
Abstract
Background
Two studies of intranasal 0.5% carbomer 980 gel were conducted evaluating nasal tolerability in healthy volunteers and safety and efficacy in adults with common cold symptoms.
Methods
Study 1 randomly assigned healthy adults to 0.5% carbomer 980 gel (n = 20) or placebo (n = 10) administered intranasally four times daily for 7 days. Nasal examinations were conducted at baseline and daily throughout the study. The primary endpoint was local nasal tolerability. Study 2 randomly assigned adults with an investigator-confirmed diagnosis of symptomatic common cold to 0.5% carbomer 980 gel (n = 87) or placebo (n = 81), administered intranasally four times daily for 7 days (except for day 1, where subjects who received their first dose mid-day administered only three doses). The primary efficacy endpoint was the average nasal symptom score over days 1‒4 (ANSS1–4). Secondary efficacy endpoints included ANSS over days 1‒7 and average total symptom score (ATSS). Adverse events (AEs) were recorded throughout the study.
Results
In study 1, subjects assigned to 0.5% carbomer 980 gel had no mucosal grading higher than grade 1B (superficial nasal mucosal erosion) and low incidences of mucosal bleeding and crusting. In study 2, there were no statistically significant differences between treatments for any efficacy endpoints. Active treatment was well-tolerated; the most commonly reported AEs were headache, myalgia, and cough.
Conclusion
While 0.5% carbomer 980 gel nasal spray demonstrated good local nasal tolerability in healthy volunteers, the spray did not significantly impact the course of infection or resolution of cold symptoms in subjects with common cold.
Similar content being viewed by others


Treatment with intranasal 0.5% carbomer 980 gel administered four times daily for 7 days was well-tolerated both in healthy adults and in adults with symptoms of the common cold. |
However, among adults with cold symptoms present for up to 72 h, intranasal 0.5% carbomer 980 gel did not effectively improve symptoms or impact the course of infection. |
1 Introduction
The common cold is one of the most frequently occurring infectious diseases in humans, with adults typically contracting two to three infections annually and children about six annually [1]. It is caused by a number of pathogens, although coronaviruses and rhinoviruses are among the most prevalent [2]. Although the pathophysiological mechanisms of these viruses may differ, it is believed that many common cold symptoms are mediated through the release of proinflammatory substances in response to viral infection [3, 4]. Rather than targeting viral infection and proliferation, most current treatments for the common cold are directed at individual symptoms, such as congestion, sneezing, rhinorrhea, fever, sore throat, and cough [5,6,7,8].
Preventing the attachment of rhinovirus to airway epithelial cells via blockade of the intercellular adhesion molecule (ICAM)-1 receptor, a mechanism that allows a virus to gain access to nasal epithelial cells and subsequently replicate, is a possible approach to reduce both the symptoms and the duration of the common cold [9,10,11]. Accordingly, pretreatment or post-treatment at 12 h after virus challenge with tremacamra, a recombinant soluble ICAM-1, was shown to be effective as prophylaxis of symptoms of experimental rhinovirus infection [10]; however, the study did not determine whether tremacamra was effective when given after symptom onset [10].
Viscous carbomer gels are believed to inhibit human rhinovirus replication in the epithelium by creating a physical barrier that reduces exposure to the nasal epithelia as well as by associating with the cationic binding sites on human rhinoviruses, thereby preventing viral interactions with nasal epithelial cells via cell surface receptors such as ICAM-1 [12]. Carbomer gels have been studied as a component of microbicides for prevention of transmission of other infectious diseases, including human immunodeficiency virus and other sexually transmitted infections [13,14,15]. The high-molecular-weight carbomer 980, a synthetic anionic acrylic acid polymer, has been shown previously to block viral replication in vitro with no deleterious effect on airway epithelial cells themselves (unpublished data) [12].
