Discovering Temporal Patterns in Longitudinal Nontargeted Metabolomics Data via Group and Nuclear Norm Regularized Multivariate Regression


Abstract
:
1. Introduction
2. Results
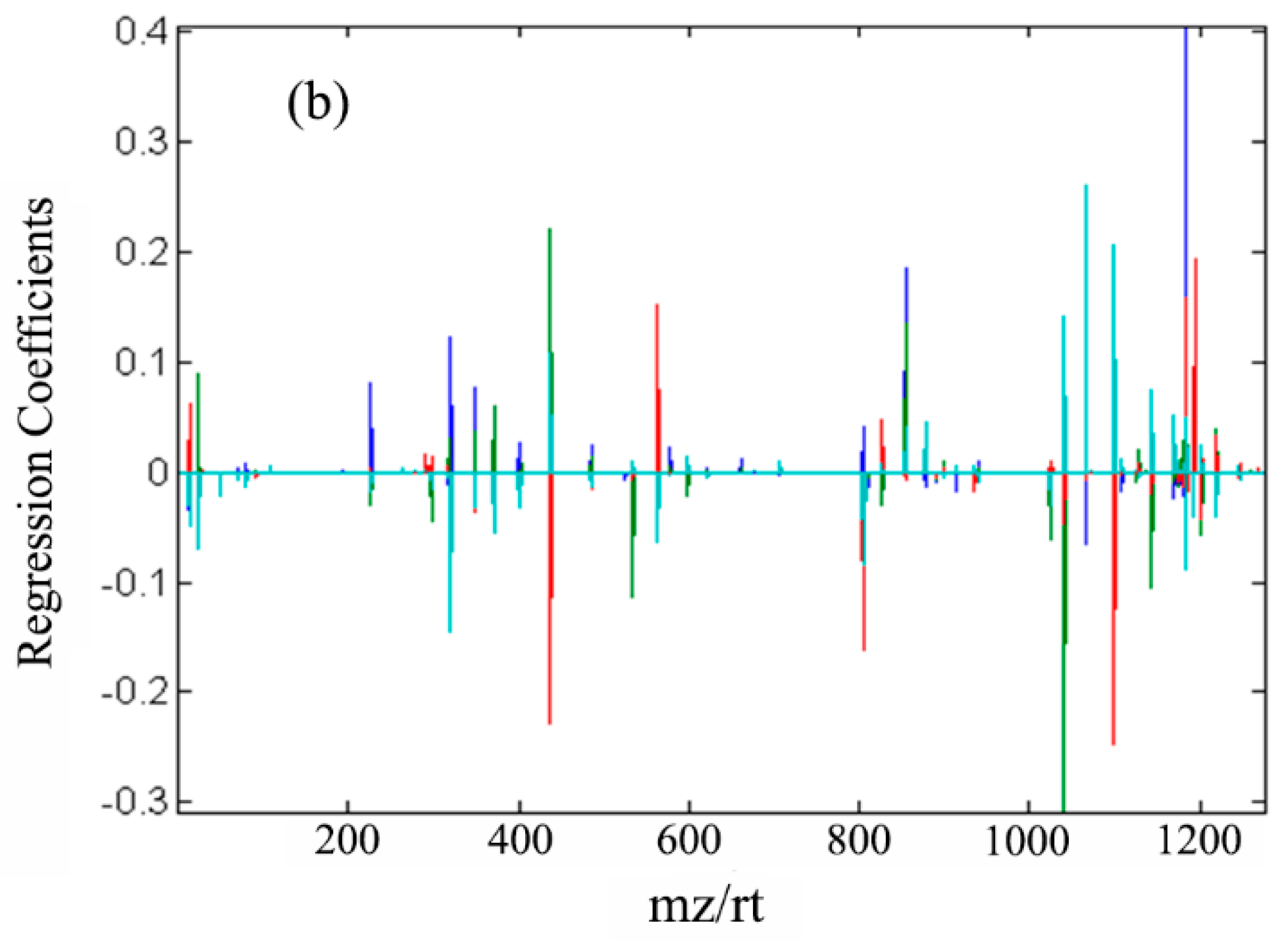
2.1. Cross-Sectional Analysis with PLS-DA
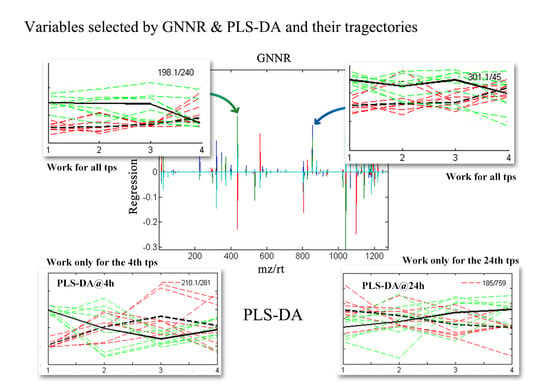
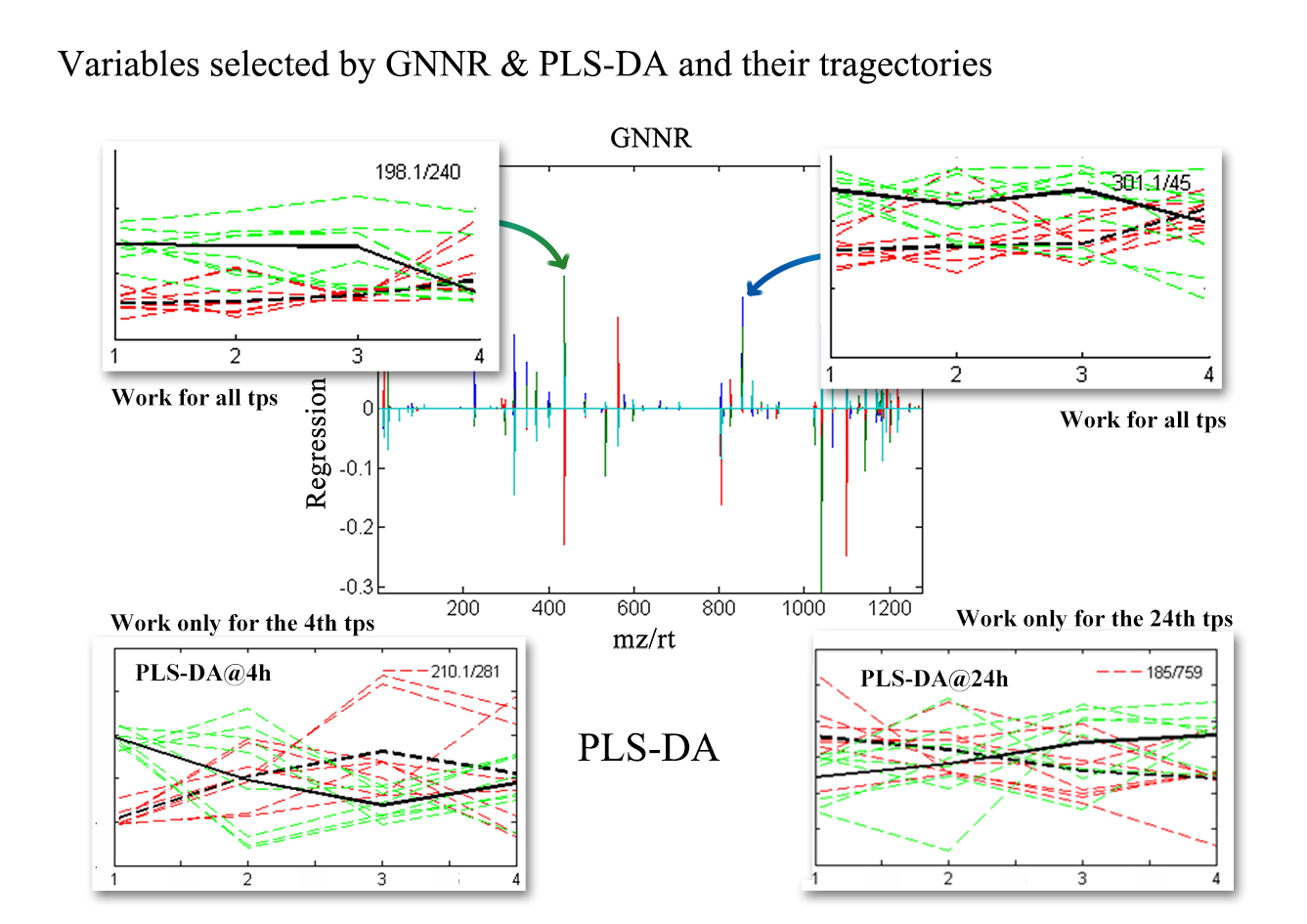
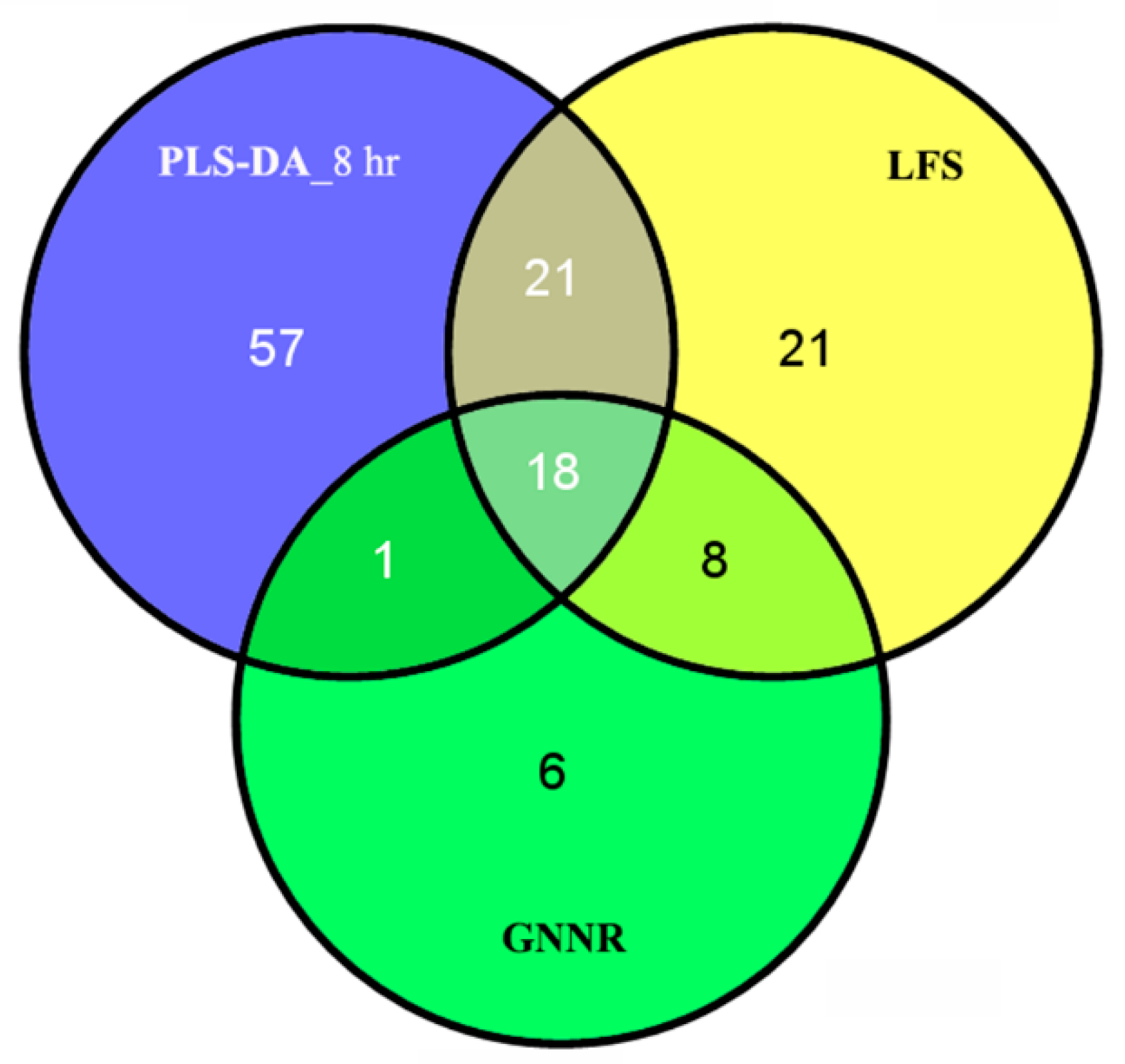
2.2. Discovering Longitudinal Metabolomics Markers
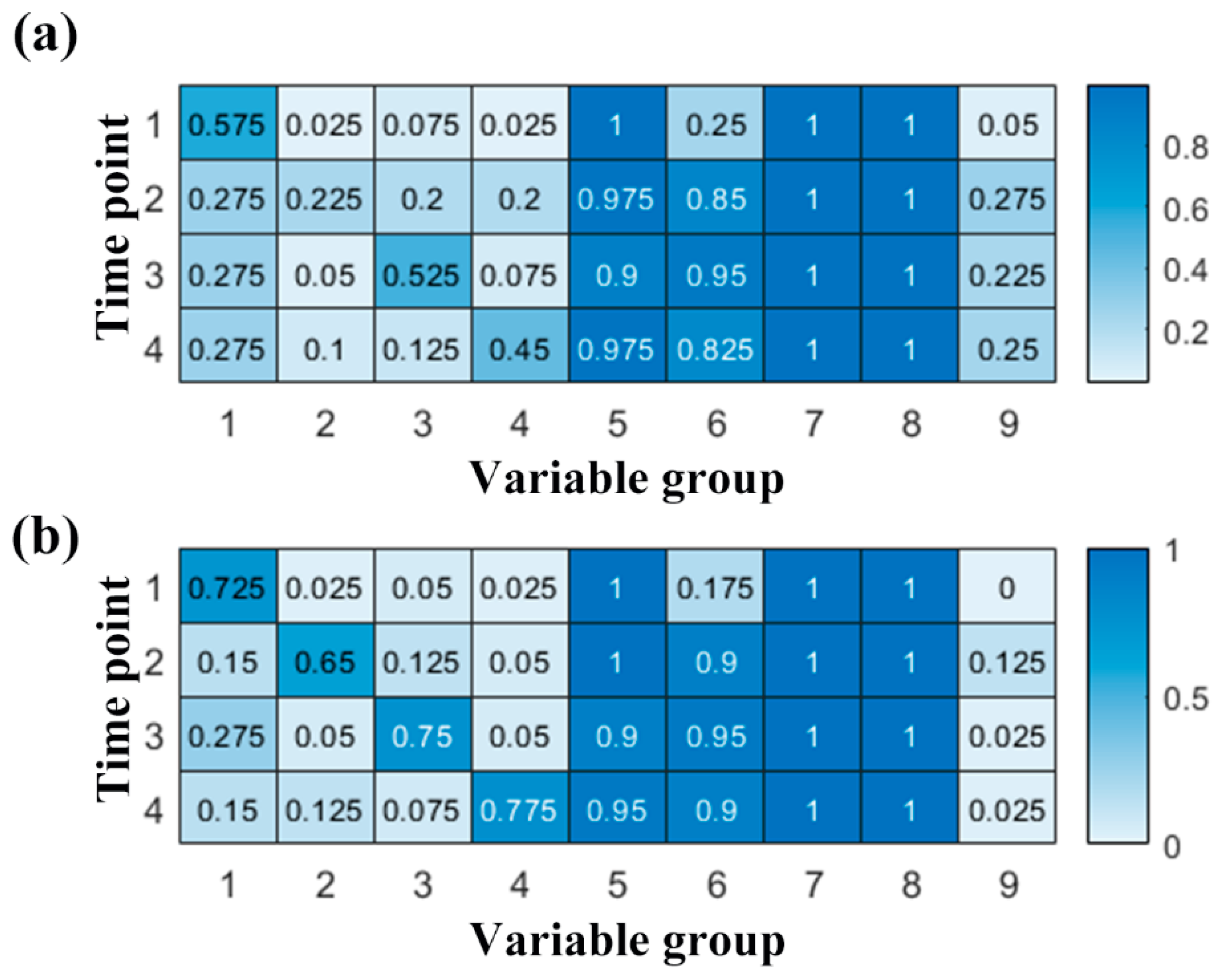
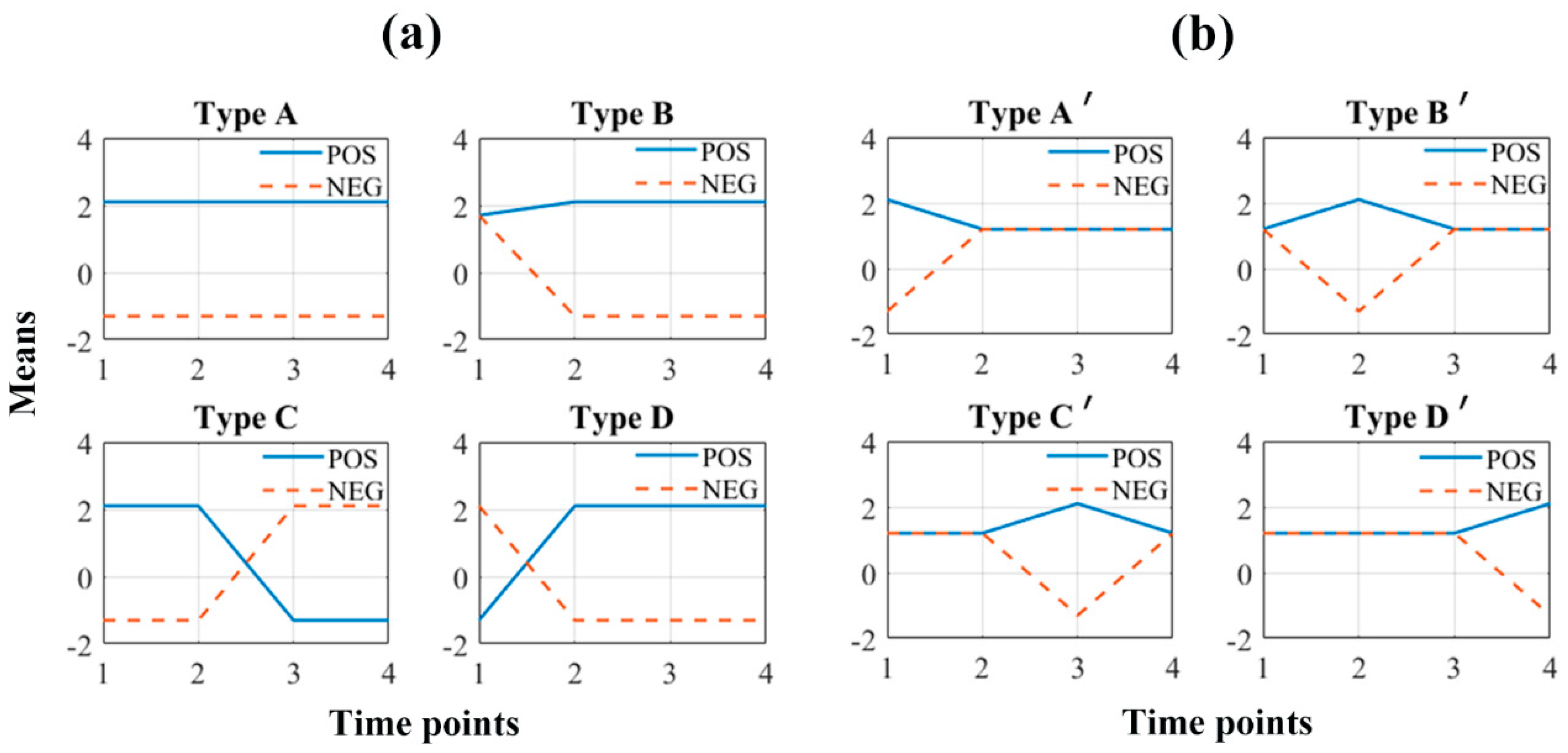
2.3. The Recovery Ability of the Two Longitudinal Methods on Simulating Data
3. Discussion
4. Materials and Methods
4.1. Methods
4.1.1. Longitudinal Feature Selection
4.1.2. Group and Nuclear Norm Regularization
4.1.3. PLS-DA
4.1.4. Performance Estimation
4.2. Sample Background and Preparation
4.3. Simulating Data
4.4. Software
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Patti, G.J.; Tautenhahn, R.; Siuzdak, G. Meta-analysis of untargeted metabolomic data from multiple profiling experiments. Nat. Protocols 2012, 7, 