In Vitro Hepatitis C Virus Infection and Hepatic Choline Metabolism

 , , , , , and
, , , , , and
Abstract
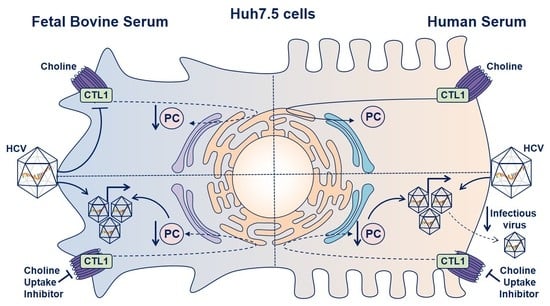
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Huh7.5 Cell Culture
2.2. JFH-1 HCV Infection Protocol
2.3. JFH-1 HCV Titration
2.4. [3H]-Choline Uptake Experiments
2.5. [3H]-Choline Incorporation into PC
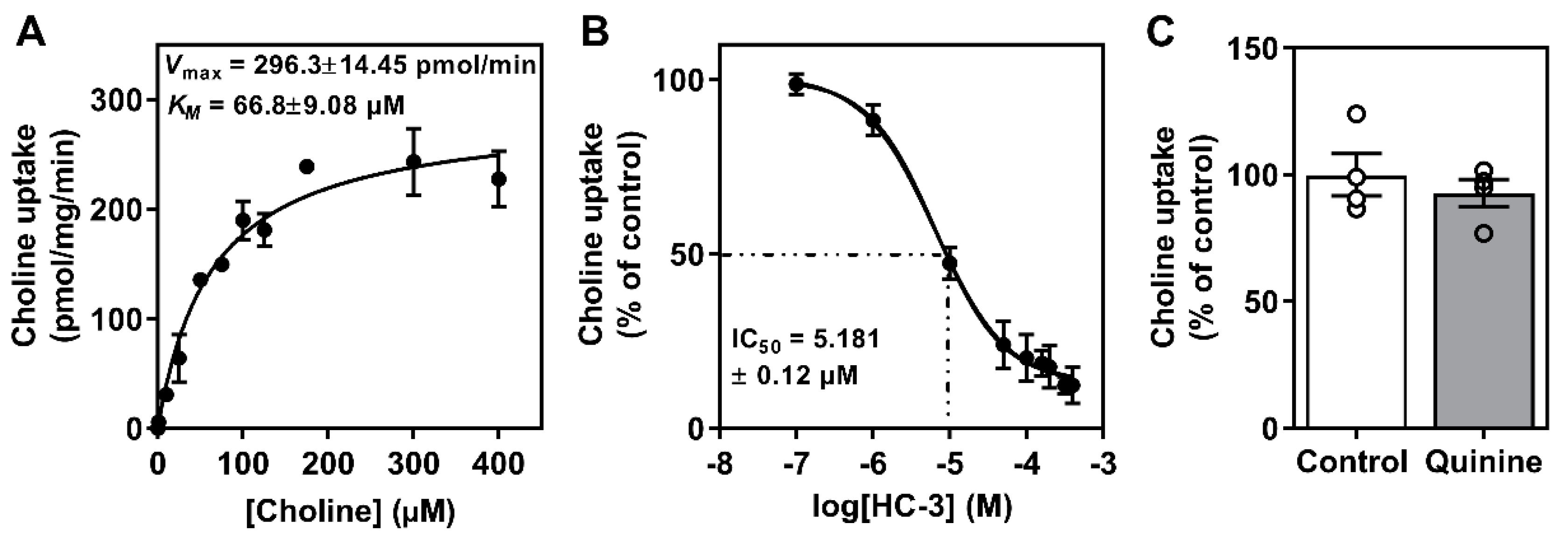
2.6. Choline Inhibition by HC3
2.7. RNA Isolation and Quantification
2.8. Immunoblotting
2.9. Statistics
3. Results
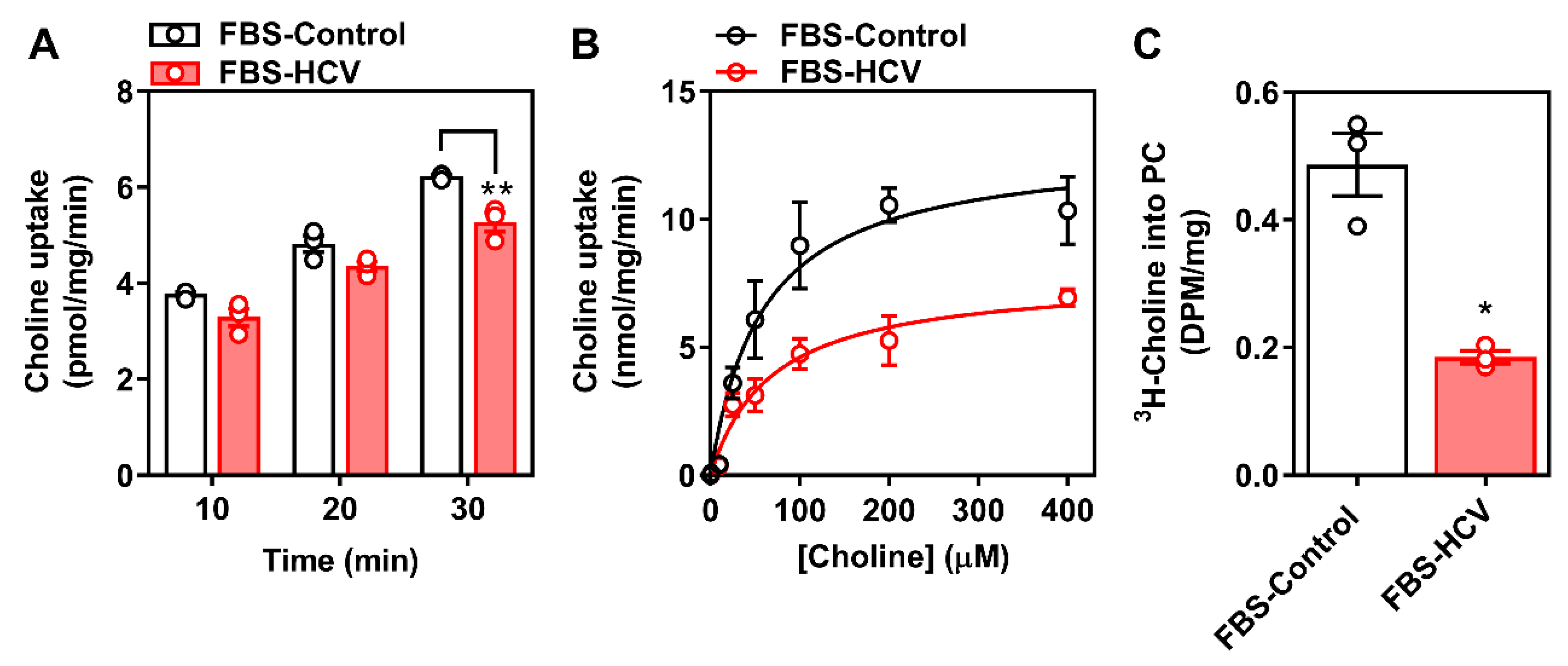
3.1. Determination of Choline Transport Kinetics
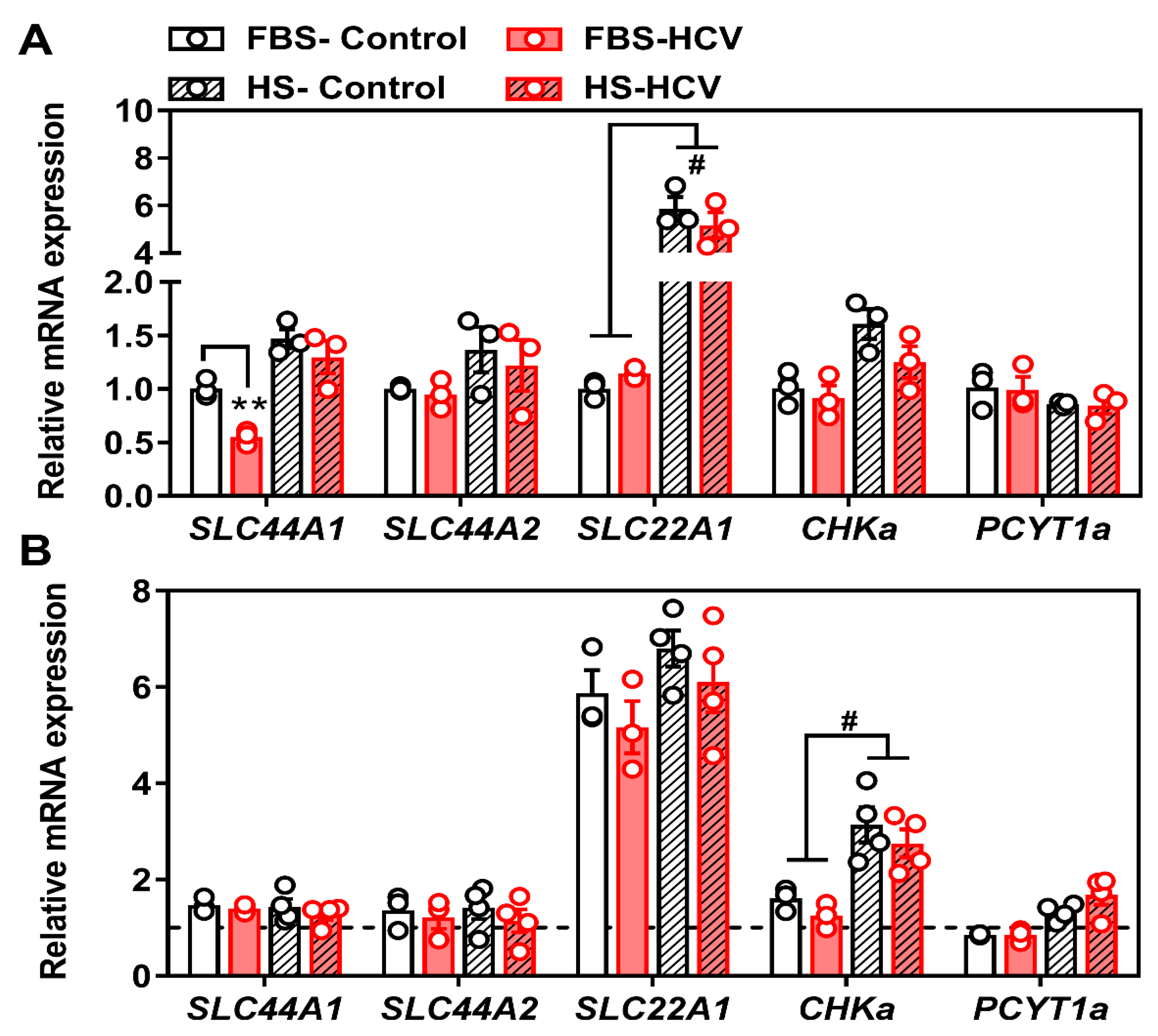
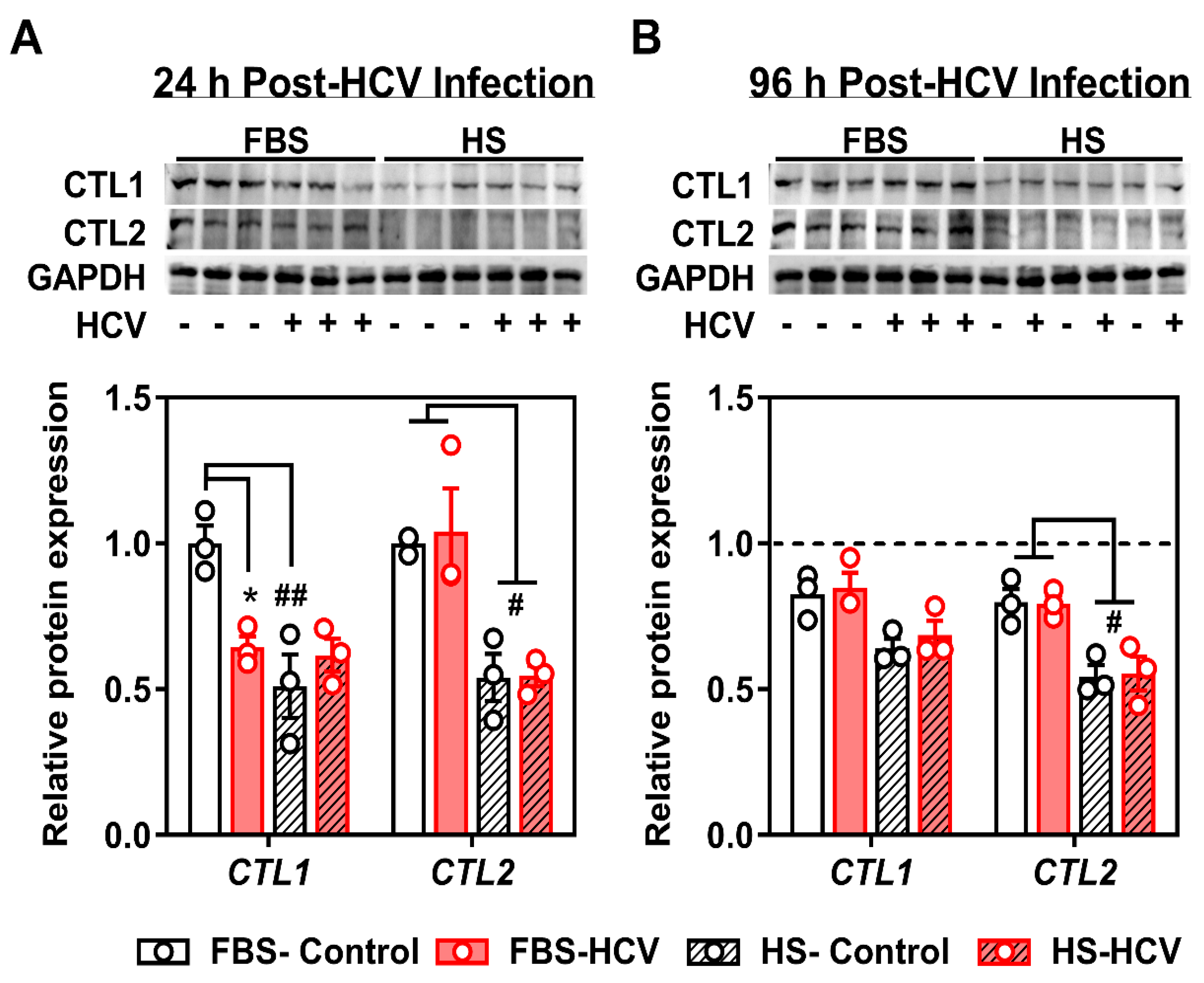
3.2. Effects of In Vitro HCV Infection on Choline Transporter Expression and Transport
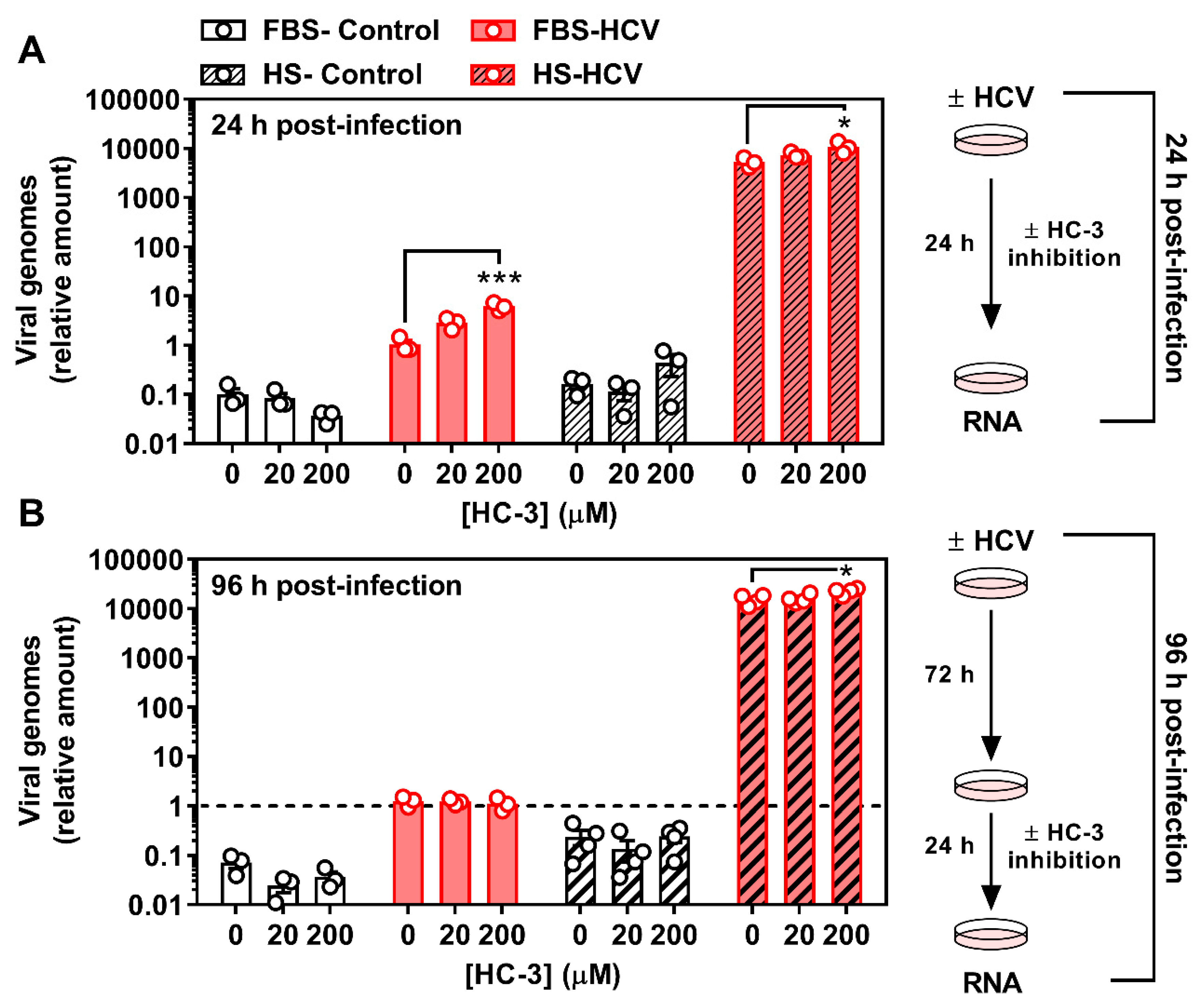
3.3. Inhibiting Choline Uptake Alters Early HCV Replication
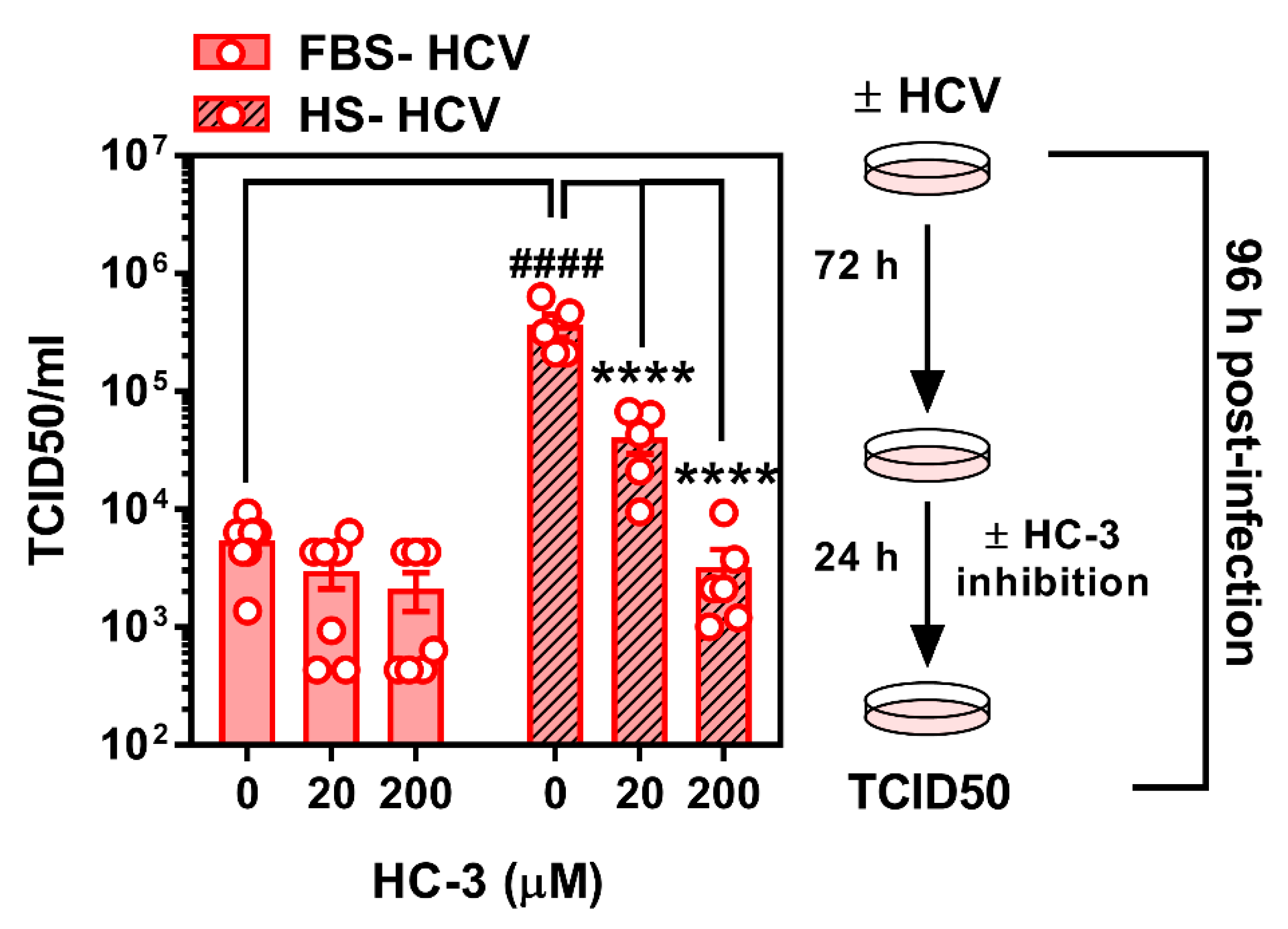
3.4. Limiting Choline Availability during HCV Infection Inhibits Viral Infectivity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- MacDonald, M.; Crofts, N.; Kaldor, J. Transmission of hepatitis C virus: Rates, routes, and cofactors. Epidemiol. Rev. 1996, 18, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.L. (Ed.) Hepatitis C Viruses: Genomes and Molecular Biology; Horizon Bioscience Press: Norfolk, UK, 2006. [Google Scholar]
