
Abstract
Diterpenoid alkaloids have neroprotective activity. Herein, three napelline-type diterpenoid alkaloids 1–3, two aconitine-type diterpenoid alkaloids 4–5, and one isoquinline-type alkaloid 6, as well as one lignan glycoside 7, have been isolated from the roots of Aconitum pendulum Busch. Compounds 1 and 7 were new compounds, and their chemical structures were determined on the basis of nuclear magnetic resonance (NMR) spectra and mass spectrometry analysis. A ThT assay revealed that compound 2 showed significant disaggregation potency on the Aβ1−42 aggregates.
Graphical Abstract

Similar content being viewed by others
1 Introduction
The plant Aconitum pendulum Busch, belonging to the Ranunculaceae family, is mainly distributed in Northwestern China at the altitude of 3000–4000 m [1]. The roots of A. pendulum have long been used as a traditional herb “Tie Bang Chui” for treating traumatic injury, fracture, rheumatism, and chilblains [2]. Previous studies showed that this plant mainly produced diterpenoid and norditerpenoid alkaloids [3], such as aconitine (AC), deoxyaconitine (DA), and mesaconitine (MA) [4], which were considered as the toxic components of this kind of folk medicine. These alkaloids have shown to cause widespread membrane excitation in cardiac, neural, and muscular tissues because of their significant activation on sodium channels [5,6,7,8]. Besides, they were also reported to show the potential as drug leads in Alzheimer diseases by targeting the neuronal nicotinic acetylcholine receptor [9,10,11].
Our research group has long been focused on the discovery of bioactive natural compounds from the traditional herbs cultivated in Northwestern China [12,13,14,15,16]. As part of this ongoing program, a phytochemical study on the roots of A. pendulum collected from Gansu province has been conducted. Three napelline-type diterpenoid alkaloids 1–3, two aconitine-type diterpenoid alkaloids 4–5, and one isoquinline-type alkaloid 6, as well as one lignan glycoside 7, were isolated (Fig. 1). Herein, we report the isolation, structural determination, and biological activity of these isolates.

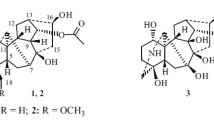
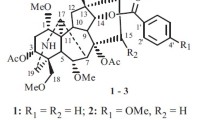
Molecular structures of compounds 1−7
2 Results and Discussion
Compound 1 was isolated as a colorless gum with a small value of specific optical rotatory value. Its chemical formula C22H32O3N was determined by the positive high-resolution MS at m/z 358.2362 (calcd. 358.2377). The IR spectrum indicated the existence of hydroxy (3396 cm−1) and olefinic (1678 cm−1) functionalities. The 1H and 13C NMR spectra of compound 1 demonstrated the presence of three oxygenated methine groups (δH 4.21, 4.14, 4.02; δC 77.2, 69.2, 68.8), one exocyclic double bond (δH 5.30, 5.16; δC 153.4, 113.3), a tertiary methyl group (δH 1.31; δC 21.3), an isolated iminium methine (δH 8.43; δC 184.8) [17], and three aliphatic quaternary carbons (δC 54.7, 52.3, 47.1). The data indicated that this compound was a C20-diterpenoid alkaloid possessing an iminium methine moiety [17]. Further HMBC and HSQC experiments determined that compound 1 shared the same molecular structure as that of aconicarmichinium A except for the relative configuration of OH-12 [17]. Specifically, three hydroxy groups were substituted at C-1, C-12, and C-15 based on the HMBC correlations from H-1 (δH 4.02) to C-2 (δC 31.9), C-3 (δC 35.5), C-5 (δC 45.0) and C-10 (δC 54.7), from H-12 (δH 4.14) to C-9 (δC 40.6), C-11 (δC 30.4), and C-13 (δC 44.9), from H-15 (δH 4.21) to C-7 (δC 52.2), C-8 (δC 52.3), C-9 (δC 40.6) and C-16 (δC 153.4). The position of Δ16,17 double bond was supported by the HMBC correlations from H2-17 (δH 5.30, 5.16) to C-13 (δC 44.9) and C-15 (δC 77.2). Moreover, the 13C NMR data of compound 1 and aconicarmichinium A [17] revealed that the major differences are the chemical shifts of C-12 (69.2 vs. 76.5), C-13 (44.9 vs. 48.6), C-14 (33.0 vs. 28.9), C-16 (153.4 vs. 158.5), and C-17 (113.3 vs. 109.6). This information suggested that the two compounds differed in the configuration of C-12, which was supported by the NOESY correlations from H-15 (δH 4.21) to H-14b (δH 1.28) and from H-14a (δH 1.78) to H-12 (δH 4.14). Therefore, the structure of 1 was named 12-epi-aconicarmichinium A chloride.