508–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinges, S.S.; Hohm, A.; Vandergrift, L.A.; Nowak, J.; Habbel, P.; Kaltashov, I.A.; Cheng, L.L. Cancer metabolomic markers in urine: Evidence, techniques and recommendations. Nat. Rev. Urol. 2019, 16, 339–362. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Liu, Y.; He, F.; An, R.; Du, Z. LC–MS based urinary metabolomics study of the intervention effect of aloe-emodin on hyperlipidemia rats. J. Pharm. Biomed. Anal. 2018, 156, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Ismail, S.N.; Maulidiani, M.; Akhtar, M.T.; Abas, F.; Ismail, I.S.; Khatib, A.; Ali, N.A.M.; Shaari, K. Discriminative Analysis of Different Grades of Gaharu (Aquilaria malaccensis Lamk.) via 1H-NMR-Based Metabolomics Using PLS-DA and Random Forests Classification Models. Molecules 2017, 22, 1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Ma, X.-M.; Wang, X.-C.; Liu, J.-H.; Huang, B.-Y.; Guo, X.-Y.; Xiong, S.-P.; La, G.-X. UPLC-QTOF analysis reveals metabolomic changes in the flag leaf of wheat (Triticum aestivum L.) under low-nitrogen stress. Plant Physiol. Biochem. 