
Abstract
In Drosophila, the fat body undergoes a massive burst of autophagy at the end of larval development in preparation for the pupal transition. To identify genes involved in this process, we carried out a microarray analysis. We found that mRNA levels of the homologs of Atg8, the coat protein of early autophagic structures, and lysosomal hydrolases were upregulated, consistent with previous results. Genes encoding mitochondrial proteins and many chaperones were downregulated, including the inhibitor of eIF2alpha kinases and the peptidyl-prolyl cis–trans isomerase FK506-binding protein of 39 kDa (FKBP39). Genetic manipulation of FKBP39 expression had a significant effect on autophagy, potentially through modulation of the transcription factor Foxo. Accordingly, we found that Foxo mutants cannot properly undergo autophagy in response to starvation, and that overexpression of Foxo induces autophagy.
Similar content being viewed by others


Main
Autophagy is the degradation of self material by lysosomes. As a primary cellular defense response, it is activated by nitrogen or amino-acid starvation from yeast to mammals, and promotes the survival of the cell or organism by recycling dispensable cellular constituents for re-use in synthetic processes. The morphology of the major pathway is well known for decades by electron microscopical studies.1 In response to starvation or other stimuli, a membranous sac called isolation membrane forms and engulfs portions of the cytosol. After sealing of its edges, the emerging double-membrane organelle is referred to as an autophagosome. Upon fusion with a lysosome an autolysosome is formed, where degradation of the sequestered material takes place. The process of autophagy is remarkably similar in all eukaryotic organisms, suggesting the involvement of an evolutionary conserved set of genes. Indeed, functional homologs of most Atg (autophagy-related) genes required for autophagy in yeast were found in multiple species including plants, worms, flies, and mammals.2 Despite the conservation of the core mechanism, there must be changes in the regulation of autophagy among different phyla, as it is involved in various cellular processes in multicellular animals. In addition to its fundamental role in starvation survival, autophagy is thought to be involved in cell death, neurodegeneration diseases, aging, immunity, growth, and cancer.1, 3, 4
In Drosophila and other insects undergoing complete metamorphosis, the larval fat body acts as a store of proteins and other materials, which are released through autophagy to provide energy and nutrients during metamorphosis and early adulthood.5, 6, 7, 8 The fat body, an analog of the human liver, is a polytenic tissue that grows in mass approximately 200-fold in feeding Drosophila larvae during the larval stages. After reaching an optimal mass, mature larvae stop eating and wander away from the food to find a suitable place for pupariation. At this time, the fat body undergoes a massive induction of autophagy, known as developmental autophagy. These changes are induced by the insect molting hormone ecdysone at a low concentration of juvenile hormone.9 Recent results showed that ecdysone induces autophagy through downregulation of phosphatidyl-inositol 3-kinase (PI3K) signaling.10
A central regulator of cell growth and autophagy is target of rapamycin (Tor) kinase. Inhibition of Tor activity rapidly results in growth arrest and induction of autophagy, which involves multiple phosphorylation and dephosphorylation events.11, 12, 13 In yeast, the phosphorylation state of a number of Atg proteins is rapamycin-sensitive, and the activity of the kinase Atg1 is regulated by Tor signaling.14 Another regulatory mechanism is the induction of genes necessary for autophagy, or repression of genes that normally inhibit the process. It is known that the gene encoding Atg8, a ubiquitin-like coat protein for autophagosomes, is upregulated in starved yeast cells15 and Drosophila larvae.16
To search for genes regulated during developmental autophagy, we carried out a microarray analysis by comparing the transcriptional profiles of fat bodies dissected from feeding and wandering third instar larvae. This analysis both demonstrated evolutionary conservation and identified additional genes with previously unknown roles in autophagy. Further characterization of a selected subset of genes in transgenic animals identified FK506-binding protein of 39 kDa (FKBP39) as an inhibitor of autophagy, which effect is likely mediated through modulation of the transcription factor Foxo.
Results
Microarray analysis of transcriptional changes during developmental autophagy
To assess gene expression changes during developmental autophagy in Drosophila larvae, we manually dissected fat bodies before and after the developmental induction of autophagy from feeding (60 h after hatching from the egg, Figure 1a) and wandering (84 h after hatching from the egg, Figure1b and e) larvae.6, 7 Samples were processed and cDNAs were hybridized to a microarray containing 3200 annotated Drosophila cDNAs.17 About 1941 of the 3200 genes investigated were expressed in the fat body. Table 1 shows the 57 genes induced by 1.65-fold or greater (estimated P-value <0.025) during autophagy. The mRNA level of the eye pigment biosynthesis gene Hn was increased, consistent with the known role of the fat body in that process during the wandering stage.18 A gene encoding Fbp2, a storage protein, was also induced; together these results provide a control for the proper developmental timing of our sample collection. Genes encoding putative lysosomal hydrolases (α-EST2, cathD, CG5932, CG1827, CG10992, CG1774) were upregulated, consistent with the expansion of the lysosomal compartment during autophagy. Ten of the 16 fly Atg gene homologs were represented on our chip, and only CG32672/Atg8a was induced significantly, in accordance with yeast and previous fly data.1, 16 A gene encoding another ubiquitin-like protein of unknown function (CG7224) was also upregulated.