Here, we report the results of two studies of intranasal 0.5% carbomer 980 gel conducted in distinct study populations to investigate nasal tolerability and subsequent efficacy and safety for treatment of the symptoms of common cold. The objective of study 1 was to assess the local nasal tolerability and safety of multiple doses of 0.5% carbomer 980 gel administered intranasally in healthy volunteers before proceeding with the efficacy study. Study 2 aimed to assess the efficacy of intranasal 0.5% carbomer 980 gel in adults with symptoms of the common cold.
2 Methods
Both studies were conducted in compliance with good clinical practice as referenced in the International Council for Harmonisation guidelines, local regulatory requirements, and the Declaration of Helsinki. The study protocols and any other relevant associated documents and informed consent forms were reviewed and approved by ethics committees (study 1: Institutional Review Board Services, Toronto, ON, Canada; study 2: Quorum Review IRB, Seattle, WA, USA) prior to the start of the studies. All subjects provided written informed consent.
2.1 Study 1: Local Nasal Tolerability in Healthy Volunteers
2.1.1 Study Design
This single-center, randomized, parallel-group, placebo-controlled study, conducted from 11 June 2016 to 18 July 2016 at inVentiv Health Clinique, Québec City, QC, Canada, was designed to evaluate the local nasal tolerability of 0.5% carbomer 980 gel in healthy adults (ClinicalTrials.gov identifier: NCT02832362). Subjects were randomized in a 2:1 ratio to receive 0.5% carbomer 980 gel or matching placebo-containing vehicle (isotonic, phosphate-buffered saline solution at pH 7.2 with 0.5% phenylethanol as the preservative) without 0.5% carbomer 980. Randomization was conducted according to a randomization schedule generated by the Biostatistics Department of GlaxoSmithKline Consumer Healthcare using validated internal software prior to the start of the study and was stratified by sex (50% each sex). Product assignment for each subject was performed in accordance with the randomized number generated for the subject. Study drug or matching placebo was provided in identical packaging to preserve treatment blinding and was administered intranasally four times daily at 8:00, 12:00, 16:00, and 20:00 for 7 days. Each dose consisted of three actuations per nostril, with 140 µL per actuation, equivalent to 140 mg and a calculated dose of 0.7 mg 0.5% carbomer 980 gel. Subjects were confined for the duration of the study, starting at screening on day -1 through the treatment period (days 1–7) until the end of the study (day 8).
Nasal examinations were conducted at baseline (day 1) prior to the first administration and daily through day 8 by blinded evaluators. Physical examinations, vital signs, and laboratory tests were conducted at screening and on day 8 prior to discharge.
2.1.2 Study Population
Study 1 enrolled healthy adults aged 18–55 years in good general and mental health who were considered to have no clinically significant and relevant abnormalities, based on medical history and physical and nasal examinations. Participants must have been free of any disease or concomitant treatment that could interfere with interpretation of study results, including history of malignancy or neoplastic disease or evidence of cardiovascular, pulmonary, renal, hepatic, gastrointestinal, hematological, endocrinological, metabolic, autoimmune, neurological, psychiatric, or other diseases at screening. Participants with clinically relevant chronic or acute infectious illnesses or febrile infections within 2 weeks prior to screening were also excluded. Women of childbearing potential must have been practicing a reliable method of contraception. All subjects must have provided written informed consent.
Subjects with any condition that prohibited use of the actuating nasal spray device, such as severe rheumatoid arthritis or deformed or missing fingers, were excluded from study participation. The study also excluded current smokers or those with a history of smoking or illicit drug use in the 5 years prior to enrollment in the study as well as subjects with evidence of alcohol abuse within 6 months of screening. Additional exclusion criteria were non-acceptance of standardized food and beverages that were provided throughout the study; evidence of any superficial or moderate nasal mucosal erosion, nasal mucosal ulceration, or nasal septum perforation during the screening visit; and women who were pregnant or breastfeeding. Subjects could not consume alcohol within 24 h before confinement and throughout the confined study period. Subjects with allergy or intolerance to any of the study medications or who had a history of drug hypersensitivity, asthma, urticaria, or other significant allergic diathesis were also excluded.