- World Health Organization. Guidelines for the Care and Treatment of Persons Diagnosed with Chronic Hepatitis C Virus Infection; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Lavanchy, D. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 2011, 17, 107–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Vance, D.E. Phosphatidylcholine and choline homeostasis. J. Lipid Res. 2008, 49, 1187–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisel, S.H. Nutritional importance of choline for brain development. J. Am. Coll. Nutr. 2004, 23, 621S–626S. [Google Scholar] [CrossRef]
- Zeisel, S.H. Choline: Critical role during fetal development and dietary requirements in adults. Annu. Rev. Nutr. 2006, 26, 229–250. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, C.J.; Chi, K.D.; Subramanian, V.; Ward, K.L.; Green, R.M. Functional expression of a high affinity mammalian hepatic choline/organic cation transporter. J. Lipid Res. 2000, 41, 1841–1848. [Google Scholar]
- Michel, V.; Yuan, Z.; Ramsubir, S.; Bakovic, M. Choline transport for phospholipid synthesis. Exp. Biol. Med. 2006, 231, 490–504. [Google Scholar] [CrossRef]
- Vance, J.E. Phospholipid synthesis and transport in mammalian cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef]
- Kaplan, C.P.; Porter, R.K.; Brand, M.D. The choline transporter is the major site of control of choline oxidation in isolated rat liver mitochondria. FEBS Lett. 1993, 321, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, E.P.; Weiss, S.B. The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar]
