
Key message
ddRAD-seq-based high-density genetic map comprising 2595 loci identified a major and consensus QTL with a linked marker in a 0.8-Mb physical interval for oil content in peanut.
Abstract
Enhancing oil content is an important breeding objective in peanut. High-resolution mapping of quantitative trait loci (QTLs) with linked markers could facilitate marker-assisted selection in breeding for target traits. In the present study, a recombined inbred line population (Xuhua 13 × Zhonghua 6) was used to construct a genetic map based on double-digest restriction-site-associated DNA sequencing (ddRAD-seq). The resulting high-density genetic map contained 2595 loci, and spanned a length of 2465.62 cM, with an average distance of 0.95 cM/locus. Seven QTLs for oil content were identified on five linkage groups, including the major and stable QTL qOCA08.1 on chromosome A08 with 10.14–27.19% phenotypic variation explained. The physical interval of qOCA08.1 was further delimited to a ~ 0.8-Mb genomic region where two genes affecting oil synthesis had been annotated. The marker SNPOCA08 was developed targeting the SNP loci associated with oil content and validated in peanut cultivars with diverse oil contents. The major and stable QTL identified in the present study could be further dissected for gene discovery. Furthermore, the tightly linked marker for oil content would be useful in marker-assisted breeding in peanut.
Similar content being viewed by others
Introduction
Peanut (Arachis hypogaea L.) is one of the word’s major sources of vegetable oil. It is grown in more than 100 countries, with the global production of 43.98 Mt harvested from 27.66 Mha (FAOSTAT 2016). China, the world’s largest peanut producer, accounts for 38% of global peanut production. However, the domestic production of peanut hardly meets the increasing demands for peanut oil. Due to the large variation of oil content in the peanut germplasm (Yol et al. 2017), it is highly feasible to develop cultivars with high oil content and yield to enhance oil productivity. In addition, an increase of 1% in the oil content in peanuts could result in a 7% increase in economic benefit for oil processing (Shasidhar et al. 2017). Therefore, high oil content is a highly desirable trait which has been one of the key breeding objectives in many countries.
The trait of oil content is polygenetically inherited and is largely influenced by the environment (Baring et al. 2013; Wilson et al. 2013). Moreover, measuring oil content requires relatively expensive and complex equipment, and can be carried out only after harvest. Traditional breeding for improvement of oil content has a low efficiency in addition to being time-consuming. Marker-assisted selection (MAS) has provided a robust and reliable tool for accelerating the development of varieties with desirable target traits in many crops including peanut (Chu et al. 2011; Janila et al. 2016b; Varshney et al. 2014; Varshney 2016). However, the availability of well-validated genetic markers for the target trait is a prerequisite to deploy MAS in peanut. Previously, four minor quantitative trait loci (QTLs) were identified for oil content based on a genetic map with 45 SSR loci (Sarvamangala et al. 2011). Subsequently, six and nine QTLs for oil content were mapped on two genetic maps with 206 and 378 SSR loci, respectively (Pandey et al. 2014). Most recently, three QTLs for oil content have been identified in an advanced backcross population with 91 SSR markers (Wilson et al. 2017). All these studies could only develop low-density genetic maps leading to low-resolution of QTL mapping, while the genetic markers also lacked information on the physical location on the genome, which restricted further investigation and discovery of candidate genes and markers controlling oil accumulation.
Several efforts have been made previously for QTL discovery using sparse genetic maps constructed using SSRs (Huang et al. 2015; Varshney et al. 2009; Wang et al. 2012, 2015). Since the genetic base of cultivated peanut is narrow (Belamkar et al. 2011; Mukri et al. 2012), available SSR markers reported in earlier studies are limited for developing saturated genetic maps. However, single nucleotide polymorphisms (SNPs) are abundant and widely distributed across the genome. Based on the next-generation sequencing platform, double-digest restriction-site-associated DNA sequencing (ddRAD-seq) has been developed for SNP mining and genotyping in biparental mapping populations (Baird et al. 2008; Peterson et al. 2012). This method combines reduced representation libraries with multiplex sequencing strategies, which could overcome the complexity of the genome and reduce the cost of sequencing. Employing this technology has led to the successful development of high-density genetic maps (HDGMs) for conducting high-resolution mapping of QTLs in many crops (Chen et al. 2017; Chutimanitsakun et al. 2011; Liu et al. 2017; Pfender et al. 2011).
In peanut, few HDGMs have been deployed in QTL analysis for oil content. In the present study, ddRAD-seq was performed for the recombinant inbred line (RIL) population to address the following objectives: (1) construction of a HDGM based on SNPs, (2) identification of stable QTLs for oil content across multiple environments and delimitation of their physical regions in reference genomes, and (3) development of markers that are highly associated with oil content.
Materials and methods
Plant materials
The mapping population consisting of 186 RILs was derived from a cross between Xuhua 13 and Zhonghua 6 through the single seed descent method. The male parent (Zhonghua 6) is a Spanish-type variety belonging to A. hypogaea ssp. fastigiata. The female parent (Xuhua 13) is an intermediated-type variety, which has higher oil content than Zhonghua 6 (Table 1). Four generations of the RIL population, i.e., from F5 (2014) to F8 (2017) were used for phenotyping and the leaf samples from the F6 generation lines were used for DNA isolation and genotyping. In addition, a panel of 42 peanut cultivars with diverse oil content was collected from 14 provinces in China (Table S1) and phenotyping data in these cultivars were measured from 2014 to 2016.
Field trials and phenotyping
The RIL population, their parents, and a panel of 42 cultivars were planted in the experimental field of OCRI-CAAS, Wuhan. Field trials were performed on a randomized complete block design with three replications. Each plot contained 12 plants at a spacing of 20 cm × 30 cm. Field management followed standard agricultural practice.