2.1.3 Endpoints
The primary endpoint was local nasal tolerability, as assessed through nasal examinations and defined by the incidence of nasal mucosal assessments of grade 1B or higher (rated on a scale of 0 = no abnormal findings; 1A = focal nasal mucosal irritation [inflammation, erythema, or hyperemia]; 1B = superficial nasal mucosal erosion; 2 = moderate nasal mucosal erosion; 3 = nasal mucosal ulceration; and 4 = nasal septum perforation), or moderate or severe mucosal bleeding or moderate or severe crusting of the mucosa. Secondary endpoints included the incidence of adverse events (AEs) and changes in vital signs and laboratory parameters.
2.1.4 Statistics
No formal sample size calculation was performed. A sample size of 24 was selected based on clinical consideration. Assuming a 20% attrition rate, 30 subjects were enrolled to ensure that 24 subjects completed the study.
All endpoints were examined using descriptive statistics in the safety population, which included all subjects who received at least one dose of study medication. Nasal examination (nasal mucosa grading, mucosal bleeding, and crusting of mucosa) findings and other categorical outcomes were summarized using frequency distributions of the number (percentage) in each category at each time point, shift tables for change from baseline to last visit and change from baseline to worst value, and the number (percentage) of subjects experiencing incidences of mucosa grading of grade 1B or higher, moderate or severe mucosal bleeding, and moderate or severe crusting of the mucosa. For continuous variables, mean, standard deviation (SD), median, and minimum and maximum values were determined.
All statistics were performed using SAS software version 9.2 (SAS Institute, Cary, NC, USA).
2.2 Study 2: Efficacy Study in Subjects with Common Cold Symptoms
2.2.1 Study Design
This multicenter, randomized, parallel-group, two-arm, placebo-controlled study, conducted from 29 December 2016 through 7 June 2017 at 21 sites in the USA, was designed to evaluate the efficacy and safety of 0.5% carbomer 980 gel in adults with symptomatic common cold (ClinicalTrials.gov identifier: NCT03005067). Subjects were randomized by site staff using interactive response technology (IRT) in a 1:1 ratio to receive 0.5% carbomer 980 gel or matching placebo-containing vehicle (isotonic, phosphate-buffered saline solution at pH 7.2 with 0.5% phenylethanol as the preservative) without 0.5% carbomer 980. Study medication was supplied in identical glass devices (to maintain the study blind) and fitted with an applicator pump. The IRT system randomized and stratified treatment assignments by site and by dosing time on day 1 and generated shipments of numbered treatment kits for each treatment arm. Study drug or matching placebo was administered intranasally four times daily for 7 days. Subjects who received their first dose between 11:01 and 13:00 on day 1 received only three doses that day. Each dose was administered as described in Sect. 2.1.1 for the safety study. With the exception of independent site personnel, everyone involved in the study was blinded to the treatment assignments, including subjects, investigators, employees of the sponsor, and study statisticians.
Common cold symptoms (headache, muscle aches, chills, sore throat, blocked nose, runny nose, cough, and sneezing) were self-evaluated daily in an e-diary at screening, on day 1 prior to dosing, and immediately prior to each succeeding dose.
2.2.2 Study Population
Adult male and female subjects aged 18–75 years, in good general and mental health with no clinically significant or relevant abnormalities in medical history or upon physical and nasal examinations, were included. Females of childbearing potential must have been practicing a reliable method of contraception. Eligible subjects had an investigator-confirmed diagnosis of symptomatic common cold with an onset of 72 h prior to randomization. The protocol was amended from onset of symptoms within 48 h to within 72 h to aid with study recruitment. Subjects were required to have a total symptom score (TSS) ≥ 9 for all eight common cold symptoms, with each of the eight symptoms rated on scale of 0 = none, 1 = mild, 2 = moderate, and 3 = severe, and a score ≥ 1 for at least one of the following symptoms: sore throat, runny nose, or blocked nose.