- Sundler, R.; Akesson, B. Regulation of phospholipid biosynthesis in isolated rat hepatocytes. Effect of different substrates. J. Biol. Chem. 1975, 250, 3359–3367. [Google Scholar] [PubMed]
- Reo, N.V.; Adinehzadeh, M.; Foy, B.D. Kinetic analyses of liver phosphatidylcholine and phosphatidylethanolamine biosynthesis using 13C NMR spectroscopy. Biochim. Biophys. Acta 2002, 1580, 171–188. [Google Scholar] [CrossRef]
- DeLong, C.J.; Shen, Y.J.; Thomas, M.J.; Cui, Z. Molecular distinction of phosphatidylcholine synthesis between the CDP-choline pathway and phosphatidylethanolamine methylation pathway. J. Biol. Chem. 1999, 274, 29683–29688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhang, Z.; Chukkapalli, V.; Nchoutmboube, J.A.; Li, J.; Randall, G.; Belov, G.A.; Wang, X. Positive-strand RNA viruses stimulate host phosphatidylcholine synthesis at viral replication sites. Proc. Natl. Acad. Sci. USA 2016, 113, E1064–E1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, D.E.; Trip, E.M.; Paddon, H.B. Poliovirus increases phosphatidylcholine biosynthesis in HeLa cells by stimulation of the rate-limiting reaction catalyzed by CTP: Phosphocholine cytidylyltransferase. J. Biol. Chem. 1980, 255, 1064–1069. [Google Scholar] [PubMed]
- Perera, R.; Riley, C.; Isaac, G.; Hopf-Jannasch, A.S.; Moore, R.J.; Weitz, K.W.; Pasa-Tolic, L.; Metz, T.O.; Adamec, J.; Kuhn, R.J. Dengue virus infection perturbs lipid homeostasis in infected mosquito cells. PLoS Pathog. 2012, 8, e1002584. [Google Scholar] [CrossRef]