Overexpression of FKBP39 inhibits developmental and starvation-induced autophagy, whereas loss of FKBP39 function leads to higher than wild-type induction of autophagy. (a–c) Lysotracker staining of fat bodies. No Lysotracker staining is seen in feeding early third instar larvae (a), whereas Lysotracker-positive granules (shown in red) accumulate in fat body cells of wandering late third instars (b). Overexpression of FKBP39 in the fat body inhibits the formation of Lysotracker-positive granules (c), and leads to an inhibition of cell growth. This effect is best seen by cortical actin staining (red) of fat bodies clonally overexpressing FKBP39, marked by co-overexpression of nuclear GFP (green) (d, top panel). Lower panel shows DNA staining. Note that the nuclei of FKBP39-overexpressing cells (green arrows) are of the same size but contain less DNA than surrounding wild-type cells (blue arrows). (e and f) Electron microscopic images show fat body cells of late third instar larvae. Developmental autophagy results in the accumulation of large autolysosomes in wild-type larvae (arrow in e; cytoplasmic dark blue granules in the inset), whereas only small autolysosomes are seen in FKBP39-overexpressing fat body cells of the same age (arrows in f). Note the greatly enlarged nucleolus (asterisk) in the nucleus (N) in f, also caused by the overexpression of FKBP39. Insets show toluidine blue staining of semi-thick sections. Again, note the greatly enlarged nucleoli (dark blue) in nuclei (arrowheads) in (f). (g) Quantitation of Lysotracker data on developmental autophagy is shown. Asterisk marks a significant change (P<0.001). (h–j) Lysotracker staining of fat bodies. Autophagy in fat body cells of early third instar larvae, induced by a 4-h starvation (h, compare to a). Fat body-specific overexpression of FKBP39 also blocks this starvation response (i). Co-overexpression of PTEN restores autophagy inhibited by FKBP39 (j). (k) Quantitation of Lysotracker data is shown. Asterisk and double asterisks mark significant differences (P<0.001 for columns 1-2, 2-3, 1-3). (l) RT-PCR of fat body samples dissected from control feeding larvae (lane 1), FKBP395-HA−2440 mutants (lane 2), and a precise excision line FKBP39exA3 (lane 3). FKBP39 expression is reduced in larval fat bodies of the mutants, but precise excision of the P-element restores wild-type mRNA expression (top panel). Bottom panel shows RT-PCR for actin as a control. (m–o) Lysotracker staining of fat bodies. Short, 80-min starvation induces higher levels of autophagy in fat bodies of FKBP395-HA−2440 mutant larvae than in wild-type controls (n, compare with m). Starvation response is restored to wild-type levels in the precise excision line FKBP39exA3 (o). (p) Quantitation of the results is shown. Asterisk marks a significant change (P=0.006 for columns 1 and 2, P<0.001 for columns 2 and 3, P=0.27 for columns 1–3). Panels a–d, h–j and m–o are of the same magnification ( × 200). Panels e and f are of the same magnification and bars equal 1 μm for electron microscopical images, 10 μm for insets. Error bars represent standard deviation in panels g, k and p. Genotypes: UASFKBP39/+ (a, b, e, h), cgGal4/UASFKBP39 (c, f, i), hsFLP; UASFKBP39/+; Act>CD2>Gal4, UASGFPnls/+ (d), cgGal4/UASFKBP39, UASPTEN (j), Df(3R)Exel6194/TM6 (m), FKBP395-HA−2440/Df(3R)Exel6194 (n), FKBP39exA3/Df(3R)Exel6194 (o). For panel l, genotypes are identical to panels m–o, respectively
The expression of 39 genes was significantly downregulated. Among them, two main subgroups could be identified: genes encoding mitochondrial proteins (CG17896, CG9140, ND42, CG6459, TRAP1, CG10664, Hsc70-5, CG2249, mRpL24, Marf) and cellular chaperones (CG8286/ Drosophila inhibitor of protein kinases (dIPK), CG4164, TRAP1, Hsc70-5, FKBP39). The expression level of Baldspot, encoding a transmembrane protein, also strongly decreased (Table 2).
Quantitation of expressional changes for a selected subset of genes
A subset of genes potentially involved in autophagy was selected for further analysis by quantitative real-time polymerase-chain reaction (QRT-PCR) to confirm and more precisely quantitate the changes in mRNA expression levels. We also analyzed expression of a few genes not represented on our chip, including Tor, CG12334/Atg8b, and CG15283, which encodes a ubiquitin-like protein highly similar to CG7224.
As shown in Table 3, these data well correlated with the results of the microarray analysis, and further confirmed the strong upregulation of both Atg8 homologs (CG32672/Atg8a: 6.96 × and CG12334/Atg8b: 3.53 × ) and cathepsin D (CG1548: 2.14 × ), a well-known lysosomal hydrolase. The two ubiquitin-related genes we tested were also induced (CG7224: 3.73 × and CG15283: 4.62 × ). Tor mRNA level did not change, whereas genes encoding the cellular chaperones FKBP39,19 CG8286/dIPK,20 and Baldspot21 were strongly repressed (FKBP39: 6.73 × , CG8286/dIPK: 4.92 × , and Baldspot: 9.91 × repression).
Analysis of candidate autophagy-related genes in transgenic animals identified FKBP39 as an inhibitor of autophagy
To establish the role of selected genes regulated in autophagy, we carried out overexpression studies using the Gal4-UAS system. Overexpression of most genes we tested (cathD, Atg8a, Baldspot, and dIPK) produced various phenotypes with different Gal4 drivers (Supplementary Table 1), but neither induced nor inhibited autophagy (not shown).