2017, 111, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Smilde, A.K.; Westerhuis, J.A.; Hoefsloot, H.C.J.; Bijlsma, S.; Rubingh, C.M.; Vis, D.J.; Jellema, R.H.; Pijl, H.; Roelfsema, F.; Greef, J. Dynamic metabolomic data analysis: A tutorial review. Metabolomics 2010, 6, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berk, M.; Ebbels, T.; Montana, G. A statistical framework for biomarker discovery in metabolomic time course data. Bioinformatics 2011, 27, 1979–1985. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Janssen, H.-G.; Vivó-Truyols, G. Trend analysis of time-series data: A novel method for untargeted metabolite discovery. Anal. Chim. Acta 2010, 663, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Shen, D.; Initiative, A.s.D.N. Predicting future clinical changes of MCI patients using longitudinal and multimodal biomarkers. PLoS ONE 2012, 7, e33182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Yuan, L.; Liu, J.; Ye, J. A multi-task learning formulation for predicting disease progression. In Proceedings of the 17th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Diego, CA, USA, 21–24 August 2011. [Google Scholar]
- Airola, A.; Pahikkala, T.; Waegeman, W.; De Baets, B.; Salakoski, T. A comparison of AUC estimators in small-sample studies. In Proceedings of the 3rd International Workshop on Machine Learning in Systems Biology (MLSB 09), Ljubljana, Slovenia, 5–6 September 2009. [Google Scholar]
- Wang, H.; Nie, F.; Huang, H.; Yan, J.; Kim, S.; Risacher, S.L.; Saykin, A.J.; Shen, L. High-Order Multi-Task Feature Learning to Identify Longitudinal Phenotypic Markers for Alzheimer’s Disease Progression Prediction. In Proceedings of the NIPS, Lake Tahoe, NV, USA, 3–8 December 2012. [Google Scholar]
- Gorodnitsky, I.F.; Rao, B.D. Sparse signal reconstruction from limited data using FOCUSS: A re-weighted minimum norm algorithm. Signal Process. IEEE Trans. 1997, 45, 600–616. [Google Scholar] [CrossRef] [Green Version]
- Perez-Enciso, M.; Tenenhaus, M. Prediction of clinical outcome with microarray data: A partial least squares discriminant analysis (PLS-DA) approach. Hum. Genet. 2003, 112, 581–592. [Google Scholar] [PubMed]