Both Oil% and H2O% in seeds were measured using nuclear magnetic resonance (PQ001; Niumag, China). Approximately 10 g seeds with less than 10% moisture content were weighed and analyzed for each of the three subsamples per entry. According to a previous study, oil content (%) was determined based on dry-weight using the formula {[oil%/(100 − H2O%)] × 100} (Pandey et al. 2014).
Statistical analysis of phenotypic data
Phenotypic data were obtained from three biological replications in each field trial. The IBM SPSS Statistics software (v.22; IBM, USA) was used to analyze the phenotypic data. The normal distribution of phenotypic values was assessed by the Shapiro–Wilk test. Significant phenotypic differences among genotypes were assessed by Bonferroni’s Multiple Comparison Test. The field trial with three replications in each year was treated as a single environment, and the univariate variance analysis was performed to evaluate the effects of genotype, environment, and the interactions between genotype and environment on the phenotypic variance of oil content.
To estimate broad-sense heritability, the phenotypic variance was resolved into the genotypic variance (\(\sigma_{\text{g}}^{2}\)), the environmental variance (\(\sigma_{\text{e}}^{2}\)), the genotype × environment interaction variance (\(\sigma_{{{\text{g}} \times {\text{e}}}}^{2}\)), and the residual variance (\(\sigma_{\varepsilon }^{2}\)) using R (version 3.4.2) package “lme4” with the linear mixed model method (https://cran.r-project.org/web/packages/lme4/index.html). The entry mean-based broad-sense heritability of oil content trait was calculated as: \(H^{2} = \sigma_{\text{g}}^{2} /(\sigma_{\text{g}}^{2} + \sigma_{{{\text{g}} \times {\text{e}}}}^{2} /r + \sigma_{\varepsilon }^{2} /rn)\), where r represents the number of environments and n represents the number of replications in each environment.
Library construction and restriction-site-associated DNA (RAD) sequencing
Genomic DNA was extracted from young leaves of the RIL population and both parents using a genomic DNA extraction kit (TIANGEN, Beijing, China). Two endonucleases EcoRI (GAATTC) and MseI (TTAA), were applied to digest the genomic DNA. Library construction procedures have been previously described in detail (Baird et al. 2008; Wu et al. 2014). Finally, 250- to 450-bp fragments were excised and purified for pair-end sequencing. RAD sequencing was performed on a Hiseq4000 platform in BGI. Raw data have been deposited in GenBank (BioProject: PRJNA520741).
SNP identification and genotyping
The software Stacks was used to identify SNPs and determine genotypes for the RIL population (Catchen et al. 2013). First, raw sequence reads were demultiplexed by their barcodes and low-quality reads were removed. Then, reads from each individual were clustered, aligned with each other, and scored for polymorphic loci. Putative loci markers with more than 50% missing data were filtered out. The remaining markers were mapped to two referenced genomes (Arachis duranensis and A. ipaensis) and only the uniquely mapped markers were used for the further construction of the genetic map.
Genetic map construction and QTL analysis
The high-density linkage map was constructed using Joinmap software (Stam 1993). The mapping algorithm was based on regression mapping, and the Kosambi mapping function was applied to transform the recombinant ratio into genetic distance. Goodness of fit to the expected segregation ratio 1:1 for each locus was assessed by Pearson’s Chi square test (P < 0.01). SNP markers in the genetic map were aligned to chromosomes of the two diploid ancestors (A. duranensis and A. ipaensis) using the BLAST tool (https://blast.ncbi.nlm.nih.gov). CIRCOS software was applied to analyze the collinearity of the SNP loci between the genetic position and the physical position (Krzywinski et al. 2009).
The HDGM with multiple-years phenotypic data was used to identify QTLs for oil content. QTL IciMapping software, with inclusive composite interval mapping, was employed in the QTL analysis (Meng et al. 2015). The mapping parameters were set to 1.0 cM step, 0.001 probability, and a LOD threshold score of 2.5.
To compare current QTLs with previously reported QTLs, the physical positions of markers in the present and previous LGs, which harbored QTLs for oil content, were analyzed. After aligning the primer sequence of markers to the referenced genomes (A. duranensis and A. ipaensis), the markers in the current and earlier genetic maps were integrated into the corresponding chromosomes. The physical position of QTLs was estimated via linked markers.
Annotation of genes in the QTL intervals
The markers linked to QTLs were aligned to the genomes of the two diploid ancestors of cultivated peanut (Bertioli et al. 2016). According to the physical position of flanking markers, annotated information on the putative genes in the QTL interval was downloaded from PeanutBase (https://www.peanutbase.org/home). Differentially expressed genes between high-oil cultivars and low-oil cultivars were obtained from Wang et al. (2018a).
To identify genes related to oil biosynthesis within the QTL interval, genes reported to be involved in oil biosynthesis in Arabidopsis were extracted from the proteome of Arabidopsis (Bates et al. 2014) and searched against the gene set in A. duranensis and A. ipaensis using the BLAST tool with an E-value cutoff of 1e−5 (https://blast.ncbi.nlm.nih.gov).
Cluster analysis of gene expression profile
The software Genesis (v.1.7.6) was used to perform hierarchical clustering of genes within the seven QTLs that were identified in the current study (Sturn et al. 2002). Unweighted pair-group average linkage was used to calculate distances of gene clusters. The information on gene abundance in 22 tissues was downloaded from Clevenger et al. (2016). Genes with expression levels below 5 FPKM in any tissues were filtered out, and the remaining genes in the QTL intervals were used to make a heat map.