In contrast to these results, fat body-specific overexpression of FKBP39, a peptidyl-prolyl cis–trans isomerase (PPiase) had a dramatic effect on developmental autophagy. Very few autolysosomes were observed in fat body cells of wandering larvae overexpressing FKBP39, and these were also smaller than in wild-type animals (Figure 1c, f and g). Interestingly, FKBP39 overexpression also inhibited cell growth. The average size of fat body cells overexpressing FKBP39 was 43±21% of wild-type neighboring cells (Figure 1d). The overexpressing cells also had a lower DNA content, suggesting a delay in endoreduplication. The fact that nuclei were about the same size as nuclei of wild-type cells is probably owing to the greatly enlarged nucleolus (Figure 1d and f).
We next tested if overexpression of FKBP39 had an effect on starvation-induced autophagy. Starving feeding larvae for 4 h in sucrose solution results in strong accumulation of Lysotracker-positive autolysosomes in larval fat body cells13 (Figure 1h). Overexpression of FKBP39 in the fat body greatly inhibited the induction of autophagy in starved animals (Figure 1i and k), similar to the case of developmental autophagy.
As a final verification, we then tested how loss of FKBP39 function influences autophagy using the hypomorphic mutant lines FKBP395-HA−2440 and FKBP395-HA−2590, isolated in the DrosDel project.22 Both lines harbor a P-element insertion 49 nucleotides upstream of the translation start codon, in the 5′ non-translated region of FKBP39.21 As expected from the insertion data, these two mutant lines behaved indistinguishable in all of our tests, therefore only data on FKBP395-HA−2440 is shown here (see Supplementary Figure 1 for data on FKBP395-HA−2590). Mutant larvae exhibited a strong reduction of FKBP39 expression, as analyzed by reverse transcription-polymerase chain reaction (RT-PCR) in dissected larval fat bodies (Figure 1l) and in whole adult flies (Supplementary Figure 1a). The mutants are viable and fertile, and show no obvious developmental abnormality or delay. Although we were unable to observe ectopic induction of autophagy in different larval tissues of these animals (not shown), we found that mutant larvae displayed a significantly higher level of autophagy than wild-type controls in response to a short, 80-min starvation (Figure 1m, n and p). Precise excision of the P-element (FKBP39exA3) restored wild-type mRNA levels in larval fat bodies (Figure 1l) and in whole adult flies (Supplementary Figure 1a), and reverted the autophagy phenotype seen in the mutants (Figure 1o).
Co-overexpression of PTEN restores autophagy inhibited by FKBP39
To identify the signal transduction pathway FKBP39 may act through, we tested several signaling cascades that were previously reported to be influenced by FKBPs.23, 24 Modulation of Ras (by overexpressing wild-type, hyperactive or dominant-negative Ras or wild-type Raf), protein kinase A (by overexpressing wild-type PKAc or its inhibitors PKI or mutant PKAr), or S6 kinase (by overexpressing hyperactive S6K) signaling had no effect on developmental autophagy, and did not restore autophagy in FKBP39-overexpressing fat bodies, as analyzed by Lysotracker staining (not shown). As Ras signaling was suggested to be involved in the induction of autophagy in other systems, and FKBP39 inhibited the Ras-mediated activation of MAPK (Supplementary Figure 2a), we analyzed autophagy by generating loss of function somatic clones of a Ras null mutation in the fat body. Although slightly smaller than surrounding wild-type cells, these cells showed normal induction of developmental autophagy (Supplementary Figure 2b), proving that Ras itself is not required for autophagy in this tissue.
In contrast to the above results, overexpression of PTEN, a phosphatase that antagonizes class I PI3K signaling, partially restored starvation-induced autophagy in FKBP39-expressing fat body cells (Figure 1j and k). Unfortunately, we could not test developmental autophagy in this case, as simultaneous expression of these two genes in the fat body led to a prolonged third instar stage and ultimately lethality, potentially owing to the cumulative growth-inhibiting effects of the two proteins.
The autophagy-inhibiting effect of FKBP39 is likely mediated by inhibition of Foxo, a novel regulator of autophagy
The above results suggested that FKBP39 may inhibit autophagy through activation of PI3K, a potent inhibitor of both starvation-induced and developmental autophagy.10, 13 To test this, we analyzed the level of PI3K signaling in FKBP39-overexpressing clones. GFP-pleckstrin homology (PH) domain fusion proteins are well-established markers of PI3K activity.25 In cells with high levels of PI3K activity, accumulation of the membrane lipid phosphatidylinositol-3,4,5-phosphate results in localization of the GFP-PH marker to the cell membrane. We found that FKBP39 overexpression led to a reduction of membrane versus cytoplasmic ratio of the signal compared to surrounding wild-type cells (Figure 2a). This result indicates that PI3K activity is reduced in FKBP39-overexpressing cells, thus discounting the possibility that FKBP39 inhibits autophagy by upregulating PI3K signaling.