Subjects were excluded if they had any condition, or used any concomitant drug, that could interfere with study evaluations or deposition of intranasal medication, such as chronic sinusitis, rhinitis medicamentosa, clinically significant polyposis, or nasal structural abnormalities, or that prevented the subject from using the actuating nasal spray device, such as severe rheumatoid arthritis or deformed or missing fingers; or if they had a fever ≥ 38 °C, reported frequent use of analgesics, or had a history of drug or alcohol abuse within 1 year. Subjects with uncontrolled cardiovascular, pulmonary, renal, hepatic, gastrointestinal, hematological, endocrine, metabolic, autoimmune, neurological, or psychiatric diseases, or asthma, were excluded, as were subjects with known or suspected intolerance or hypersensitivity to any of the study treatments. Women who were pregnant or breastfeeding were also excluded from study participation.
2.2.3 Endpoints
The primary efficacy endpoint was the average nasal symptom score (NSS) over days 1–4 (ANSS1-4). At each assessment, the individual were summed, and the scores for each assessment (excluding baseline NSS) were averaged to provide a daily NSS. Subjects assessed nasal symptoms (blocked nose, runny nose, and sneezing) on a 4-point scale (0 = none, 1 = mild, 2 = moderate, and 3 = severe). The ANSS1-4 was calculated as the mean of NSS scores for days 1 to 4.
Secondary efficacy endpoints included ANSS over days 1–7 (ANSS1-7), ATSS over days 1–4 (ATSS1-4), and ATSS over days 1–7 (ATSS1-7). The ATSS1-4 was the mean of the daily total symptom score across study days 1–4. Similarly, the ATSS1-7 was the mean of total symptom scores across days 1–7. The TSS was calculated in a manner similar to the ANSS but also included the symptoms headache, muscle aches, chills, sore throat, and cough. Exploratory endpoints included the mean daily NSS, TSS, and individual symptom scores on days 1–7.
AEs were recorded throughout the study, either by the investigator or in the subject’s e-diary. Vital sign assessments and physical examinations were conducted at screening and on day 8.
2.2.4 Statistics
An approximate sample size of 170 (85 subjects in each treatment group) was considered sufficient based on a review of the recent literature, which found that similar studies employing the use of nasal spray products with a similar purported mechanism of action had sample sizes ranging from 153 to 211 subjects [16,17,18]. Continuous data were summarized using descriptive statistics (mean [SD] and median [range]), and categorical data were summarized by frequency (n [%]). The primary and secondary efficacy analyses for each treatment group were summarized descriptively, and the differences were compared using analysis of covariance (ANCOVA) at a significance level of 0.05, including factors for treatment, center, day 1 dosing stratification (three vs. four doses), and baseline NSS as covariates. Efficacy analyses were conducted on the modified intent-to-treat (mITT) population, which included subjects who received at least one dose of study drug and at least one post-baseline efficacy assessment. The primary efficacy endpoint used a two-sided test at a type I error rate of 0.05. p-Values were not adjusted for multiplicity and were considered nominal.
The frequency of treatment-emergent AEs (TEAEs) was reported for the safety population, which included all subjects who received at least one dose of study medication. Changes in vital signs were summarized using descriptive statistics.
All analyses were conducted with SAS software version 9.3.
3 Results
3.1 Study 1: Local Nasal Tolerability in Healthy Volunteers
3.1.1 Baseline Characteristics
A total of 30 subjects were randomized (20 in the 0.5% carbomer 980 gel arm and ten in the placebo arm), and 29 completed the study (Fig. 1). One subject in the 0.5% carbomer 980 gel arm withdrew consent on day 7. The study groups were well-matched in terms of baseline characteristics (Table 1). Study treatment compliance was 97.3% with 0.5% carbomer 980 gel nasal spray and 90.9% with matched placebo.