Compound 7 was purified as a white powder. The molecular formula C26H34O11 was determined by the positive high resolution MS at m/z 545.1992 ([M+Na]+, calcd. 545.1999). The IR spectrum suggested the presence of hydroxy (3360 cm−1) and benzene unit (1646, 1452, 1379 cm−1). Acid hydrolysis of 7 with 1 M HCl afforded d-glucose, based on gas chromatography analysis following treatment with l-cysteine methyl ester hydrochloride and trimethylsilylimidazole derivatization, and coupling pattern of the anomeric proton (d, J = 7.8 Hz) indicated a β configuration for the glucose unit. The 1H and 13C NMR data indicated the presence of a glucose unit (δH 4.86, 3.85, 3.67, 3.46, 3.46, 3.38, 3.38; δC 103.0, 78.4, 78.0, 75.1, 71.5, 62.7), two methoxyl groups (δH 3.85, 3.82; δC 56.9, 56.5), two tri-substituted phenolic rings (δH 7.12, 6.98, 6.87, 6.78, 6.70, 6.63; δC 151.0, 149.2, 147.4, 146.0, 139.7, 133.6, 122.3, 119.7, 118.1, 116.4, 113.5, 111.5), one oxygenated methine (δH 4.81; δC 84.0), and two oxygenated methylene (δH 3.99, 3.83, 3.72, 3.64; δC 73.8, 60.6). Detailed analysis of the NMR data indicated that compound 7 shared a similar structure with that of 3-(2,4-dihydroxy-3-methoxybenzyl)-4-(4-hydroxy-3-methoxybenzy1)tetrahydrofuran [18]. However, compound 7 had an additional sugar unit, which was substituted at C-4′ based on the HMBC correlation from H-1′' (δH 4.86) to C-4′ (δC 147.4). Due to the limited amount, the relative configuration at C-7′ remained undetermined. Therefore, compound 7 was determined as 3-(2,4-dihydroxy-3-methoxybenzyl-4-O-glucopyranosyl)-4-(4-hydroxy-3-methoxybenzy1)tetrahydrofuran.
On the basis of the literature data, the known compounds were determined as songorine (2) [19], napelline (3) [20, 21], ducloudine C (4) [22], aconine (5) [23], magnoflorine (6) [24].
Due to the available amounts, compounds 1–4 were evaluated for their anti-AD potential based on their effect on the copper-mediated Aβ1−42 disaggregation by using ThT assay [25] with resveratrol as positive control. All compounds were treated at 25 µM. As shown in Table 1, the disaggregate potency of compounds 1–4 were ranging from 10.2 ± 3.8 to 28.6 ± 2.9%, while the datum for the positive control resveratrol was 46.9 ± 4.6%. Compound 2 showed the most significant disaggregation effect on the Aβ1−42 aggregates.