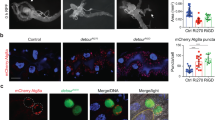
The autophagy-inhibiting effect of FKBP39 overexpression is potentially mediated by inhibition of Foxo. (a) The membrane localization of a probe used as an indicator of PI3K activity (GFP-PH, green) is greatly reduced compared to the cytoplasmic signal in fat body cells of feeding larvae that clonally overexpress FKBP39 (marked by punctate myrRFP expression, red). (a′) shows the green channel separately. (b) Foxo staining (red) in fat bodies of wild-type mid-third instar larvae shows both nuclear and cytoplasmic labeling. In cells overexpressing FKBP39 (and GFP, green), the nuclear pool of Foxo is completely abolished, and only cytoplasmic signal is seen. (b′) shows the red channel separately. (c) Null mutation of Foxo (Foxo21/Foxo25) strongly decreases starvation-induced autophagy in the larval fat body, as shown by Lysotracker staining (red) (compare to Figure 1h). (d) Overexpression of FKBP39 in a Foxo null mutant background results in a reduction similar to that seen in case of Foxo mutation or FKBP39 overexpression alone, also shown by quantitation of Lysotracker data in panel e. Asterisks mark significant changes (P<0.001 for columns 1–2, 1–3, 1–4, P>0.2 for columns 2–3, 2–4, 3–4). (f) Overexpression of an activated form of Foxo, Foxo™ strongly induces autophagy in fat body cells of feeding larvae. Inset shows the red channel (Lysotracker staining) in the overexpressing cells (delineated by a yellow line). Magnification is × 300 for panel a and × 200 for panels b–d and f. Error bars represent standard deviation in panel e. Genotypes: hsFLP; UASFKBP39/UASmyrRFP; Act>CD2>Gal4, tGPH/+ (a), hsFLP; UASFKBP39/+; Act>CD2>Gal4, UASGFPnls/+ (b), Foxo21/Foxo25 (c), cgGal4/UASFKBP39; Foxo21/Foxo25 (d), hsFLP; UASFoxo™/+; Act>CD2>Gal4, UASGFPnls/+ (f)
As another potential measure of PI3K signaling, we also examined the localization of the transcription factor Foxo in FKBP39 overexpressing clones. Elevated PI3K signaling results in phosphorylation of Foxo by the serine/threonine kinase Akt.26 In response to this phosphorylation event, Foxo changes its intracellular localization: the nuclear pool translocates to the cytoplasm.26 Wild-type mid-third instar fat body cells displayed a moderate level of nuclear Foxo. Overexpression of FKBP39 led to nearly complete exclusion of Foxo from the nucleus (Figure 2b), despite the reduced level of PI3K signaling in these cells, suggesting that the autophagy-inhibiting effect of FKBP39 may be mediated through inhibition of Foxo.
To test this hypothesis, we carried out overexpression of FKBP39 in a Foxo null mutant background. Null mutation of Foxo alone strongly interfered with starvation-induced autophagy, indicating for the first time a role for Foxo in this process (Figure 2c and e). Overexpression of FKBP39 in a Foxo null mutant background failed to further suppress autophagy compared to mutation of Foxo or overexpression of FKBP39 alone (Figure 2d and e), suggesting that the autophagy-inhibiting effect of FKBP39 is at least partly mediated by inhibition of Foxo.
Finally, we also expressed an activated version of Foxo (Foxo™)26 in fat body cells of feeding larvae. We found that Foxo™ expression was sufficient to strongly induce autophagy, further confirming the important role of Foxo in regulating autophagy (Figure 2f). Together, our results indicate that Foxo is necessary and sufficient for proper induction of autophagy, and they identify Foxo as a relevant target of FKBP39 action.
Discussion
We have used microarrays to identify genes that are regulated at the time of developmental autophagy in the larval fat body. Upregulated genes whose protein products likely function in developmental autophagy included various lysosomal hydrolases like cathepsin D and cathepsin B, and the ubiquitin-like coat proteins for autophagosomes, Atg8a and Atg8b. Expansion of the lysosomal system is usually observed during autophagy, and the upregulation of hydrolase genes is consistent with the demand for increased degrading capacity during massive induction of autophagy. Of the fly homologs of yeast autophagy genes, Atg8a and Atg8b showed induction, similarly to starved yeast cells or Drosophila larvae.1, 16 Genes of two additional small ubiquitin-like proteins, CG7224 and CG15283, were also strongly upregulated. Although the potential role of these genes in autophagy requires further studies, it is interesting to note that they share a strongly conserved human homolog, LOC135154.
Genes encoding various mitochondrial proteins were downregulated, which is consistent with the observation that a large number of mitochondria are degraded during developmental autophagy.5, 6, 7 mRNA levels of many cellular chaperones also decreased, as reported earlier (see below).
Gene expression profiling analyses under a variety of experimental conditions or developmental transitions in which induction of autophagy is observed have been reported previously.16, 27, 28 Although it is impossible to isolate autophagy from the numerous other cellular processes with which cells respond to environmental or developmental conditions that also induce autophagy, careful comparison of our results and the results from these studies enabled us to identify the most promising candidate genes that may play a common role during autophagy. Of the genes we identified as being induced during developmental autophagy in the larval fat body, several were previously found to be also upregulated in response to sucrose or no-sugar starvation, conditions that provoke a strong autophagic response in most polyploid larval tissues. These included CG7224, CG15309, CG13603, PAIP2, Atg8a and CG10992/cathepsin B.16 Moreover, all but one of these genes were also identified as upregulated during salivary gland cell death that likely involves autophagy.27, 28 The expression of Atg8, the coat protein for autophagosomes, which was identified as a rate-limiting factor for the main pathway of autophagy in yeast,15 showed only a slight induction in these studies. It may suggest that apart from the main pathway of autophagy, other forms of lysosomal degradation are potentially involved in salivary gland cell death. In fact, studies of Drosophila null mutants for dark, the fly homolog of the apoptosis gene Apaf-1, suggest that autophagy is not affected in mutant salivary glands that fail to undergo histolysis,29 similar to hypomorphic Hid mutants or to flies overexpressing the caspase inhibitor p35.30
The expression of the genes Nop56, CG8286/dIPK, CG4164/Hsp40, CG6459, Hsc70-5, CG3902, His2Av and FKBP39 was also reduced both during developmental autophagy and in response to sucrose or no-sugar starvation.16 Three of these, CG8286/dIPK, CG4164/Hsp40 and Hsc70-5 were also downregulated in dying salivary glands.27, 28
In this work, we chose three downregulated genes for detailed analysis. Overexpression of dIPK, a cellular chaperone, and Baldspot, a transmembrane protein, did not inhibit autophagy in our tests. However, it is important to note that overexpression of Atg8a or cathD had no effect on autophagy either, despite their well-established role during autophagy. Indeed, the hypomorphic Baldspot mutation l(3)02281 results in darker than usual body color.21 As pigment granules are lysosome-related organelles, it may suggest that Baldspot normally inhibits the biogenesis of these pigment granules, and its mutation results in generation of more granules.