Results
Phenotypic analysis of oil content in RIL population
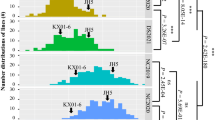
The oil content trait for 186 RILs and their parents were measured across four consecutive years (2014–2017). The oil content of the female parent (Xuhua 13) was consistently higher than that of the male parent (Zhonghua 6) across multiple environments (Table 1). The phenotypic data of the RIL population varied from 43.19 to 51.80% in 2014, from 47.90 to 56.37% in 2015, from 45.30 to 54.18% in 2016, and from 45.18 to 52.97% in 2017. Continuous distributions with transgressive segregation of phenotypic values of the RIL population are shown in Fig. 1. The Shapiro–Wilk (w) normality test indicates that the phenotypic data of the RIL population across four consecutive years were normally distributed (Table 1). Variance analysis across all four environments showed that genetic and environmental factors both significantly influenced oil content in RIL population at the P < 0.001 level (Table 2). However, interaction between genotype and environment only showed a moderate influence on the phenotype (P = 0.03). The broad sense heritability of oil content was estimated to be 0.84 in the RIL population.

Phenotypic distribution of oil content in the RIL population across four consecutive years (2014–2017). The y-axis represents the density and the x-axis represents the oil content of seeds (%)
Genotyping the RIL population through ddRAD-seq
After filtration of adapter and low-quality sequences, a total of 359.96 Gb of clean data (containing 3712.28 million reads) was obtained from RAD sequencing for 186 RILs and their parents. The Q20 ratio was 96.67% and the average GC content was 38.85% across the samples. High-quality reads of each individual were further aligned with each other and were subsequently clustered to RAD tags. The numbers of RAD tags in the female parent (Xuhua 13) and the male parent (Zhonghua 6) were 4.68 million and 4.65 million, respectively. Their corresponding sequencing depths were 55.33 and 53.13, respectively. In the RIL population, the RAD tag numbers ranged from 402,454 to 1,110,512 and the sequencing depths varied from 10.39 to 22.77 (Figs. S1A, S1B). These data were used to analyze SNPs and to catalog polymorphic loci. In total, 16,042 polymorphic loci across the whole RIL population were obtained. Considering that the two parents of the RIL population are homozygous, only aa × bb type loci containing 8064 SNPs markers were used in the construction of the genetic map. In each individual line, the number of genotyping aa × bb type loci ranged from 5377 to 7714 (Fig. S1C). These SNPs loci were further filtered using the stringent criterion described in “Materials and methods”, and 2595 SNP markers were mapped on the final genetic linkage map (Table S2).
Construction of high-density genetic map
The final genetic map harbored 2595 SNP loci which covered a total distance of 2465.62 cM with an average length of 0.95 cM/locus (Fig. 2a; Table 3). LG A01–A10 were assigned to subgenome A, which contained 1235 loci with a total length of 1256.94 cM. LG B01–B10 were assigned to subgenome B, which contained 1360 loci spanning 1208.68 cM in length. Among the 20 LGs in the genetic map, 14 LGs have more than 100 loci. B10 was the largest LG, which harbored 218 loci with an average marker interval of 0.91 cM/locus. A04 and B09 harbored less than 50 loci and were the two shortest LGs in the genetic map. The average marker interval ranged from 0.51 cM/locus in B05 to 3.71 cM/locus in B09, and more than half of the LGs (12) had marker densities of below 1 cM/locus. The index of “Gap ≤ 5”, which was used to evaluate the degree of linkage of between markers, was shown to have an average value of 96.11%. The largest gaps in the genetic map were 23.54 cM on LG A06, followed by 23.43 cM on LG B10. Among the 2595 loci in the genetic map, 18.6% of loci (483) were shown to have distorted segregation (P < 0.01). LG B05 harbored the largest number of distorted loci (152), whereas LG A08, B09, and B10 contained less than four distorted loci.

Overview of the genetic map constructed using SNP markers. a Distribution of SNP markers on 20 linkage groups. Black bars denote SNP markers. The linkage group numbers are shown on the x-axis. b Collinearity analysis between the genetic map and the physical map. a01–a10 and b01–b10 represent the 20 linkage groups. A01–A10 and B01–B10 indicate physical maps of Arachis duranensis and Arachis ipaensis, respectively
To further assess the quality of the genetic map, collinearity analysis was conducted to compare between the genetic and physical positions of the SNP loci. Each locus in the genetic map was mapped to the referenced genomes. The physical distance between the start and end loci of each LG were estimated to cover 80% of the total genomes of the two diploid ancestors. Most loci in the genetic map had the same order as those on the corresponding chromosomes of the two reference genome generally. LGs assigned to subgenome A had better compatibility with the physical map than LGs assigned to subgenome B (Fig. 2b). LGs A03, B06, B07 and B09 showed a degree of deviation in the collinearity analysis.
Identification of quantitative loci for oil content trait
In total, seven QTLs for oil content were mapped to the HDGM across four environments (Fig. S2; Table 4). The seven QTLs with 6.07–27.19% explained phenotypic variation (PVE), located on five LGs, namely, LG A04 (qOCA04.1), LG A05 (qOCA05.1), LG A08 (qOCA08.1 and qOCA08.2), LG B05 (qOCB05.1 and qOCB05.2) and LG B06 (qOCB06.1) (Table 4). The genetic distance of the QTL intervals ranged from 0.8 cM for qOCB06.1 to 7.1 cM for qOCB05.2, and the average distance of QTL interval was below 2.6 cM. Based on the sequences of flanking markers, these QTLs were mapped to the reference genomes of A. duranensis and A. ipaensis. The physical intervals of QTLs ranged from 0.3 Mb to 2.5 Mb, and the number of genes within these intervals varied from 12 to 217 (Tables 4, S3). When BLAST was performed with these genes against homologous genes in Arabidopsis (Bates et al. 2014), a total of 22 putative genes were found to be involved in 11 oil-related pathways. These pathways included plastidial glycerolipid, galactolipid and sulfolipid synthesis, sphingolipid synthesis, aromatic suberin synthesis, lipid-related synthesis, lipid signaling, eukaryotic phospholipid synthesis, cuticular wax synthesis, lipase synthesis, cutin synthesis, triacylglycerol biosynthesis and triacylglycerol degradation. In addition, 21 putative transcription factor genes belonged to 15 families, specifically FAR1, bHLH, MYB-related, G2-like, WRKY, TALE, NF-Y, AP2, HSF, BES1, HD-ZIP, bZIP, C2H2, MYB, and NAC (Wilson et al. 2008). Based on publicly released transcriptome data for tetraploid peanut (Arachis hypogaea cv. Tifrunner, Clevenger et al. 2016), genes abundances in 22 tissues were obtained including complete seed developmental stages. Genes in the seven QTL intervals could be classified into four subgroups, i.e., Sub A, B, C and D, based on their expression profiles (Fig. S3, Table S3). In Sub A, most genes were highly expressed in leaves. In Sub B, most genes were highly expressed in shoot tips. In Sub C, the expression levels of most genes were relatively higher in roots and reproductive tissues than in leaves, flowers, and pistils. In Sub D, more than half of the genes were predominantly expressed in seeds at different developmental stages. In addition, 36 genes in the QTL intervals have been reported to be differentially expressed between high-oil cultivars and low-oil cultivars (Wang et al. 2018a). Of these 36 genes, three oil-related genes and one transcription factor gene were upregulated in high-oil cultivars (Table S3).