- Zhaozhou, L.; Yanling, P.; Zhao, C.; Xinyuan, S.; Yanjiang, Q. Improving the creditability and reproducibility of variables selected from near infrared spectra. In Proceedings of the IEEE 2013 Ninth International Conference on Natural Computation (ICNC), Shenyang, China, 23–25 July 2013. [Google Scholar]
- Westerhuis, J.; Van Velzen, E.J.; Hoefsloot, H.J.; Smilde, A. Discriminant Q2 (DQ2) for improved discrimination in PLSDA models. Metabolomics 2008, 4, 293–296. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Guo, M.; Peng, L.; Zhao, B.; Su, J.; Liu, H.; Zhang, L.; Bai, X.; Qiao, Y. UPLC Q-TOF/MS-Based Metabolic Profiling of Urine Reveals the Novel Antipyretic Mechanisms of Qingkailing Injection in a Rat Model of Yeast-Induced Pyrexia. Evid.-Based Complement. Altern. Med. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Tautenhahn, R.; Böttcher, C.; Neumann, S. Annotation of LC/ESI-MS Mass Signals. In Bioinformatics Research and Development; Hochreiter, S., Wagner, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 371–380. [Google Scholar]
- Kuhl, C.; Tautenhahn, R.; Böttcher, C.; Larson, T.R.; Neumann, S. CAMERA: An Integrated Strategy for Compound Spectra Extraction and Annotation of Liquid Chromatography/Mass Spectrometry Data Sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Ji, S.; Ye, J. SLEP: Sparse learning with efficient projections. Ariz. State Univ. 2009, 6, 7. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Full PLS-DA | N of Vars. | Reduced PLS | |||||
|---|---|---|---|---|---|---|---|
| Time Points | CE_AUC | ACC | DQ2 | - | CE_AUC | ACC | DQ2 |
| 4 h | 0.7814 | 0.70 | 0.3769 | 49 | 1 | 1 | 0.9857 |
| 8 h | 0.7501 | 0.72 | 0.1990 | 97 | 1 | 1 | 0.9150 |
| 12 h | 0.7968 | 0.87 | 0.0553 | 54 | 1 | 1 | 0.7981 |
| 24 h | 0.8749 | 0.88 | 0.6542 | 34 | 1 | 1 | 0.9876 |
| CE_AUC | DQ2 | |||||||
|---|---|---|---|---|---|---|---|---|
| Time Points | 4 h | 8 h | 12 h | 24 h | 4 h | 8 h | 12 h | 24 h |
| 4 h * | 1 | 0.9344 | 0.7808 | 0.8576 | 0.9857 | 0.6122 | −0.3148 | 0.4112 |
| 8 h * | 0.9344 | 1 | 0.9088 | 0.8768 | 0.6969 | 0.9150 | 0.3671 | 0.3085 |
| 12 h * | 0.7360 | 0.8 | 1 | 0.9536 | −0.1339 | −0.3079 | 0.7981 | 0.7672 |
| 24 h * | 0.6720 | 0.7967 | 0.9216 | 1 | −1.0532 | 0.1240 | 0.5987 | 0.9876 |