Subject flow in study 1
3.1.2 Nasal Tolerability
Nasal mucosal gradings ranged from grade 0 to grade 1B during the treatment period; no incidences of grades 2–4 occurred during the study. All cases of nasal mucosal irritation/erosion were mild in severity, and all subjects remained on treatment with no worsening of the condition. The incidence of grade 1A mucosal grading was similar for both treatment groups and, at each time point, occurred in up to 80% of subjects receiving 0.5% carbomer 980 gel nasal spray versus up to 90% receiving placebo. The incidence of grade 1B mucosal grading was numerically higher in subjects who received 0.5% carbomer 980 gel nasal spray, with up to 35% of subjects per time point, compared with up to 10% of subjects per time point in the placebo arm. The majority of cases of grade 1B mucosal grading occurred near the end of treatment (days 6–8), with 35.0% of subjects receiving 0.5% carbomer 980 gel nasal spray and 10.0% of those receiving placebo on day 7 having a mucosal grade of 1B. Most subjects in both treatment groups with a reported grade 1B erosion did not have the expected associated clinical sequelae (e.g., epistaxis, nasal crusting, pain, etc.), making the clinical relevance of these findings uncertain.
Figure 2 shows the shift from baseline to last visit for the nasal mucosal grading scale. Focal nasal mucosal irritation (grade 1A) was the highest mucosal grade recorded in 55% of subjects randomized to 0.5% carbomer 980 gel and 80% of those randomized to placebo, and superficial mucosal irritation (grade 1B) was the highest grade recorded in 45% and 20%, respectively (Fig. 3).

Shift from baseline to last visit in nasal mucosal grade in healthy volunteers (study 1)

Highest nasal mucosal grade recorded during treatment period in healthy volunteers (study 1)
Low incidences of mucosal bleeding and crusting occurred during the treatment period; all were of mild severity, with no relevant differences between groups. Mucosal bleeding was experienced by no more than 10% of subjects at any time point in either arm. Mucosal crusting was experienced by no more than 5% of subjects at any time point in the 0.5% carbomer 980 gel arm, and no crusting was observed in the placebo arm. No relevant differences in nasal tolerability were observed between males and females.
3.1.3 Safety
The most common drug-related TEAEs in the 0.5% carbomer 980 gel nasal spray arm were headache, nasal mucosal erosion, and nausea (Table 2). Most AEs were mild in severity. No serious AEs, severe TEAEs, or nasal mucosal assessments of grade 3 or 4 were reported over the course of the study, and no TEAEs led to study withdrawal. Two subjects, one in the 0.5% carbomer 980 gel arm and one in the placebo arm, experienced AEs leading to interruption and/or withdrawal of study drug. One subject receiving 0.5% carbomer 980 gel experienced moderate nausea and vomiting and mild abdominal pain that led to drug interruption on days 1 and 2 and later experienced mild nasal mucosal erosion and nasal congestion on day 6 that led to drug withdrawal. One subject in the placebo arm experienced moderate nausea and vomiting and mild headache, which led to study drug interruption.
No safety issues were observed for clinical laboratory measures or vital signs.
3.2 Study 2: Efficacy in Adults with Common Cold Symptoms
3.2.1 Baseline Characteristics
A total of 171 subjects were randomized (90 in the 0.5% carbomer 980 gel arm and 81 in the placebo arm; Fig. 4). Three subjects randomized to 0.5% carbomer 980 gel did not receive study medication and were not included in the safety population or the mITT population. Overall, 80 (88.9%) subjects randomized to 0.5% carbomer 980 gel and 74 (91.4%) of those randomized to placebo completed the study. Demographic characteristics were similar across both treatment groups (Table 1). Mean common cold symptoms, including nasal symptoms, headache, muscle aches, chills, sore throat, and cough, were comparable between treatment groups at screening (Table 3).

Subject flow in study 2. mITT modified intent-to-treat
3.2.2 Efficacy
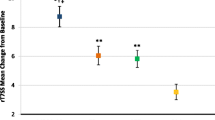
There was no statistically significant difference between 0.5% carbomer 980 gel nasal spray and placebo for the primary endpoint of ANSS1-4 (p = 0.516; Table 4). A sensitivity analysis, in which NSSs were recorded within 4 h of using other cough/cold medications set to missing and imputed, yielded similar results. Results for subjects stratified by number of doses on study day 1 were similar for those who received three doses and those who received four doses.
