Article Text

Statistics from Altmetric.com
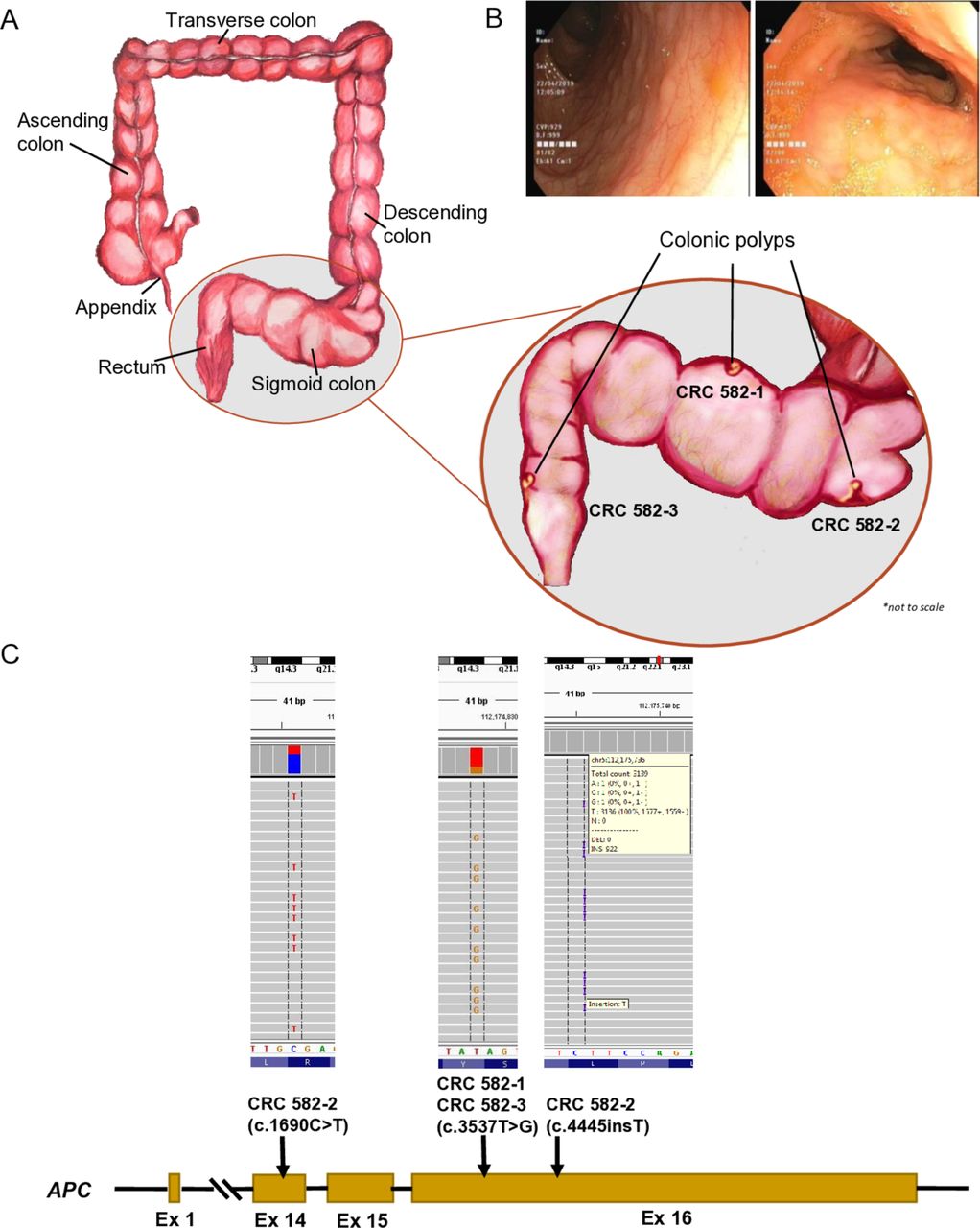
We read with interest the study by Møller and colleagues.1 The isolation and relative homogeneity of the Saudi population enhance the potential to discover founder mutations, while its high rates of consanguinity enhances homozygosity even for typically dominant disease genes.2 3 We have previously explored the contribution of Lynch syndrome (LS) to colorectal cancer (CRC) based on ~800 Saudi patients.4 We have since expanded our cohort to 1207 CRC patients. Of the 112 mismatch repair deficient (dMMR) cases, MMR gene mutations were identified in 26 cases and these varied in their cancer risk by age 50 years (figure 1). Three founder mutations were observed, accounting for 42.3% (n=11) of LS cases, and none is listed in gnomAD. The PMS2:c.1376C>G;p.S459X variant was the most commonly encountered LS variant (n=6), and we calculated its minor allele frequency at 0.0001. Importantly, we were able to identify a homozygous individual for PMS2:c.1376C>G;p.S459X through a large scale whole-exome sequencing (WES) study.2 This individual presented at age 8 years with haematochezia and a typical picture of familial adenomatous polyposis (FAP) with thousands of polyps but negative adenomatous polyposis coli (APC) testing for germline mutations and dMMR (figure 2). High depth capture sequencing of APC on three independent polyps showed at least one pathogenic APC variant per polyp (one had two variants, while one variant was shared by two polyps) completely absent in blood (figure 2). We also performed WES on one of the polyps and compared it to the WES performed on blood-derived DNA and observed no apparent enrichment for dinucleotide or trinucleotide repeat-derived mutations.