- Lykidis, A.; Jackowski, S. Regulation of mammalian cell membrane biosynthesis. Prog. Nucleic Acid Res. Mol. Biol. 2001, 65, 361–393. [Google Scholar]
- Reiss, S.; Rebhan, I.; Backes, P.; Romero-Brey, I.; Erfle, H.; Matula, P.; Kaderali, L.; Poenisch, M.; Blankenburg, H.; Hiet, M.S.; et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 2011, 9, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.T.; Chen, S.S. Hepatitis C Virus Subverts Human Choline Kinase-alpha To Bridge Phosphatidylinositol-4-Kinase IIIalpha (PI4KIIIalpha) and NS5A and Upregulates PI4KIIIalpha Activation, Thereby Promoting the Translocation of the Ternary Complex to the Endoplasmic Reticulum for Viral Replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Su, B.; Jacobs, R.L.; Kennedy, B.; Francis, G.A.; Waddington, E.; Brosnan, J.T.; Vance, J.E.; Vance, D.E. Lack of phosphatidylethanolamine N-methyltransferase alters plasma VLDL phospholipids and attenuates atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1349–1355. [Google Scholar] [CrossRef] [Green Version]
- Pécheur, E.I. Lipoprotein receptors and lipid enzymes in hepatitis C virus entry and early steps of infection. Science 2012, 2012, 709853. [Google Scholar] [CrossRef] [PubMed]
- Grassi, G.; Di Caprio, G.; Fimia, G.M.; Ippolito, G.; Tripodi, M.; Alonzi, T. Hepatitis C virus relies on lipoproteins for its life cycle. World J. Gastroenterol. 2016, 22, 1953–1965. [Google Scholar] [CrossRef] [PubMed]