Among the seven QTLs, qOCA08.1 with 10.14–27.19% PVE could be repeatedly detected across 3 years. The consensus QTL qOCA08.1 between the markers AhMXZ190701 and AhEXZ283046 was aligned to a ~ 0.8-Mb (41.9–42.7 Mb) genomic region of pseudomolecule A08 of A. duranensis (Fig. 3). After resequencing the genomes of the two parents (Appendix S1), 38 SNPs were found to be within the physical region of qOCA08.1 (Table S4). Two SNPs were located on the introns of two genes that encoded a calcium-dependent protein, Annexin (Aradu.N8MUP), and a flavin-binding monooxygenase family protein (Aradu.1F2UP). Six SNPs were located upstream or downstream of six genes that encoded the proline-rich family protein (Aradu.VWM5Q), Ulp1 protease family proteins (Aradu.VZ1DU and Aradu.D7VM0), NF-Y transcription factor (Aradu.S0YHI), unknown proteins (Aradu.F5MYE), and flavin-binding monooxygenase family protein (Aradu.1F2UP). The remaining 30 SNPs were located in intergenic regions.

Location of QTLs on LG A08 and the corresponding physical map. The different colored boxes represent the QTLs detected during different years. The markers that tightly link to the QTL are highlighted in red. Gene density represents gene number per 80-kb interval
Development and validation of markers for oil content
The flanking markers of QTL qOCA08.1 were AhMXZ190701 and AhEXZ283046. For the AhMXZ190701 locus, oil contents of the lines that harbored the homozygous allele of the female parent were significantly higher than that of the male parent across four consecutive years (P < 0.01; Fig. 4a; Table S5). Similarly, a significant difference in oil content was found between lines with alleles from the female parent and the male parent at the AhEXZ283046 locus (Table S5). Values for agronomic traits, 100-pod weight, and plant height of lines with alleles from the male parent did not significantly differ from lines with alleles from the female parent at two loci (AhMXZ190701 and AhEXZ283046) (P < 0.05; Table S6). Since QTL qOCA08.1 had a large effect on oil content across multiple environments, the tightly linked SNP markers, AhMXZ190701 and AhEXZ283046, are a potentially valuable application in MAS for a high-oil trait.

Validation of the AhMXZ190701 SNP locus for high-oil content selection. a Phenotypic difference between two genotypes at the AhMXZ190701 SNP locus in the RIL population. The AA homozygous allele originated from Xuhua 13 (female parent) and the aa homozygous allele originated from Zhonghua 6 (male parent). b Geographic distribution of the 42 cultivars in China. Cultivars carrying nucleotide T at the SNP site are represented by a red circle. Cultivars carrying nucleotide C at the SNP site are denoted by a blue triangle. c Phenotypic difference between accessions with nucleotide C and accessions with nucleotide T at the AhMXZ190701 site. d Validation of SNP-based markers in high-oil and low-oil cultivars and in the parents of RIL population (Xuhua 13 and Zhonghua 6). Within plots, the bars with different letters are significantly different according to Bonferroni’s Multiple Comparison Test (P < 0.001)
Both SNP sites were then validated in a panel of 42 cultivars collected from 14 provinces across China (Fig. 4b; Table S1). The oil contents of these 42 cultivars ranged from 47.09 to 57.19% on average over a 3-year period (2014–2016), with mean of 51.61% (Table S1). The resequencing of the genomes of these 42 cultivars (Appendix S1) indicated that the AhMXZ190701 site was polymorphic. Seven accessions had the nucleotide T at the AhMXZ190701 site, while the remaining accessions had the nucleotide C at this site (Fig. 4b; able S1). Of accessions with nucleotide T at the AhMXZ190701 site, the average oil content exceeded 54.9%, whereas the oil content of accessions with nucleotide C at this site was significantly lower, with an average of only 50.9% (P < 0.01; Fig. 4c). It is worth noting that nucleotide T at the AhMXZ190701 site did not occur in any low-oil accessions (oil content < 50%). These results suggested that the AhMXZ190701 SNP locus was highly associated with the trait of oil content and may be a suitable diagnostic marker candidate. Therefore, the marker SNPOCA08 was developed to amplify 561-bp fragments, containing the AhMXZ190701 site (Table S7), from five high-oil cultivars (Zhh7684, 7719, 7789, 7689, and 7688), five low-oil cultivars (Zhh7704, 7642, 7693, 7669, 7671), and the two parents of the RIL population (Xuhua 13 and Zhonghua 6). Sanger sequencing showed that this marker could distinguish between high-oil varieties with 54.1–57.2% oil content and low-oil varieties with 47.1–48.6% oil content, based on nucleotide variation at the AhMXZ190701 site. These results demonstrated that the marker SNPOCA08 could be useful for selecting high-oil lines in the peanut breeding process.