3 Experimental Section
3.1 General
The optical rotation values were measured on a 241 polarimeter (Perkin-Elmer). The infrared spectra were measured by a FTS 165-IR instrument (Bio-Rad, USA). A Varian INOVA-400 FT-NMR spectrometer (USA) and a Bruker APEX II spectrometer were used to record the NMR and HRESIMS data, respectively. Different types of chromatographic materials were used for the fractionation of natural compounds, including Sephadex LH-20 (Amersham Biosciences), silica gel (200–300 mesh, Qingdao Haiyang Chemical Co., Ltd), and ODS (YMC Co., Ltd). Prep-HPLC separation was performed on a prep-HPLC manufactured by Hanbon Sci & Tech of China using a Megres C18 column (250 mm × 20 mm).
3.2 Plant Materials
The roots of A. pendulum Busch (Ranunculaceae), collected from Gansu Province in China, were purchased from Lanzhou Huanghe Herbal Medicines Market in 2017. The materials were identified by Dr. Huan-Yang Qi at Lanzhou Institute of Chemical Physics (LICP), and a voucher specimen (ZY2017HTBC) was deposited at the CAS Key Laboratory of Chemistry of Northwestern Plant Resources.
3.3 Extraction and Isolation
The air-dried roots (200 g) of A. pendulum were extracted with 90% ethanol/water (v/v) at room temperature (72 h × 3). After evaporated under reduced pressure, the residue (16.1 g) was suspended in water and successively partitioned with petroleum ether (PE), EtOAc, and n-BuOH (each 1.0 L × 3). The dried EtOAc part (3.9 g) was chromatographed over silica gel by eluting with gradient CH2Cl2/methanol (v/v, from 20:1 to 1:1) to yield 10 fractions (A1–A10) based on the TLC analysis. Fraction A3 (137 mg) was subjected to a Sephadex LH-20 column eluting with methanol to afford compounds 4 (16.3 mg) and 5 (2.2 mg). Fraction A5 (67 mg) was purified over ODS with methanol/water (v/v, from 50 to 90%) to afford compounds 1 (13.5 mg), 2 (10.0 mg), and 3 (15.1 mg). Fraction A7 (32 mg) was purified by HPLC eluting with methanol in water from 50 to 90% to yield compounds 6 (1.7 mg) and 7 (2.2 mg).
3.4 12-epi-Aconicarmichinium A chloride (1)
Colorless gum; \([\alpha ]_{D}^{20}\) − 2.3 (c 0.26, methanol); IR (neat) vmax 3396, 2923, 1678, 1200, 1132 cm−1; 1H (methanol-d4, 400 MHz) and 13C NMR (methanol-d4, 100 MHz) see Table 2; HRESIMS m/z 358.2362 [M]+ (calcd for C22H32NO3, 358.2377).
3.5 3-(2,4-Dihydroxy-3-methoxybenzyl-4-O-glucopyranosyl)-4-(4-hydroxy-3-methoxybenzy1)tetrahydrofuran (7)
White powder; \([\alpha ]_{D}^{20}\) − 2.9 (c 0.37, methanol); IR (neat) vmax 3360, 2926, 1646, 1452, 1379, 1101, 1048 cm−1; 1H (methanol-d4, 400 MHz) and 13C NMR (methanol-d4, 100 MHz see Table 3; HRESIMS m/z 545.1992 [M+Na]+ (calcd for C26H34O11Na, 545.1993).
3.6 Acid Hydrolysis of Compound 7
This process was performed according to the literature [26]. The detailed process can be found in Supporting Information.
3.7 ThT Assay
The procedures of ThT assay were reported in the literature [25]. The detailed information can be found in Supporting Information.