CG8286/dIPK is a cellular chaperone containing a heat shock protein DnaJ domain, a tetratricopeptide repeat that mediates protein–protein interactions, and a protein prenyltransferase domain. The human ortholog of dIPK is the 58 kDa inhibitor of protein kinases (p58IPK), which strongly inhibits eIF2alpha protein kinases PKR and PERK.31, 32 These kinases function in mediating various stress signals such as the presence of double-stranded RNA viruses, interferon gamma, endoplasmic reticulum stress or starvation.33, 34 p58IPK was shown to inhibit virus- or TNFα-induced cell death by both eIF2alpha kinase-dependent and independent mechanisms.35 The yeast eIF2alpha kinase GCN2 is required for starvation-induced autophagy, and PKR null or non-phosphorylatable eIF2alpha mutant murine embryonic fibroblasts are defective in virus-induced autophagy.36 In our system, overexpression of dIPK did not interfere with autophagy, suggesting that eIF2alpha kinase signaling is not the major pathway functioning during this process in Drosophila. Co-overexpression of dIPK partially rescued the phenotypes caused by expression of the proapoptotic protein reaper or the caspase dronc (G.J. unpublished), proving the evolutionary conserved anti-apoptotic function of dIPK.
FKBP39 belongs to the family of PPiases with 21 members in Drosophila.21 PPiases catalyze the isomerization of the peptide bond between a proline and a bulky residue like phosphoserine or phosphothreonine. This motif is generated through the action of proline-directed kinases, the best characterized examples of which are kinases downstream of Ras. FKBPs are also the intracellular receptors of the immunosuppressive drugs rapamycin and FK506. The FKBP12-rapamycin complex binds to Tor and inhibits cell growth and induces autophagy. Physiological roles of FKBPs are diverse and not fully characterized yet, but FKBP12 is known to bind to various receptor kinases and the phosphatase calcineurin and inhibit their activity. FKBP12 also binds to ER-resident calcium channels, modulating calcium release regulation by protein kinase A and calcineurin.23
We identified FKBP39 as a downregulated gene during developmental autophagy in Drosophila, suggesting that FKBP39 is an inhibitor of autophagy. Indeed, overexpression of FKBP39 led to a strong inhibition of both developmental and starvation-induced autophagy in the larval fat body, whereas loss of FKBP39 function resulted in a higher than wild-type induction of autophagy in response to a short starvation.
PI3K signaling is a major regulator of cell growth and autophagy, and it was recently shown that during developmental reprogramming of the fat body preceding metamorphosis, ecdysone induces autophagy in part by downregulating PI3K signaling.10 In that study, expression of a dominant-negative ecdysone receptor inhibited developmental autophagy, and co-overexpression of PTEN, the phosphatase that antagonizes PI3K activity, reversed this effect. We identified FKBP39 as a potential physiological inhibitor of autophagy, its overexpression causing effects similar to inhibition of ecdysone signaling. Co-overexpression of PTEN also reversed the inhibitory effect of FKBP39 overexpression on autophagy. However, in a more direct test of PI3K activity, we found that overexpression of FKBP39 decreased the membrane localization of a probe that reflects PI3K activity. These results indicate that autophagy is inhibited in FKBP39-overexpressing cells despite a reduction in PI3K signaling. Further, they suggest that the small size of these cells may be due in part to decreased PI3K activity.
Interestingly, overexpression of FKBP39 resulted in nuclear exclusion of Foxo, suggesting that FKBP39 may inhibit autophagy through inhibition of Foxo. Foxo is a Forkhead family transcription factor activated by decreased PI3K or increased stress signaling, and has been shown to be required for growth inhibition, increased stress resistance, and lifespan extension provoked by modulation of these signaling pathways, respectively.37, 38 Mutation of Foxo caused a similar reduction of starvation-induced autophagy as overexpression of FKBP39, and overexpression of activated Foxo was sufficient to induce autophagy in feeding larvae, demonstrating that Foxo is indeed involved in the regulation of autophagy. Overexpression of FKBP39 in a Foxo mutant background did not significantly decrease autophagy compared to cells in which Foxo was mutated or FKBP39 was overexpressed alone. This result suggests that at least part of the effect of FKBP39 overexpression on autophagy is mediated by its inhibition of Foxo, as one would expect additive effects of autophagy inhibition in case of independent signaling pathways. Other effects caused by overexpression of FKBP39 (enlarged nucleolus and larval-prepupal lethality) were not affected by the Foxo mutant background.