Distribution of cases in the cohort (n=1207), with MSS and non-mutated MMR genes (n=1095); MSI, but without mutated MMR genes (n=86); and cases with MSI and mutated MMR genes (Lynch Syndrome, n=26). Mutations in these genes varied in their cancer risk by age 50 years; MLH1 14%, MSH2 35%, MSH6 20% and PMS2 increased 26%, EPCAM 7%. EPCAM, epithelial cell adhesion molecule; MMR, mismatch repair; MLH1, mutL homolog 1; MSI, microsatellite instable; MSH2, mutS homolog 2; MSH6, mutS homolog 6; MSS, microsatellite stable; PMS2, postmeiotic segregation 2.
{kind=link}
{kind=link}
Illustration (A) and colonoscopy findings (B) showing the approximate location in the sigmoid colon and rectum, from where the multiple polyps from case CRC 582, with familial adenomatous polyposis, were identified. (C) Schematic representation of the APC gene (NM_000038) exons, with indicated mutations and integrative genomics viewer (IGV) representation, for each of the polyps biopsied.
Thus, while our study shows a comparable prevalence of LS and associated lifetime risk of CRC between Saudi and other populations, the genetic landscape of LS was distinct. First, the distribution of mutations among the MMR genes is unique since majority of LS cases in the literature are caused by mutations in MLH1 and MSH2 with less than 5% being caused by PMS2.5–7 Second, we note a strong founder effect in our population likely due to the heritage of tribal affiliation. The founder mutation (c.1376C>G;p.S459X) in PMS2 was previously reported in a single family in which the parents developed LS and one child developed T-cell acute lymphoblastic leukaemia.8 9 Here, we show homozygosity for this variant in a young child with typical colonic appearance for FAP. A study of APC-negative FAP cases found that biallelic inactivating mutations in MutS Homolog 3 (MSH3) induce somatic APC mutations that are largely consistent with dinucleotide and trinucleotide repeat instability.10 That study also showed that biallelic PMS2 mutations cause FAP but did not analyse APC in those patients. Our patient with a homozygous PMS2 truncation, therefore, offers the first evidence to date that PMS2-related FAP is mediated by somatic APC mutations as demonstrated by the finding of at least one APC mutation in each of the three polyps. But why was one somatic mutation shared between two physically distinct polyps? This may hint at an earlier developmental onset of that somatic mutation such that a segment of the colon is affected. The resulting FAP phenotype and the unravelling of multiple somatic APC mutations that are not dinucleotide or trinucleotide repeat-derived raise important questions about the underlying mechanism, which should be the subject of future investigations. In the interim, patients with biallelic mutations in MMR genes may benefit from targeted APC sequencing in their colon as part of their cancer screening.
Footnotes
AKS, TM and RB contributed equally.
Contributors FSA and KSA designed and implemented the study. TM, AKS and SS performed WES, critical data analyses, interpretation of results and wrote the manuscript. RB performed DNA extraction and analysed the data. SKP, AA and FAD contributed samples and analysed clinical data. FSA and KSA wrote the original draft and critically reviewed the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; internally peer reviewed.