For the secondary endpoints of ANSS1-7, ATSS1-4, and ATSS1-7, no statistically significant differences were reported between the 0.5% carbomer 980 gel nasal spray and placebo groups (Table 4). Similar trends were observed for the exploratory endpoints of mean daily NSS, TSS, and individual symptom scores: mean change from baseline gradually increased in absolute magnitudes across the treatment period, and results were comparable between the groups.
Overall, compliance with treatment was good, with 63.7% of subjects taking 80–120% of the target number of actuations during study days 1–7. No subject took more than 120% of the target number of actuations.
3.2.3 Safety
The 0.5% carbomer 980 gel nasal spray was well-tolerated. Overall, 11 (12.6%) TEAEs in the 0.5% carbomer 980 gel arm and 16 (19.8%) in the placebo arm were considered related to treatment. The most commonly reported TEAEs occurring in ≥ 5% of subjects were headache (9.2 and 7.4% in the 0.5% carbomer 980 gel and placebo arms, respectively), myalgia (5.7 and 4.9%), and cough (5.7 and 1.2%). Most AEs were mild to moderate in intensity.
Four (4.6%) subjects in the 0.5% carbomer 980 gel arm and three (3.7%) in the placebo arm experienced AEs that led to study discontinuation. Dizziness was the AE that most frequently led to study withdrawal in the 0.5% carbomer 980 gel group (n = 2). No AEs resulted in dose reduction or dose delay. One subject in the 0.5% carbomer 980 gel group had a serious AE of bipolar affective disorder, which was not considered related to study drug.
There were no clinically notable differences in vital signs or physical examinations between groups.
4 Discussion
Although 0.5% carbomer 980 gel nasal spray was well-tolerated, it failed to reduce symptoms of the common cold. The failure to demonstrate any clinical benefit in the efficacy study was not likely due to a lack of statistical power, as a sample size of 150–200 subjects was considered sufficient based on prior studies using products with a similar proposed mechanism of action [16,17,18]. The results were flat (i.e., not even a trend in favor of 0.5% carbomer). The NSS and TSS, which comprised the primary and secondary endpoints of the efficacy study, have been widely used in studies of allergic rhinitis and have been shown to be sensitive tools [19,20,21,22,23]. These scores have been less extensively used in the common cold, but their use in some studies has demonstrated significant improvements in symptoms [5, 18, 24,25,26]. While the common cold is generally more prevalent in colder months, it can occur all year round. It is therefore possible that the lack of clinical benefit of carbomer 980 in our study may be due to the seasonal variability of the pathogens responsible for the common cold [27, 28]. For example, rhinoviruses, which are among the predominate causes of the common cold [1, 2], are genetically diverse and appear to circulate with seasonal variation [27]. Moreover, although subjects with seasonal allergic rhinitis were excluded, subjects with springtime allergies may not have mentioned them, particularly since screening for study 2 began in winter, when seasonal allergies are not top of mind [1, 2, 27].
To aid with recruitment of study participants, the protocol for study 2 was amended to allow inclusion of subjects with onset of symptoms within 72 h instead of 48 h. Cold symptoms typically peak by the third day of illness and then begin to decline [29]. Considering that, once activated, the inflammatory response continues and prolongs cold symptoms even when viral replication is reduced [29], preventing viral attachment and replication once cold symptoms are well-established may not be an effective strategy. It is also possible that preventing further viral replication with 0.5% carbomer 980 gel nasal spray would have been more effective at preventing or reducing symptoms if given earlier in the course of illness. In addition, the natural decline in cold symptoms after 72 h of illness may have made it difficult to detect differences between the 0.5% carbomer 980 gel nasal spray and placebo groups.
The 0.5% carbomer 980 gel nasal spray was well-tolerated in both study populations: healthy volunteers (study 1) and adults with symptoms of common cold (study 2). Across the two studies, the incidence of TEAEs was lower in the active treatment arm than in the placebo arm. Multiple administrations of 0.5% carbomer 980 gel were well-tolerated with regard to local nasal tolerability, with no mucosal grading higher than grade 1B and low incidences of mild mucosal bleeding and crusting. The use of a conservative grading scale in the local nasal tolerability study likely led to a higher reporting of nasal irritation than observed in the efficacy study. Overall, most AEs were mild or moderate in intensity. The only serious AE that occurred in either study was unrelated to study treatment. Laboratory variables, vital signs, and physical examinations did not reveal any clinically significant effects of 0.5% carbomer 980 gel treatment.