- Steenbergen, R.H.; Joyce, M.A.; Thomas, B.S.; Jones, D.; Law, J.; Russell, R.; Houghton, M.; Tyrrell, D.L. Human serum leads to differentiation of human hepatoma cells, restoration of very-low-density lipoprotein secretion, and a 1000-fold increase in HCV Japanese fulminant hepatitis type 1 titers. Hepatology 2013, 58, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Steenbergen, R.; Oti, M.; Ter Horst, R.; Tat, W.; Neufeldt, C.; Belovodskiy, A.; Chua, T.T.; Cho, W.J.; Joyce, M.; Dutilh, B.E.; et al. Establishing normal metabolism and differentiation in hepatocellular carcinoma cells by culturing in adult human serum. Sci. Rep. 2018, 8, 11685. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B., Jr.; Chisari, F.V. Production of infectious hepatitis C virus by well-differentiated, growth-arrested human hepatoma-derived cells. J. Virol. 2006, 80, 10253–10257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fullerton, M.D.; Wagner, L.; Yuan, Z.; Bakovic, M. Impaired trafficking of choline transporter-like protein-1 at plasma membrane and inhibition of choline transport in THP-1 monocyte-derived macrophages. Am. J. Physiol.-Cell Physiol. 2006, 290, C1230–C1238. [Google Scholar] [CrossRef] [Green Version]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Fullerton, M.D.; Hakimuddin, F.; Bonen, A.; Bakovic, M. The development of a metabolic disease phenotype in CTP:phosphoethanolamine cytidylyltransferase-deficient mice. J. Biol. Chem. 2009, 284, 25704–25713. [Google Scholar] [CrossRef] [Green Version]