Given the known targets of PPiases, the effect of FKBP39 on Foxo localization is probably indirect. In Drosophila, Foxo localization and activity has been shown to be regulated through phosphorylation by PI3K/Akt and Jun N-terminal kinase (JNK) signaling.26, 38 On the basis of our results, PI3K signaling is unlikely to mediate the effects of FKBP39 overexpression on Foxo localization, although it remains possible that FKBP39 may affect Akt independently of PI3K. Therefore, JNK signaling is a promising candidate for mediating at least some of the effects of FKBP39 overexpression,24 especially considering the strong effect of FKBP39 overexpression on kinases downstream of Ras that are closely related to JNK family kinases. This issue clearly warrants future studies.
In summary, we have identified numerous genes regulated during developmental autophagy in larval Drosophila fat body, and we have shown that FKBP39, a gene downregulated during the process, encodes a relevant physiological inhibitor of autophagy. We have also identified Foxo as a novel regulator of autophagy, potentially mediating the inhibitory effect of FKBP39 on autophagy.
Materials and methods
Drosophila lines and methods
The following Drosophila lines were used in our study: EP362 (Atg8a), EP2151 (cathD) (Szeged Stock Center, Hungary), UASmyrRFP, hsGal4, cgGal4, GMRGal4, actGal4, tubGal4, eyGal4, Df(3R)Exel6174 (Bloomington Stock Center, IN, USA), FKBP395-HA−2440 and FKBP395-HA−2590 (kindly provided by Gunther Reuter), FRT82B Rasc40b (kindly provided by Celeste Berg), hsFLP, Act>CD2>Gal4, UASGFPnls and Act>CD2>Gal4, tGPH (kindly provided by Bruce A. Edgar), Foxo25 and Foxo21 (kindly provided by Heinrich Jasper and Ernst Hafen), and UASFoxo™ (kindly provided by Marc Tatar). Fruit flies were maintained at room temperature (23–25°C). Heat shocks were carried out by immersing vials in a 37°C water bath for 1 h. Gain of function clones in the fat body were spontaneously generated by the leaky expression of FLP recombinase in hsFLP; Act>CD2>Gal4, UASGFPnls/UASFKBP39 animals.25 We generated Ras null mutant clones in the fat body by heat-shocking 0–8 h old embryos of the genotype hsFLP; cgGal4/+; FRT82B, UASGFP/FRT82B, Rasc40b. As expected, no Ras null mutant cells were recovered in the eyes of eyFLP; FRT82B, M(3)95A/FRT82B, Rasc40b animals. For P element excision experiments, transposase was reintroduced to FKBP395-HA−2440 and FKBP395-HA−2590 animals (both lines have a P element inserted at the same place in the 5′ UTR of FKBP39, 49 bases upstream of the translation start codon). Multiple clean excision lines were identified by PCR and analyzed for each mutant as described in Theopold et al.29 FKBP39exA3 is a revertant for FKBP395-HA−2440, and FKBP39exB1 is a revertant for FKBP395-HA−2590.
Molecular cloning and establishment of transgenic lines
Full-length cDNAs encoding Baldspot, dIPK and FKBP39 (EST# GH11554, LD25575 and LD30817, kindly provided by Miklós Erdélyi) were cloned into the pUAST fly transformation vector using EcoRI and XhoI restriction sites. The resulting plasmids were sequenced and injected into fruit fly embryos using standard microinjection techniques. Multiple transgenic lines were established and analyzed for each construct with similar results.
Microarrays, probe preparations, hybridizations, scanning, data analysis
Construction and use of microarrays were performed as described.39 Briefly, 3200 amplified cDNA inserts from Drosophila melanogaster were purified with MultiScreen-PCR plate (Millipore), resuspended in 50% dimethylsulfoxide/water, and arrayed on amino-silanized slides (Sigma-Aldrich, Germany) by using a MicroGrid Total Array System (BioRobotics, Genomic Solutions Ltd., UK) spotter with 16 pins with a 4 × 4 format. All clones were spotted in duplicate. Before to hybridization, the slides were blocked in 1 × SSC, 0.2% SDS, 1% BSA for 30 min at 42°C, washed with water and dried with high pressure air. Fat bodies were manually dissected from 150 to 200 carefully staged feeding and wandering animals. All tissues except the embedded gonad disks were dissected away before freezing fat bodies in liquid nitrogen. Fifteen microgram of total RNA from each sample was amplified by a linear antisense RNA amplification method, and labeled with Cy3 or Cy5 fluorescent dye during reverse transcription as described previously.40 Probes were mixed, reconstituted in 12 μl hybridization buffer (50% formamide, 5 × SSC, 0.1% SDS, 100 mg/ml salmon sperm DNA) and applied onto the array after denaturation by heating for 1 min at 90°C. The slide was covered and incubated at 42°C for 20 h in a humid hybridization chamber. After hybridization the arrays were washed by submersion and agitation for 10 min in 1 × SSC with 0.1% SDS, for 10 min in 0.1% × SSC with 0.1% SDS and for 10 min in 0.1 × SSC at room temperature, then rinsed briefly in deionized water and dried. Each array was scanned under a green laser (532 nm) (for Cy3 labeling) and under a red laser (660 nm) (for Cy5 labeling) by using a ScanArray Lite (GSI Lumonics, UK) scanning confocal fluorescent scanner with 10 μm resolution. Scanned output files were analyzed using the GenePix Pro 3.0 software (Axon Instruments Inc, Foster City, CA, USA). Each spot was defined by automatic positioning of a grid of circles over the image. The average and median pixel intensity ratios calculated from both channels and the local background of each spot were determined. An average expression ratio (MeaR, denotes the average of local background corrected pixel intensity ratios) was determined for each spot. Data analysis was done by the significance analysis of microarrays (SAM) method and visualization of scatter images were performed with the Microsoft EXCEL software.