References
P.G. Xiao, F.P. Wang, F. Gao, L.P. Yan, D.L. Chen, Y. Liu, Acta Phytotaxon. Sin. 44, 1–46 (2006)
M. El-Shazly, C.J. Tai, T.Y. Wu, D. Csupor, J. Hohmann, F.R. Chang, Y.C. Wu, J. Food Drug Anal. 24, 29–45 (2016)
Y.J. Wang, C.J. Zeng, Z. Yao, J. Zhang, Y. Zhang, F. Zhang, Chin. Tradit. Herb. Drugs 41, 347–351 (2010)
D.G. Barceloux, Medical Toxicology of Natural Substances, Foods, Fungi, Medicinal Herbs, Toxic Plants, and Venomous Animals (Wiley, Hoboken, 2008), pp. 736–742
C.C. Lin, T.Y.K. Chan, J.F. Deng, Ann. Emerg. Med. 43, 574–579 (2004)
F. Moritz, P. Compagnon, I.G. Kaliszczak, Y. Kaliszczak, V. Caliskan, C. Girault, Clin. Toxicol. 43, 873–876 (2005)
S.W. Smith, R.R. Shah, C.A. Herzog, Ann. Emerg. Med. 45, 100–101 (2005)
T.Y.K. Chan, Toxins 6, 2605–2611 (2014)
A.M. Bello-Ramirez, J. Buendia-Orozco, A.A. Nava-Ocampo, Fundam. Clin. Pharmacol. 17, 575–580 (2003)
S.R. Breining, Curr. Top. Med. Chem. 4, 609–629 (2004)
Y.Z. Zhu, K.H. Tan Benny, B.H. Bay, C.H. Liu, Natural Products: Essential Resources for Human Survival (World Scientific, Singapore, 2007), p. 85100 (2007).
H.L. Jiang, W. Ha, J.L. Yang, Y.P. Shi, Phytochem. Lett. 17, 1–5 (2016)
J.L. Yang, Y.M. Zhao, Y.P. Shi, Nat. Prod. Bioprospect. 6, 211–216 (2016)
X.Y. Xie, R. Wang, Y.P. Shi, Planta Med. 78, 1010–1014 (2012)
J.L. Yang, Y.P. Shi, Phytochemistry 76, 124–132 (2012)
J.L. Yang, R. Wang, L.L. Liu, Y.P. Shi, Planta Med. 77, 362–367 (2011)
X.H. Meng, Z.B. Jiang, C.G. Zhu, Q.L. Guo, C.B. Xu, J.G. Shi, Chin. Chem. Lett. 27, 993–1003 (2016)
A. Estévez-braun, R. Estévez-reyes, J.A. González-pérez, A.G. González, J. Nat. Prod. 58, 887–892 (1995)
Z.C. Fan, Z.Q. Zhang, J Chem. Crystallogr. 38, 635 (2008)
J. Chen, Res. Pract. Chin. Med. 27, 33–35 (2013)
Z.G. Chen, H.C. Wang, A.N. Lao, B.D. Wang, S.H. Hong, Chin. J. Org. Chem. 9, 490–497 (1989)
T.P. Yin, L. Cai, J.M. He, J.W. Dong, H.X. Fang, H. Zhou, Z.T. Ding, J. Asian Nat. Prod. Res. 16, 345–350 (2014)
S.W. Pelletier, N.V. Mody, R.S. Sawhney, Can. J. Chem. 57, 1652–1655 (1979)
J.C. Holzbach, L.M.X. Lopes, Molecules 15, 9462–9472 (2010)
T. Chai, X.-B. Zhao, W.-F. Wang, Y. Qiang, X.-Y. Zhang, J.-L. Yang, Molecules 23, 2663 (2018)
J. Wang, J.-L. Yang, P.-P. Zhou, X.-H. Meng, Y.-P. Shi, J. Agric. Food Chem. 65, 5926–5934 (2017)
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (Nos. 81673325 and 81711540311), the International Partnership Program of CAS (No. 153631KYSB20160004), Gansu Province Key International S&T Cooperation Project (No. 18YF1WA127), and CAS Pioneer Hundred Talents Program.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing financial interest.
Additional information
The article is dedicated to Professor Han-Dong Sun on the occasion of his 80th birthday.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wang, J., Meng, XH., Chai, T. et al. Diterpenoid Alkaloids and One Lignan from the Roots of Aconitum pendulum Busch. Nat. Prod. Bioprospect. 9, 419–423 (2019). https://doi.org/10.1007/s13659-019-00227-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-019-00227-y