- Horie, A.; Ishida, K.; Watanabe, Y.; Shibata, K.; Hashimoto, Y. Membrane transport mechanisms of choline in human intestinal epithelial LS180 cells. Biopharm. Drug Dispos. 2014, 35, 532–542. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, V.; Bakovic, M. The solute carrier 44A1 is a mitochondrial protein and mediates choline transport. FASEB J. 2009, 23, 2749–2758. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Y.; Salem, N.; Corn, D.J.; Erokwu, B.; Tian, H.; Wang, F.; Lee, Z. Transport and metabolism of radiolabeled choline in hepatocellular carcinoma. Mol. Pharm. 2010, 7, 2077–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwao, B.; Yara, M.; Hara, N.; Kawai, Y.; Yamanaka, T.; Nishihara, H.; Inoue, T.; Inazu, M. Functional expression of choline transporter like-protein 1 (CTL1) and CTL2 in human brain microvascular endothelial cells. Neurochem. Int. 2016, 93, 40–50. [Google Scholar] [CrossRef]
- Nies, A.T.; Koepsell, H.; Winter, S.; Burk, O.; Klein, K.; Kerb, R.; Zanger, U.M.; Keppler, D.; Schwab, M.; Schaeffeler, E. Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology 2009, 50, 1227–1240. [Google Scholar] [CrossRef]
- Chen, L.; Shu, Y.; Liang, X.; Chen, E.C.; Yee, S.W.; Zur, A.A.; Li, S.; Xu, L.; Keshari, K.R.; Lin, M.J.; et al. OCT1 is a high-capacity thiamine transporter that regulates hepatic steatosis and is a target of metformin. Proc. Natl. Acad. Sci. USA 2014, 111, 9983–9988. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.Q.; Saksena, M.M.; Soriano, V.; Vispo, E.; Saksena, N.K. Differential regulation of cytotoxicity pathway discriminating between HIV, HCV mono- and co-infection identified by transcriptome profiling of PBMCs. Virol. J. 2015, 12, 4. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.; Skibbe, K.; Steinmann, E.; Pfaender, S.; Kuntzen, T.; Megger, D.A.; Groten, S.; Sitek, B.; Lauer, G.M.; Kim, A.Y.; et al. Distinct Escape Pathway by Hepatitis C Virus Genotype 1a from a Dominant CD8+ T Cell Response by Selection of Altered Epitope Processing. J. Virol. 2016, 90, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Cassol, E.; Misra, V.; Holman, A.; Kamat, A.; Morgello, S.; Gabuzda, D. Plasma metabolomics identifies lipid abnormalities linked to markers of inflammation, microbial translocation, and hepatic function in HIV patients receiving protease inhibitors. BMC Infect. Dis. 2013, 13, 203. [Google Scholar] [CrossRef] [Green Version]
- Puri, P.; Sanyal, A.J. Role of obesity, insulin resistance, and steatosis in hepatitis C virus infection. Clin. Liver Dis. 2006, 10, 793–819. [Google Scholar] [CrossRef]
- Sheikh, M.Y.; Choi, J.; Qadri, I.; Friedman, J.E.; Sanyal, A.J. Hepatitis C virus infection: Molecular pathways to metabolic syndrome. Hepatology 2008, 47, 2127–2133. [Google Scholar] [CrossRef] [PubMed]
- Popescu, C.I.; Riva, L.; Vlaicu, O.; Farhat, R.; Rouillé, Y.; Dubuisson, J. Hepatitis C virus life cycle and lipid metabolism. Biology 2014, 3, 892–921. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, E.A.; Chung, R.T. HCV and host lipids: An intimate connection. Semin. Liver Dis. 2013, 33, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.T.; Chen, S.S. Human Choline Kinase-α Promotes Hepatitis C Virus RNA Replication through Modulation of Membranous Viral Replication Complex Formation. J. Virol. 2016, 90, 9075–9095. [Google Scholar] [CrossRef] [Green Version]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010, 6, e1000719. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Choi, B.H.; Jang, K.L.; Min, D.S. Phospholipase D activity is elevated in hepatitis C virus core protein-transformed NIH3T3 mouse fibroblast cells. Exp. Mol. Med. 2004, 36, 454–460. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gobeil Odai, K.; O’Dwyer, C.; Steenbergen, R.; Shaw, T.A.; Renner, T.M.; Ghorbani, P.; Rezaaifar, M.; Han, S.; Langlois, M.-A.; Crawley, A.M.; et al. In Vitro Hepatitis C Virus Infection and Hepatic Choline Metabolism. Viruses 2020, 12, 108. https://doi.org/10.3390/v12010108
Gobeil Odai K, O’Dwyer C, Steenbergen R, Shaw TA, Renner TM, Ghorbani P, Rezaaifar M, Han S, Langlois M-A, Crawley AM, et al. In Vitro Hepatitis C Virus Infection and Hepatic Choline Metabolism. Viruses. 2020; 12(1):108. https://doi.org/10.3390/v12010108
Chicago/Turabian StyleGobeil Odai, Kaelan, Conor O’Dwyer, Rineke Steenbergen, Tyler A. Shaw, Tyler M. Renner, Peyman Ghorbani, Mojgan Rezaaifar, Shauna Han, Marc-André Langlois, Angela M. Crawley, and et al. 2020. "In Vitro Hepatitis C Virus Infection and Hepatic Choline Metabolism" Viruses 12, no. 1: 108. https://doi.org/10.3390/v12010108