Google Scholar] [CrossRef]
- Tognarelli, E.I.; Palomino, T.F.; Corrales, N.; Bueno, S.M.; Kalergis, A.M.; Gonzalez, P.A. Herpes Simplex Virus Evasion of Early Host Antiviral Responses. Front. Cell Infect. Microbiol. 2019, 9, 127. [Google Scholar] [CrossRef]
- Cunningham, A.L.; Diefenbach, R.J.; Miranda-Saksena, M.; Bosnjak, L.; Kim, M.; Jones, C.; Douglas, M.W. The cycle of human herpes simplex virus infection: Virus transport and immune control. J. Infect. Dis. 2006, 194 (Suppl. 1), S11–S18. [Google Scholar] [CrossRef] [Green Version]
- Beck, K.; Meyer-Konig, U.; Weidmann, M.; Nern, C.; Hufert, F.T. Human cytomegalovirus impairs dendritic cell function: A novel mechanism of human cytomegalovirus immune escape. Eur. J. Immunol. 2003, 33, 1528–1538. [Google Scholar] [CrossRef]
- Mikloska, Z.; Danis, V.A.; Adams, S.; Lloyd, A.R.; Adrian, D.L.; Cunningham, A.L. In vivo production of cytokines and beta (C-C) chemokines in human recurrent herpes simplex lesions--do herpes simplex virus-infected keratinocytes contribute to their production? J. Infect. Dis. 1998, 177, 827–838. [Google Scholar] [CrossRef]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef]
- Alcami, A.; Koszinowski, U.H. Viral mechanisms of immune evasion. Trends Microbiol. 2000, 8, 410–418. [Google Scholar] [CrossRef]
- Kruse, M.; Rosorius, O.; Kratzer, F.; Stelz, G.; Kuhnt, C.; Schuler, G.; Hauber, J.; Steinkasserer, A. Mature dendritic cells infected with herpes simplex virus type 1 exhibit inhibited T-cell stimulatory capacity. J. Virol. 2000, 74, 7127–7136. [Google Scholar] [CrossRef] [Green Version]
- Mikloska, Z.; Bosnjak, L.; Cunningham, A.L. Immature monocyte-derived dendritic cells are productively infected with herpes simplex virus type 1. J. Virol. 2001, 75, 5958–5964. [Google Scholar] [CrossRef] [Green Version]
- Goldwich, A.; Prechtel, A.T.; Mühl-Zürbes, P.; Pangratz, N.M.; Stossel, H.; Romani, N.; Steinkasserer, A.; Kummer, M. Herpes simplex virus type I (HSV-1) replicates in mature dendritic cells but can only be transferred in a cell-cell contact-dependent manner. J. Leukoc. Biol. 2011, 89, 973–979. [Google Scholar] [CrossRef]
- Prechtel, A.T.; Turza, N.M.; Theodoridis, A.A.; Steinkasserer, A. CD83 knockdown in monocyte-derived dendritic cells by small interfering RNA leads to a diminished T cell stimulation. J. Immunol. 2007, 178, 5454–5464. [Google Scholar] [CrossRef] [Green Version]
- Heilingloh, C.S.; Mühl-Zürbes, P.; Steinkasserer, A.; Kummer, M. Herpes simplex virus type 1 ICP0 induces CD83 degradation in mature dendritic cells independent of its E3 ubiquitin ligase function. J. Gen. Virol. 2014, 95, 1366–1375. [Google Scholar] [CrossRef]
- Morrow, G.; Slobedman, B.; Cunningham, A.L.; Abendroth, A. Varicella-zoster virus productively infects mature dendritic cells and alters their immune function. J. Virol. 2003, 77, 4950–4959. [Google Scholar] [CrossRef] [Green Version]
- Allan, R.S.; Waithman, J.; Bedoui, S.; Jones, C.M.; Villadangos, J.A.; Zhan, Y.; Lew, A.M.; Shortman, K.; Heath, W.R.; Carbone, F.R. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity 2006, 25, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprecher, E.; Becker, Y. Langerhans cell density and activity in mouse skin and lymph nodes affect herpes simplex type 1 (HSV-1) pathogenicity. Arch. Virol. 1989, 107, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Bedoui, S.; Greyer, M. The role of dendritic cells in immunity against primary herpes simplex virus infections. Front. Microbiol. 2014, 5, 533. [Google Scholar] [CrossRef]
- Eidsmo, L.; Allan, R.; Caminschi, I.; van Rooijen, N.; Heath, W.R.; Carbone, F.R. Differential migration of epidermal and dermal dendritic cells during skin infection. J. Immunol. 2009, 182, 3165–3172. [Google Scholar] [CrossRef]