Quantitative real-time PCR and reverse transcription PCR
QRT-PCR was performed on a RotorGene 2000 instrument (Corbett Research, Australia) with gene-specific primers and SybrGreen protocol to confirm the gene expression changes observed by using microarrays. Twenty micrograms of total RNA from each pool was reverse-transcribed in the presence of poly(dT) sequences in a total volume of 20 μl. After dilution of the mix with 80 μl of water, 2 μl of this mix was used as template in the QRT-PCR reaction. Relative expression ratios were normalized to actin. For RT-PCR, total RNA was isolated from fat bodies dissected from feeding L3 larvae or from whole adult flies, using RNAlater and RNeasy Micro Kit (Qiagen, Germany). cDNA synthesized with RevertAid First Strand cDNA Synthesis Kit (Fermentas, Lithuania) was used in RT-PCR reactions with a control primer pair specific for actin, and three different primer pairs for FKBP39 that gave identical results. We performed 25 amplification cycles in these reactions to avoid reaching saturation phase. PCR primer sequences used in this study are available upon request. All PCRs were performed in triplicates.
Histology
Lysotracker red staining of dissected larval fat bodies, cortical actin-phalloidin staining and electron microscopy was done as described previously.8, 10, 13 Anti-Foxo antibody26 was used at a dilution of 1 : 300. Student's t-test was used to determine probability values in quantification experiments.
Western blots
Western blots were carried out as described in Juhasz et al.8 using an antibody specific to di-phosphorylated MAPK (Sigma) at a dilution of 1 : 1000.
Abbreviations
- dIPK:
-
Drosophila inhibitor of protein kinases
- FKBP39:
-
FK506-binding protein of 39 kDa
- JNK:
-
Jun N-terminal kinase
- PI3K:
-
phosphatidyl-inositol 3-kinase
- Ppiase:
-
peptidyl-prolyl cis-trans isomerase
- QRT-PCR:
-
quantitative real-time polymerase chain reaction
- RT-PCR:
-
reverse transcription polymerase chain reaction
- Tor:
-
target of rapamycin
References
Klionsky DJ, Emr SD . Autophagy as a regulated pathway of cellular degradation. Science 2000; 290: 1717–1721.
Klionsky DJ, Cregg JM, Dunn Jr WA, Emr SD, Sakai Y, Sandoval IV et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell 2003; 5: 539–545.
Levine B, Klionsky DJ . Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004 Apr; 6: 463–477.
Mizushima N . The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ 2005 Nov; 12 (Suppl 2): 1535–1541.
Sass M, Kovacs J . The effect of ecdysone on the fat body cells of the penultimate larvae of Mamestra brassicae. Cell Tissue Res 1977; 180: 403–409.
Butterworth FM, Forrest EC . Ultrastructure of the preparative phase of cell death in the larval fat body of Drosophila melanogaster. Tissue Cell 1984; 16: 237–250.
Butterworth FM, Emerson L, Rasch EM . Maturation and degeneration of the fat body in the Drosophila larva and pupa as revealed by morphometric analysis. Tissue Cell 1988; 20: 255–268.
Juhasz G, Csikos G, Sinka R, Erdelyi M, Sass M . The Drosophila homolog of Aut1 is essential for autophagy and development. FEBS Lett 2003; 543: 154–158.
Sass M, Kovacs J . Ecdysterone and an analogue of juvenile hormone on the autophagy in the cells of fat body of mamestra brassicae. Acta Biol Acad Sci Hung 1975; 26: 189–196.
Rusten TE, Lindmo K, Juhasz G, Sass M, Seglen PO, Brech A et al. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev Cell 2004 Aug; 7: 179–192.
Noda T, Kim J, Huang WP, Baba M, Tokunaga C, Ohsumi Y et al. Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J Cell Biol 2000 Feb 7; 148: 465–480.
Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ . Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem 1995; 270: 2320–2326.
Scott RC, Schuldiner O, Neufeld TP . Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell 2004 Aug; 7: 167–178.
Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y . Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 2000; 150: 1507–1513.
Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T et al. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol 1999; 147: 435–446.
Zinke I, Schutz CS, Katzenberger JD, Bauer M, Pankratz MJ . Nutrient control of gene expression in Drosophila: microarray analysis of starvation and sugar-dependent response. EMBO J 2002; 21: 6162–6173.
Szuperak M, Zvara A, Erdelyi M . Identification of germ plasm-enriched mRNAs in Drosophila melanogaster by the cDNA microarray technique. Gene Exp Patterns 2005 Jun; 5: 717–723.
Tearle R . Tissue specific effects of ommochrome pathway mutations in Drosophila melanogaster. Genet Res 1991 Jun; 57: 257–266.
Theopold U, Dal Zotto L, Hultmark D . FKBP39, a Drosophila member of a family of proteins that bind the immunosuppressive drug FK506. Gene 1995 Apr 24; 156: 247–251.
Melville MW, Katze MG, Tan SL . P58IPK, a novel cochaperone containing tetratricopeptide repeats and a J-domain with oncogenic potential. Cell Mol Life Sci 2000 Feb; 57: 311–322.
Drysdale RA, Crosby MA . FlyBase: genes and gene models. Nucleic Acids Res 2005 Jan 1; 33: D390–D395.