Discussion
Improving oil content of seeds is an important objective for peanut breeding. MAS is an effective tool for accelerating the breeding process, which has been widely deployed in the development of varieties with disease resistance and desirable fatty acid composition in peanut (Chu et al. 2011; Janila et al. 2016a). Employment of MAS for high-oil breeding requires identification of QTLs with closely linked makers for oil content. However, the allotetraploid peanut has a narrow genetic base, which hinders development of a dense genetic map for QTL fine mapping. Based on the next generation sequencing platform, ddRAD-seq could be applied for large-scale SNP mining and genotyping. It is an effective and relatively inexpensive method to construct HDGM. Hence, it has been widely used in many crops for QTL mapping (Baird et al. 2008; Chen et al. 2017; Pei et al. 2018; Zhang et al. 2016; Zhou et al. 2018). In 2014, the first HDGM was constructed for allotetraploid peanut through ddRAD-seq. Subsequently, based on SNP markers, a number of HDGMs were applied for the QTL identification of several agronomic traits, such as fatty acid composition and disease resistance (Agarwal et al. 2018; Hu et al. 2018; Vishwakarma et al. 2016; Wang et al. 2018b; Zhou et al. 2014). In the present study, we constructed and deployed an HDGM in QTL identification and marker development for oil content in peanut. This genetic map consisted of 2595 loci, spanning 2465.62 cM with an average distance of 0.95 cM/locus (Table 3). To our knowledge, the number of independent genetic loci in this map approaches or even exceeds that of other recently reported HDGMs for peanut (Agarwal et al. 2018; Hu et al. 2018; Wang et al. 2018b). Dependent on the same RIL population, a genetic map, consisting of 817 SSR markers, has been developed with a marker interval of 2.1 cM/locus (Luo et al. 2017). The density of the genetic map in the present study has been considerably improved, which could provide more tightly linked markers. In addition, each SNP locus of the HDGM in this study could be precisely mapped to the reference genomes of diploid progenitors, which enables deeper insight into the target genomic location. In the current study, the physical length of QTL intervals ranged from 0.3 to 2.5 Mb, which was identified by locating flanking SNP markers on reference genomes of A. duranensis and A. ipaensis (Table 4). The average number of genes in the QTL intervals was below 75, and lowest number was only 12, for qOCA08.2 (Table S3).
A total of seven QTLs on five LGs were identified in the current study (Table 4). Previous studies have also observed that the trait of oil content was controlled by multiple QTLs, indicating that oil content is polygenetically inherited (Pandey et al. 2014; Shasidhar et al. 2017; Wang et al. 2018a; Wilson et al. 2017). According to the physical position of linked markers in the reference genomes of the two diploids, seven QTLs in the present study were mapped to pseudomolecules A04, A05, and A08 in A. duranensis, and to pseudomolecules B05 and B04 in A. ipaensis. Previously reported QTLs with linked markers for oil content were also located on A04, A05, and A08 (Fig. S4; Pandey et al. 2014; Sarvamangala et al. 2011; Shasidhar et al. 2017; Wilson et al. 2017). For instance, marker PMc348 has been reported to be linked to QTL for oil content and was mapped to 82.3 Mb of A04 (Wilson et al. 2017). However, in the present study, qOCA04.1 was located on a 3.6- to 6.1-Mb interval of A04. Two additional and previously identified QTLs for oil content were found to be located within the marker interval of GM1878 and GM1890, and the marker interval of TC2b09 and RN16F05, which were mapped to the 6.4- to 10.9-Mb and 15.0- and 21.7-Mb regions of A05, respectively (Pandey et al. 2014; Sarvamangala et al. 2011). In the current study, qOCA05.1 was located on the 102.0- to 102.6-Mb regions of A05. It is our understanding that the two parents of the current RIL population (Xuhua 13 × Zhonghua 6) did not have a close genetic relationship with populations previously evaluated for oil content, such as RIL from SunOleic 97R × NC94022, RIL from TG 26 × GPBD 4, and BC3F6 from Florunner × TxAG-6. Thus, the qOCA04.1 and qOCA05.1 in the present study seem not to be the same as previously described QTLs on A04 and A05. However, using linked markers sometimes did not accurately estimate the physical position of the QTLs. The discrepancies in positions between current and previous QTLs may also be due to the relatively low density of previous genetic maps. On chromosome A08, markers Seq 2A05 (Alias 2A5) and TC1E05, that have been previously reported, were found to be adjacent to qOCA08.1 and qOCA08.2 on A08 (Fig. S4). According to the flanking markers, qOCA08.1 was mapped to the 41.9- to 42.7-Mb position of A08. The QTL qOCA08.2 was located approximately 1.3 Mb upstream of qOCA08.1 (Fig. 3). Among the different accessions of A. duranensis, 2A5 was a polymorphic marker and was found to be highly correlated with the oil content trait (Huang et al. 2012). This marker was located at the 39.9-Mb position on chromosome A08, which is close to qOCA08.1 and qOCA08.2. TC1E05 was a flanked marker of mqOC for oil content which was detected in the RIL population from SunOleic 97R × NC94022 (Pandey et al. 2014). This marker was mapped to ~ 1.1 Mb downstream of QTL qOCA08.1 (~ 43.8 Mb of A08). Although it cannot yet be confirmed that qOCA08.1 and qOCA08.2 are the same as previously detected QTLs based on the flanking markers, it is reasonable to assume that the physical region of 39.9–43.8 Mb on chromosome A08 is a hot spot for genes controlling oil content.