- Hor, J.L.; Whitney, P.G.; Zaid, A.; Brooks, A.G.; Heath, W.R.; Mueller, S.N. Spatiotemporally Distinct Interactions with Dendritic Cell Subsets Facilitates CD4+ and CD8+ T Cell Activation to Localized Viral Infection. Immunity 2015, 43, 554–565. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.K.; Zamora, M.; Linehan, M.M.; Iijima, N.; Gonzalez, D.; Haberman, A.; Iwasaki, A. Differential roles of migratory and resident DCs in T cell priming after mucosal or skin HSV-1 infection. J. Exp. Med. 2009, 206, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Villadangos, J.A.; Schnorrer, P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat. Rev. Immunol. 2007, 7, 543–555. [Google Scholar] [CrossRef]
- Zhao, X.; Deak, E.; Soderberg, K.; Linehan, M.; Spezzano, D.; Zhu, J.; Knipe, D.M.; Iwasaki, A. Vaginal submucosal dendritic cells, but not Langerhans cells, induce protective Th1 responses to herpes simplex virus-2. J. Exp. Med. 2003, 197, 153–162. [Google Scholar] [CrossRef]
- Schönrich, G.; Raftery, M.J. Dendritic cells as Achilles’ heel and Trojan horse during varicella zoster virus infection. Front. Microbiol. 2015, 6, 417. [Google Scholar] [CrossRef]
- Johansson, S.; Svineng, G.; Wennerberg, K.; Armulik, A.; Lohikangas, L. Fibronectin-integrin interactions. Front. Biosci. 1997, 2, 126–146. [Google Scholar] [CrossRef] [Green Version]
- Harris, E.S.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A. The leukocyte integrins. J. Biol. Chem. 2000, 275, 23409–23412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Kooyk, Y.; Figdor, C.G. Avidity regulation of integrins: The driving force in leukocyte adhesion. Curr. Opin. Cell Biol. 2000, 12, 542–547. [Google Scholar] [CrossRef]
- Quast, T.; Tappertzhofen, B.; Schild, C.; Grell, J.; Czeloth, N.; Förster, R.; Alon, R.; Fraemohs, L.; Dreck, K.; Weber, C.; et al. Cytohesin-1 controls the activation of RhoA and modulates integrin-dependent adhesion and migration of dendritic cells. Blood 2009, 113, 5801–5810. [Google Scholar] [CrossRef] [Green Version]
- Kolanus, W. Guanine nucleotide exchange factors of the cytohesin family and their roles in signal transduction. Immunol. Rev. 2007, 218, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Wegener, K.L.; Partridge, A.W.; Han, J.; Pickford, A.R.; Liddington, R.C.; Ginsberg, M.H.; Campbell, I.D. Structural basis of integrin activation by talin. Cell 2007, 128, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Critchley, D.R.; Gingras, A.R. Talin at a glance. J. Cell Sci. 2008, 121, 1345–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kliche, S.; Nagel, W.; Kremmer, E.; Atzler, C.; Ege, A.; Knorr, T.; Koszinowski, U.; Kolanus, W.; Haas, J. Signaling by human herpesvirus 8 kaposin A through direct membrane recruitment of cytohesin-1. Mol. Cell 2001, 7, 833–843. [Google Scholar] [CrossRef]
- Benedict, C.A.; Arens, R.; Loewendorf, A.; Janssen, E.M. Modulation of T-Cell Mediated Immunity by Cytomegalovirus. In Control of Innate and Adaptive Immune Responses during Infectious Diseases; Aliberti, J., Ed.; Springer: New York, NY, USA, 2012; pp. 121–139. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grosche, L.; Mühl-Zürbes, P.; Ciblis, B.; Krawczyk, A.; Kuhnt, C.; Kamm, L.; Steinkasserer, A.; Heilingloh, C.S. Herpes Simplex Virus Type-2 Paralyzes the Function of Monocyte-Derived Dendritic Cells. Viruses 2020, 12, 112. https://doi.org/10.3390/v12010112
Grosche L, Mühl-Zürbes P, Ciblis B, Krawczyk A, Kuhnt C, Kamm L, Steinkasserer A, Heilingloh CS. Herpes Simplex Virus Type-2 Paralyzes the Function of Monocyte-Derived Dendritic Cells. Viruses. 2020; 12(1):112. https://doi.org/10.3390/v12010112
Chicago/Turabian StyleGrosche, Linda, Petra Mühl-Zürbes, Barbara Ciblis, Adalbert Krawczyk, Christine Kuhnt, Lisa Kamm, Alexander Steinkasserer, and Christiane Silke Heilingloh. 2020. "Herpes Simplex Virus Type-2 Paralyzes the Function of Monocyte-Derived Dendritic Cells" Viruses 12, no. 1: 112. https://doi.org/10.3390/v12010112