5 Conclusions
The 0.5% carbomer 980 gel nasal spray was well-tolerated in separate studies of healthy volunteers and subjects with common cold symptoms. However, an efficacy study in subjects who had experienced cold symptoms for ≤ 72 h provided no evidence that the spray had any impact on the course of infection and resolution of symptoms.
Change history
20 July 2019
The original publication of the article contained the following errors.
20 July 2019
The original publication of the article contained the following errors.
References
Allan GM, Arroll B. Prevention and treatment of the common cold: making sense of the evidence. CMAJ. 2014;186:190–9.
Hull JD, Barton IP, Torgersen J, McNeil CM. A survey of the experience and impact of acute upper respiratory tract infections on people in six countries in the 2011/2012 common cold and flu season. Open J Respir Dis. 2013;3:175–87.
Kennedy JL, Turner RB, Braciale T, Heymann PW, Borish L. Pathogenesis of rhinovirus infection. Curr Opin Virol. 2012;2:287–93.
Turner RB, Hendley JO, Gwaltney JM Jr. Shedding of infected ciliated epithelial cells in rhinovirus colds. J Infect Dis. 1982;145:849–53.
Sperber SJ, Sorrentino JV, Riker DK, Hayden FG. Evaluation of an alpha agonist alone and in combination with a nonsteroidal antiinflammatory agent in the treatment of experimental rhinovirus colds. Bull N Y Acad Med. 1989;65:145–60.
Gwaltney JM Jr, Druce HM. Efficacy of brompheniramine maleate for the treatment of rhinovirus colds. Clin Infect Dis. 1997;25:1188–94.
Hayden FG, Diamond L, Wood PB, Korts DC, Wecker MT. Effectiveness and safety of intranasal ipratropium bromide in common colds. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1996;125:89–97.
Sperber SJ, Hendley JO, Hayden FG, Riker DK, Sorrentino JV, Gwaltney JM, Jr. Effects of naproxen on experimental rhinovirus colds. A randomized, double-blind, controlled trial. Ann Intern Med. 1992;117:37–41.
Staunton DE, Merluzzi VJ, Rothlein R, Barton R, Marlin SD, Springer TA. A cell adhesion molecule, ICAM-1, is the major surface receptor for rhinoviruses. Cell. 1989;56:849–53.
Turner RB, Wecker MT, Pohl G, Witek TJ, McNally E, St George R, et al. Efficacy of tremacamra, a soluble intercellular adhesion molecule 1, for experimental rhinovirus infection: a randomized clinical trial. JAMA. 1999;281:1797–804.
Winther B, Arruda E, Witek TJ, Marlin SD, Tsianco MM, Innes DJ, et al. Expression of ICAM-1 in nasal epithelium and levels of soluble ICAM-1 in nasal lavage fluid during human experimental rhinovirus infection. Arch Otolaryngol Head Neck Surg. 2002;128:131–6.
WO2017212422A1—Topical compositions comprising carbomer for the treatment and prevention of viral infections and allergic conditions: Google Patents; 2017. https://patents.google.com/patent/WO2017212422A1/en.
Ndesendo VM, Pillay V, Choonara YE, Buchmann E, Bayever DN, Meyer LC. A review of current intravaginal drug delivery approaches employed for the prophylaxis of HIV/AIDS and prevention of sexually transmitted infections. AAPS Pharm Sci Tech. 2008;9:505–20.
Van Damme L, Wright A, Depraetere K, Rosenstein I, Vandersmissen V, Poulter L, et al. A phase I study of a novel potential intravaginal microbicide, PRO 2000, in healthy sexually inactive women. Sex Transm Infect. 2000;76:126–30.
Balzarini J, Van Damme L. Microbicide drug candidates to prevent HIV infection. Lancet. 2007;369:787–97.