The QTL qOCA08.1 with 10.14–27.19% PVE in this region was repeatedly detected in multiple environments, which indicated that it may be a valuable candidate for further fine mapping. In the 0.8-Mb physical region of qOCA08.1, eight SNPs between two parents were found to be located either upstream or downstream, or as an intron of seven genes (Table S4). Of these, Aradu.S0YHI was predominantly expressed in a series of reproductive tissues, in which triacylglycerol was synthesized and accumulated (Fig. S3; Table S3). Aradu.S0YHI encoded a transcription factor that belongs to the NF-Y family. Several members of the NF-Y transcription factor family (e.g., LEC1, ABI3, and FUS3) have been shown to play important roles in the regulation of oil accumulation in seeds (Lu et al. 2016; Mu et al. 2008; Tang et al. 2018). One SNP was detected 38 bp upstream of the start codon for Aradu.S0YHI. However, the link between this variant and gene function remains unclear. In addition to Aradu.S0YHI, Aradu.1F2UP has been reported to be differentially expressed between high-oil cultivars and low-oil cultivars (Wang et al. 2018a). Aradu.1F2UP encodes a flavin-binding monooxygenase family protein, which plays a key role in auxin biosynthesis (Yi et al. 2013). Auxin exerts significant effects on plant development and metabolism, which includes oil accumulation (Zhao 2010, Jusoh et al. 2015). Two SNPs were found to be located 72 bp upstream of the intron of Aradu.1F2UP. It will be interesting to further investigate whether the variant in the promoter of Aradu.1F2UP would lead to a difference in the transcript abundance between both parents of the RIL population. Although SNP analysis, expression profile analysis, and gene annotation indicate these putative genes in the QTL interval as candidates for controlling oil content, it is still possible that key genes may sometimes exist outside the QTL interval between two significant markers. Further studies that include transgenic assays should be performed in the future to verify these putative genes for oil content.
In the present study, the SNP marker AhMXZ190701 was tightly linked to the major and stable QTL qOCA08.1. Whether in the RIL population or in a panel of cultivars, there was a significant difference in oil content between the allelotypes with nucleotide T and allelotypes with nucleotide C at site AhMXZ190701 (Fig. 4; Table S5). Agronomic traits for the RIL population, such as 100-pod weight and plant height, have been previously assessed (Lv et al. 2018; Luo et al. 2017). Phenotypic differences for these additional agronomic traits across multiple environments were not observed among different genotypes of the SNP marker AhMXZ190701 (Table S6). These results suggested that the SNP AhMXZ190701 locus may be highly associated with the oil content trait, while it may not be linked with additional agronomic traits. Based on the AhMXZ190701 locus, the marker SNPOCA08 was developed to distinguish high-oil and low-oil cultivars through Sanger sequencing (Fig. 4d). Since the oil content trait is affected by the environment, the deployment of the marker SNPOCA08 for trait selection can help to enhance the efficiency of high-oil breeding. It is notable that most peanut cultivars (35 of 42) selected from the main production areas in China did not have the high-oil allele (nucleotide A) at the AhMXZ190701 locus. Consequently, this marker may be valuable for introducing the superior allele into peanut cultivars for improvement of oil content.
In summary, we constructed a high dense genetic map covering 2595 loci with an average distance of 0.95 cM/locus. Using 4-year phenotypic data of the RIL population, seven QTLs for the oil content were identified by high-resolution mapping. The consensus QTL qOCA08.1 with up to 27.19% PVE was located in a 0.8-Mb interval of chromosome A08, which contained two genes influencing oil synthesis. This stable and major QTL allowed us to develop a marker SNPOCA08 for the flanked SNP site AhMXZ19070 associated with the trait of oil content. These results will facilitate the further investigation of the genetic basis of oil synthesis, and may also provide a useful marker in MAS for a high-oil trait in peanut.
References
Agarwal G, Clevenger J, Pandey MK, Wang H, Shasidhar Y, Chu Y, Fountain JC, Choudhary D, Culbreath AK, Liu X et al (2018) High-density genetic map using whole-genome resequencing for fine mapping and candidate gene discovery for disease resistance in peanut. Plant Biotechnol J 16:1954–1967
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 3:e3376
Baring MR, Wilson JN, Burow MD, Simpson CE, Ayers JL, Cason JM (2013) Variability of total oil content in peanut across the state of texas. J Crop Improv 27:125–136
Bates PD, Johnson SR, Cao X, Li J, Nam JW, Jaworski JG, Ohlrogge JB, Browse J (2014) Fatty acid synthesis is inhibited by inefficient utilization of unusual fatty acids for glycerolipid assembly. Proc Natl Acad Sci U S A 111:1204–1209
Belamkar V, Selvaraj MG, Ayers JL, Payton PR, Puppala N, Burow MD (2011) A first insight into population structure and linkage disequilibrium in the US peanut minicore collection. Genetica 139:411–429
Bertioli DJ, Cannon SB, Froenicke L, Huang G, Farmer AD, Cannon EKS, Liu X, Gao D, Clevenger J, Dash S et al (2016) The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat Genet 48:438–446
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140
Chen J, Wang B, Zhang Y, Yue X, Li Z, Liu K (2017) High-density ddRAD linkage and yield-related QTL mapping delimits a chromosomal region responsible for oil content in rapeseed (Brassica napus L.). Breed Sci 67:296–306