Ryder E, Blows F, Ashburner M, Bautista-Llacer R, Coulson D, Drummond J et al. The DrosDel collection: a set of P-element insertions for generating custom chromosomal aberrations in Drosophila melanogaster. Genetics 2004 Jun; 167: 797–813.
Harrar Y, Bellini C, Faure JD . FKBPs: at the crossroads of folding and transduction. Trends Plant Sci 2001 Sep; 6: 426–431.
Klettner A, Baumgrass R, Zhang Y, Fischer G, Burger E, Herdegen T et al. The neuroprotective actions of FK506 binding protein ligands: neuronal survival is triggered by de novo RNA synthesis, but is independent of inhibition of JNK and calcineurin. Brain Res Mol Brain Res 2001 Dec 16; 97: 21–31.
Britton JS, Lockwood WK, Li L, Cohen SM, Edgar BA . Drosophila's insulin/PI3-kinase pathway coordinates cellular metabolism with nutritional conditions. Dev Cell 2002; 2: 239–249.
Hwangbo DS, Gersham B, Tu MP, Palmer M, Tatar M . Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature 2004 Jun 3; 429: 562–566.
Lee CY, Clough EA, Yellon P, Teslovich TM, Stephan DA, Baehrecke EH . Genome-wide analyses of steroid- and radiation-triggered programmed cell death in Drosophila. Curr Biol 2003 Feb 18; 13: 350–357.
Gorski SM, Chittaranjan S, Pleasance ED, Freeman JD, Anderson CL, Varhol RJ et al. A SAGE approach to discovery of genes involved in autophagic cell death. Curr Biol 2003 Feb 18; 13: 358–363.
Akdemir F, Farkas R, Chen P, Juhasz G, Medved'ova L, Sass M et al. Autophagy occurs upstream or parallel to the apoptosome during histolytic cell death. Development 2006 Apr; 133: 1457–1465.
Juhasz G, Sass M . Hid can induce, but is not required for autophagy in polyploid larval Drosophila tissues. Eur J Cell Biol 2005 Apr; 84: 491–502.
Yan W, Frank CL, Korth MJ, Sopher BL, Novoa I, Ron D et al. Control of PERK eIF2alpha kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc Natl Acad Sci USA 2002 Dec 10; 99: 15920–15925.
Tan SL, Gale Jr MJ, Katze MG . Double-stranded RNA-independent dimerization of interferon-induced protein kinase PKR and inhibition of dimerization by the cellular P58IPK inhibitor. Mol Cell Biol 1998 May; 18: 2431–2443.
Pain VM . Translational control during amino acid starvation. Biochimie 1994; 76: 718–728.
Williams BR . PKR; a sentinel kinase for cellular stress. Oncogene 1999 Nov 1; 18: 6112–6120.
Tang NM, Korth MJ, Gale Jr M, Wambach M, Der SD, Bandyopadhyay SK et al. Inhibition of double-stranded RNA- and tumor necrosis factor alpha-mediated apoptosis by tetratricopeptide repeat protein and cochaperone P58(IPK). Mol Cell Biol 1999 Jul; 19: 4757–4765.
Talloczy Z, Jiang W, Virgin HWt, Leib DA, Scheuner D, Kaufman RJ et al. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA 2002 Jan 8; 99: 190–195.
Junger MA, Rintelen F, Stocker H, Wasserman JD, Vegh M, Radimerski T et al. The Drosophila Forkhead transcription factor FOXO mediates the reduction in cell number associated with reduced insulin signaling. J Biol 2003; 2: 20.
Wang MC, Bohmann D, Jasper H . JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell 2005 Apr 8; 121: 115–125.
Puskas LG, Zvara A, Hackler Jr L, Micsik T, van Hummelen P . Production of bulk amounts of universal RNA for DNA microarrays. Biotechniques 2002 Oct; 33: 898–900, 902, 904..
Puskas LG, Zvara A, Hackler Jr L, Van Hummelen P . RNA amplification results in reproducible microarray data with slight ratio bias. Biotechniques 2002 Jun; 32: 1330–1334, 1336, 1338, 1340.
Acknowledgements
We thank Zsoltné Pálfia, Mariann Saródy and Emese Léder for the excellent technical assistance, our colleagues listed in the Materials and Methods section for providing reagents, and our anonymous reviewers for their helpful comments. This work was supported by NIH grant RO1 GM62509 provided to TPN and by the Hungarian Ministry of Education grant MEDICHEM 1/047 NKFP provided to MS.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by E Baehrecke
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Rights and permissions
About this article
Cite this article
Juhász, G., Puskás, L., Komonyi, O. et al. Gene expression profiling identifies FKBP39 as an inhibitor of autophagy in larval Drosophila fat body. Cell Death Differ 14, 1181–1190 (2007). https://doi.org/10.1038/sj.cdd.4402123
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402123
Keywords
This article is cited by
-
Identification, characterization and spatiotemporal expression analysis of the FKBP family genes in Locusta migratoria
Scientific Reports (2023)
-
Early-adult methionine restriction reduces methionine sulfoxide and extends lifespan in Drosophila
Nature Communications (2023)
-
FKBP39 controls nutrient dependent Nprl3 expression and TORC1 activity in Drosophila
Cell Death & Disease (2021)
-
Proteostasis-associated aging: lessons from a Drosophila model
Genes & Genomics (2021)
-
FOXO1 promotes HIV latency by suppressing ER stress in T cells
Nature Microbiology (2020)

{kind=link}
{kind=link}