| Methods | Metrics | 4 h | 8 h | 12 h | 24 h |
|---|---|---|---|---|---|
| LFS | CE_AUC | 0.8 | 0.7808 | 0.8448 | 0.9088 |
| DQ2 | 0.4074 | 0.3658 | 0.5024 | 0.6301 | |
| GNNR | CE_AUC | 0.8768 | 0.7168 | 0.8256 | 0.9216 |
| DQ2 | 0.4614 | 0.2843 | 0.5112 | 0.6740 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Z.; Zhang, Q.; Dai, S.; Gao, X. Discovering Temporal Patterns in Longitudinal Nontargeted Metabolomics Data via Group and Nuclear Norm Regularized Multivariate Regression. Metabolites 2020, 10, 33. https://doi.org/10.3390/metabo10010033
Lin Z, Zhang Q, Dai S, Gao X. Discovering Temporal Patterns in Longitudinal Nontargeted Metabolomics Data via Group and Nuclear Norm Regularized Multivariate Regression. Metabolites. 2020; 10(1):33. https://doi.org/10.3390/metabo10010033
Chicago/Turabian StyleLin, Zhaozhou, Qiao Zhang, Shengyun Dai, and Xiaoyan Gao. 2020. "Discovering Temporal Patterns in Longitudinal Nontargeted Metabolomics Data via Group and Nuclear Norm Regularized Multivariate Regression" Metabolites 10, no. 1: 33. https://doi.org/10.3390/metabo10010033