Chu Y, Wu CL, Holbrook CC, Tillman BL, Person G, Ozias-Akins P (2011) Marker-assisted selection to pyramid nematode resistance and the high oleic trait in peanut. Plant Genome 4:110–117
Chutimanitsakun Y, Nipper RW, Cuestamarcos A, Cistué L, Corey A, Filichkina T, Johnson EA, Hayes PM (2011) Construction and application for QTL analysis of a restriction site associated DNA (RAD) linkage map in barley. BMC Genom 12:4
Clevenger J, Chu Y, Scheffler B, Ozias-Akins P (2016) A developmental transcriptome map for Allotetraploid Arachis hypogaea. Front Plant Sci 7:1446
FAOSTAT (2016) Statistical database FAOSTAT. http://faostat3.fao.org
Hu XH, Zhang SZ, Miao HR, Cui FG, Shen Y, Yang WQ, Xu TT, Chen N, Chi XY, Zhang ZM, Chen J (2018) High-density genetic map construction and identification of QTLs controlling oleic and linoleic acid in peanut using SLAF-seq and SSRs. Sci Rep 8:5479
Huang L, Jiang HF, Ren XP, Chen YN, Xiao YJ, Zhao XY, Tang M, Huang JQ, Upadhyaya HD, Liao BS (2012) Abundant microsatellite diversity and oil content in wild Arachis species. PLoS ONE 7:e50002
Huang L, He HY, Chen WG, Ren XP, Chen YN, Zhou XJ, Xia YL, Wang XL, Jiang XG, Liao BS et al (2015) Quantitative trait locus analysis of agronomic and quality-related traits in cultivated peanut (Arachis hypogaea L.). Theor Appl Genet 128:1103–1115
Janila P, Pandey MK, Shasidhar Y, Variath MT, Sriswathi M, Khera P, Manohar SS, Nagesh P, Vishwakarma MK, Mishra GP (2016a) Molecular breeding for introgression of fatty acid desaturase mutant alleles (ahFAD2A and ahFAD2B) enhances oil quality in high and low oil containing peanut genotypes. Plant Sci 242:203–213
Janila P, Variath MT, Pandey MK, Desmae H, Motagi BN, Okori P, Manohar SS, Rathnakumar AL, Radhakrishnan T, Liao B et al (2016b) Genomic tools in groundnut breeding program: status and perspectives. Front Plant Sci 7:289
Jusoh M, Loh SH, Chuah TS, Aziz A, Cha TS (2015) Indole-3-acetic acid (IAA) induced changes in oil content, fatty acid profiles and expression of four fatty acid biosynthetic genes in Chlorella vulgaris at early stationary growth phase. Phytochemistry 111:65–71
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645
Liu N, Li M, Hu X, Ma Q, Mu Y, Tan Z, Xia Q, Zhang G, Nian H (2017) Construction of high-density genetic map and QTL mapping of yield-related and two quality traits in soybean RILs population by RAD-sequencing. BMC Genom 18:466
Lu X, Li QT, Xiong Q, Li W, Bi YD, Lai YC, Liu XL, Man WQ, Zhang WK, Ma B et al (2016) The transcriptomic signature of developing soybean seeds reveals the genetic basis of seed trait adaptation during domestication. Plant J 86:530–544
Luo H, Guo J, Ren X, Chen W, Huang L, Zhou X, Chen Y, Liu N, Xiong F, Lei Y, Liao B, Jiang H (2017) Chromosomes A07 and A05 associated with stable and major QTLs for pod weight and size in cultivated peanut (Arachis hypogaea L.). Theor Appl Genet 131:1–16
Lv J, Liu N, Guo J, Xu Z, Li X, Li Z, Luo H, Ren X, Huang L, Zhou X, Chen Y, Chen W, Lei Y, Tu J, Jiang H, Liao B (2018) Stable QTLs for plant height on chromosome A09 identified from two mapping populations in peanut (Arachis hypogaea L.). Front Plant Sci 9:684
Men L, Li H, Zhang L, Wang J (2015) QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J 3:269–283
Mu J, Tan H, Zheng Q, Fu F, Liang Y, Zhang J, Yang X, Wang T, Chong K, Wang XJ, Zuo J (2008) LEAFY COTYLEDON1 is a key regulator of fatty acid biosynthesis in Arabidopsis. Plant Physiol 148:1042–1054
Mukri G, Nadaf HL, Bhat RS, Gowda MVC, Upadhyaya HD, Sujay V (2012) Phenotypic and molecular dissection of ICRISAT mini core collection of peanut (Arachis hypogaea L.) for high oleic acid. Plant Breed 131:418–422
Pandey MK, Wang ML, Qiao L, Feng S, Khera P, Wang H, Tonnis B, Barkley NA, Wang J, Holbrook CC et al (2014) Identification of QTLs associated with oil content and mapping FAD2 genes and their relative contribution to oil quality in peanut (Arachis hypogaea L.). BMC Genet 15:133
Pei R, Zhang J, Tian L, Zhang S, Han F, Yan S, Wang L, Li B, Sun J (2018) Identification of novel QTL associated with soybean isoflavone content. Crop J 6:244–252
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7:e37135
Pfender WF, Saha MC, Johnson EA, Slabaugh MB (2011) Mapping with RAD (restriction-site associated DNA) markers to rapidly identify QTL for stem rust resistance in Lolium perenne. Theor Appl Genet 122:1467–1480
Sarvamangala C, Gowda MVC, Varshney RK (2011) Identification of quantitative trait loci for protein content, oil content and oil quality for groundnut (Arachis hypogaea L.). Field Crops Res 122:49–59
Shasidhar Y, Vishwakarma MK, Pandey MK, Janila P, Variath MT, Manohar SS, Nigam SN, Guo B, Varshney RK (2017) Molecular mapping of oil content and fatty acids using dense genetic maps in groundnut (Arachis hypogaea L.). Front Plant Sci 8:794
Stam P (1993) Construction of integrated genetic linkage maps by means of a new computer package: join map. Plant J 3:739–744
Sturn A, Quackenbush J, Trajanoski Z (2002) Genesis: cluster analysis of microarray data. Bioinformatics 18(1):207–208
Tang G, Xu P, Ma W, Wang F, Liu Z, Wan S, Shan L (2018) Seed-specific expression of AtLEC1 increased oil content and altered fatty acid composition in seeds of peanut (Arachis hypogaea L.). Front Plant Sci 9:260
Varshney RK (2016) Exciting journey of 10 years from genomes to fields and markets: some success stories of genomics-assisted breeding in chickpea, pigeonpea and groundnut. Plant Sci 242:98–107