Fazekas T, Eickhoff P, Pruckner N, Vollnhofer G, Fischmeister G, Diakos C, et al. Lessons learned from a double-blind randomised placebo-controlled study with a iota-carrageenan nasal spray as medical device in children with acute symptoms of common cold. BMC Complement Altern Med. 2012;12:147.
Ludwig M, Enzenhofer E, Schneider S, Rauch M, Bodenteich A, Neumann K, et al. Efficacy of a carrageenan nasal spray in patients with common cold: a randomized controlled trial. Respir Res. 2013;14:124.
Eccles R, Winther B, Johnston SL, Robinson P, Trampisch M, Koelsch S. Efficacy and safety of iota-carrageenan nasal spray versus placebo in early treatment of the common cold in adults: the ICICC trial. Respir Res. 2015;16:121.
Berger WE, Jacobs RL, Amar NJ, Tantry SK, Li J, Small CJ. Efficacy and safety of beclomethasone dipropionate nasal aerosol in children with perennial allergic rhinitis. Ann Allergy Asthma Immunol. 2015;115:130–6.
Takahashi G, Matsuzaki Z, Okamoto A, Ito E, Matsuoka T, Nakayama T, et al. A randomized control trail of stepwise treatment with fluticasone propionate nasal spray and fexofenadine hydrochloride tablet for seasonal allergic rhinitis. Allergol Int. 2012;61:155–62.
Mi J, Chen X, Lin X, Guo J, Chen H, Wei L, et al. Treatment of persistent allergic rhinitis via acupuncture at the sphenopalatine acupoint: a randomized controlled trial. Trials. 2018;19:28.
Okubo K, Baba K. Therapeutic effect of montelukast, a cysteinyl leukotriene receptor 1 antagonist, on Japanese patients with seasonal allergic rhinitis. Allergol Int. 2008;57:247–55.
Grubbe RE, Lumry WR, Anolik R. Efficacy and safety of desloratadine/pseudoephedrine combination vs its components in seasonal allergic rhinitis. J Investig Allergol Clin Immunol. 2009;19:117–24.
Belongia EA, Berg R, Liu K. A randomized trial of zinc nasal spray for the treatment of upper respiratory illness in adults. Am J Med. 2001;111:103–8.
Doyle WJ, Riker DK, McBride TP, Hayden FG, Hendley JO, Swarts JD, et al. Therapeutic effects of an anticholinergic-sympathomimetic combination in induced rhinovirus colds. Ann Otol Rhinol Laryngol. 1993;102:521–7.
Adam P, Stiffman M, Blake RL Jr. A clinical trial of hypertonic saline nasal spray in subjects with the common cold or rhinosinusitis. Arch Fam Med. 1998;7:39–43.
Miller EK, Mackay IM. From sneeze to wheeze: what we know about rhinovirus Cs. J Clin Virol. 2013;57:291–9.
Worrall G. Common cold. Can Fam Physician. 2011;57:1289–90.
Gwaltney JM. Clinical significance and pathogenesis of viral respiratory infections. Am J Med. 2002;112(Suppl 6A):13s–8s.
Acknowledgements
Medical writing assistance was provided by Adrienne Drinkwater, PhD, of Peloton Advantage, Parsippany, NJ, USA, funded by GlaxoSmithKline Consumer Healthcare.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by GlaxoSmithKline Consumer Healthcare, Warren, NJ, USA. The study sponsor contributed to the study design; the analysis and interpretation of the data; the writing of the report; and the decision to submit the paper for publication.
Conflicts of interest
Gilbert Shanga and Lara Dennie are employees of GlaxoSmithKline Consumer Healthcare.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in these studies.
Data sharing
Anonymized individual participant data and study documents can be requested for further research from http://www.clinicalstudydatarequest.com.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dennie, L. Safety and Efficacy of 0.5% Carbomer 980 Gel for Treatment of Symptoms of Common Cold: Results of 2 Randomized Trials. Drugs R D 19, 191–200 (2019). https://doi.org/10.1007/s40268-019-0270-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-019-0270-3