Varshney RK, Bertioli DJ, Moretzsohn MC, Vadez V, Krishnamurthy L, Aruna R, Nigam SN, Moss BJ, Seetha K, Ravi K (2009) The first SSR-based genetic linkage map for cultivated groundnut (Arachis hypogaea L.). Theor Appl Genet 118:729–739
Varshney RK, Pandey MK, Janila P, Nigam SN, Sudini H, Gowda VC et al (2014) Marker-assisted introgression of a QTL region to improve rust resistance in three elite and popular varieties of peanut (Arachis hypogaea L.). Theor Appl Genet 127:1771–1781
Vishwakarma MK, Pandey MK, Shasidhar Y, Manohar SS, Nagesh P, Janila P, Varshney RK (2016) Identification of two major quantitative trait locus for fresh seed dormancy using the diversity arrays technology and diversity arrays technology-seq based genetic map in Spanish-type peanuts. Plant Breed 135:367–375
Wang H, Penmetsa RV, Yuan M, Gong L, Zhao Y, Guo B, Farmer AD, Rosen BD, Gao J, Isobe S et al (2012) Development and characterization of BAC-end sequence derived SSRs, and their incorporation into a new higher density genetic map for cultivated peanut (Arachis hypogaea L.). BMC Plant Biol 12:1–11
Wang ML, Khera P, Pandey MK, Wang H, Qiao L, Feng S, Tonnis B, Barkley NA, Pinnow D, Holbrook CC et al (2015) Genetic mapping of QTLs controlling fatty acids provided insights into the genetic control of fatty acid synthesis pathway in peanut (Arachis hypogaea L.). PLoS ONE 10:e0119454
Wang X, Xu P, Yin L, Ren Y, Li S, Shi Y, Alcock TD, Xiong Q, Qian W, Chi X et al (2018a) Genomic and transcriptomic analysis identified gene clusters and candidate genes for oil content in peanut (Arachis hypogaea L.). Plant Mol Biol Rep 36:518–529
Wang Z, Huai D, Zhang Z, Cheng K, Kang Y, Wan L, Yan L, Jiang H, Lei Y, Liao B (2018b) Development of a high-density genetic map based on specific length amplified fragment sequencing and its application in quantitative trait loci analysis for yield-related traits in cultivated peanut. Front Plant Sci 9:827
Wilson D, Charoensawan V, Kummerfeld SK, Teichmann SA (2008) DBD—taxonomically broad transcription factor predictions: new content and functionality. Nucleic Acids Res 36:D88–D92
Wilson J, Baring M, Burow M, Rooney W, Simpson C (2013) Generation means analysis of oil concentration in peanut. J Crop Improv 27:85–95
Wilson JN, Chopra R, Baring MR, Selvaraj MG, Simpson CE, Chagoya J, Burow MD (2017) Advanced backcross quantitative trait loci (QTL) analysis of oil concentration and oil quality traits in peanut (Arachis hypogaea L.). Trop Plant Biol 10:1–17
Wu J, Li LT, Li M, Khan MA, Li XG, Chen H, Yin H, Zhang SL (2014) High-density genetic linkage map construction and identification of fruit-related QTLs in pear using SNP and SSR markers. J Exp Bot 65:5771–5781
Yi J, Liu L, Cao Y, Li J, Mei M (2013) Cloning, characterization and expression of OsFMO(t) in rice encoding a flavin monooxygenase. J Genet 92:471–480
Yol E, Ustun R, Golukcu M, Uzun B (2017) Oil content, oil yield and fatty acid profile of groundnut germplasm in mediterranean climates. J Am Oil Chem Soc 94:787–804
Zhang Z, Shang H, Shi Y, Huang L, Li J, Ge Q, Gong J, Liu A, Chen T, Wang D et al (2016) Construction of a high-density genetic map by specific locus amplified fragment sequencing (SLAF-seq) and its application to quantitative trait loci (QTL) analysis for boll weight in upland cotton (Gossypium hirsutum.). BMC Plant Biol 16:79
Zhao YD (2010) Auxin biosynthesis and its role in plant development. Annu Rev Plant Biol 61(1):49–64
Zhou XJ, Xia YL, Ren XP, Chen YL, Huang L, Huang SM, Liao BS, Lei Y, Yan LY, Jiang HF (2014) Construction of a SNP-based genetic linkage map in cultivated peanut based on large scale marker development using next-generation double-digest restriction-site-associated DNA sequencing (ddRADseq). BMC Genom 15:14
Zhou GF, Jian JB, Wang PH, Li CD, Tao Y, Li X, Renshaw D, Clements J, Sweetingham M, Yang HA (2018) Construction of an ultra-high density consensus genetic map, and enhancement of the physical map from genome sequencing in Lupinus angustifolius. Theor Appl Genet 131:209–223
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (31471534, 31601340, 31801403, and 31871666), the National Peanut Industry Technology System Construction (CARS-13), the Plant Germplasm Resources Sharing Platform (NICGR2018-36), the National Program for Crop Germplasm Protection of China (2018NWB033), the Science and technology innovation Project of Chinese Academy of Agricultural Sciences, and the Fundamental Research Funds for Central Non-profit Scientific Institution (No. Y2018PT52).
Author information
Authors and Affiliations
Contributions
NL, JG, XZ, LH, HL, YL, BL, and HJ conceived and designed the research. HJ and YC developed the RIL population. JG and WC planted the materials and conducted field management. NL and JG performed the measurement of oil content and statistical analysis of the phenotyping data. NL, XZ and BW performed ddRAD-seq, QTL analysis, and development and validation of the marker associated with oil content. NL and HL performed SNP analysis. NL and JG wrote the manuscript, YH, BL, and HJ revised the manuscript and improved the English writing. All the authors read and approved the final manuscript.
Corresponding author
Additional information
Communicated by Brent Hulke.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Liu, N., Guo, J., Zhou, X. et al. High-resolution mapping of a major and consensus quantitative trait locus for oil content to a ~ 0.8-Mb region on chromosome A08 in peanut (Arachis hypogaea L.). Theor Appl Genet 133, 37–49 (2020). https://doi.org/10.1007/s00122-019-03438-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-019-03438-6