
Abstract
Background
Two phase I studies assessed the pharmacokinetics of buprenorphine, its metabolite norbuprenorphine, and naloxone following administration of buprenorphine/naloxone sublingual tablets in Chinese participants.
Methods
In the first phase I, open-label, single ascending-dose (SAD) study, 82 opioid-naïve volunteers received a single buprenorphine/naloxone dose ranging from 2 mg/0.5 mg to 24 mg/6 mg while under naltrexone block. In a second phase I, open-label, multiple ascending-dose (MAD) study, 27 patients with opioid dependence in withdrawal received buprenorphine/naloxone doses of either 16 mg/4 mg or 24 mg/6 mg for 9 consecutive days. Serial blood samples were collected after a single dose (SAD study) and at steady-state (MAD study). Pharmacokinetic parameters were calculated using non-compartmental analysis. Safety assessments included adverse events monitoring and laboratory tests.
Results
The pharmacokinetic profiles of buprenorphine and naloxone were consistent between single- and multiple-dose studies. Peak plasma concentrations (Cmax) were reached early for buprenorphine (0.75–1.0 h) and naloxone (0.5 h), supporting rapid absorption. In the SAD study, increases in plasma exposures to buprenorphine and naloxone were less than dose proportional, in line with previous observations in Western populations. Buprenorphine-to-naloxone ratios for Cmax and area under the curve (AUC) were constant over the dose range investigated and also consistent with Western populations data. Steady state was reached within 7 days of daily dosing, with slight accumulation over repeated doses. No serious adverse events were observed.
Conclusions
The present data suggest that buprenorphine/naloxone pharmacokinetic profiles in Chinese participants are consistent, overall, with those in Western populations, supporting no differences in dosing.
Clinical Trial Registration
The protocols were registered on the official website of the China Food and Drug Administration (CFDA): http://www.chinadrugtrials.org.cn/; Registration numbers CTR20132963 (RB-CN-10-0012), CTR20140153 (RB-CN-10-0015).
Similar content being viewed by others

The current studies suggest that buprenorphine/naloxone pharmacokinetic profiles in Chinese participants are consistent, overall, with those in Western populations. |
These findings suggest that no differences in buprenorphine dosing are needed for patients with opioid use disorder in China. |
1 Introduction
Opioid abuse and opioid use disorder (OUD) are serious health issues in countries around the world, including China [1,2,3,4,5]. In 2013, there were approximately 2.5 million registered drug users in China. Users in China are registered if they seek treatment or come in contact with law enforcement; as such, estimates place the actual number of users closer to 14 million, although not all are using opioids [6]. Compared with other countries, medical interventions for OUD are limited in China [1, 2, 4, 7]. Most commonly, traditional Chinese medicines (e.g., Fukang tablets, Xuanxia detoxification capsules) consisting of primarily herbal mixtures are used. A recent meta-analysis has demonstrated that these medicines are less effective than pharmacological interventions in the critical early days of withdrawal and less effective than methadone overall [8]. Methadone, a full agonist at the µ-opioid receptor, is also approved for maintenance treatment in China, but requires specialty clinics that are licensed, and is associated with an increased risk of respiratory depression and fatal overdose [3, 4, 9, 10]. Further, patients may misuse and abuse methadone, possibly becoming addicted and suffering from withdrawal [8, 11, 12].
Based on robust randomized clinical trials, buprenorphine/naloxone (SUBOXONE®) sublingual film and sublingual tablets have been approved in more than 40 countries worldwide for the treatment of OUD, but are not yet approved in China [13,14,15,16,17]. Buprenorphine is a partial agonist at the µ-opioid receptor that can attenuate withdrawal symptoms while lowering the risk of respiratory depression and fatal overdose associated with full agonists [16]. Naloxone is a full antagonist at the µ-opioid receptor that is poorly absorbed when administered sublingually; however, when injected, naloxone precipitates withdrawal in opioid-dependent individuals currently taking full agonist opioids. As a result, the buprenorphine/naloxone formulation deters parenteral abuse of the sublingual film and tablet, which is an important factor to consider in the treatment of OUD, though misuse and abuse remain a concern here as well. Of note, the co-administration of naloxone has no impact on the pharmacokinetics (PK) of buprenorphine [18].
A phase III, randomized, placebo-controlled study evaluating the efficacy and safety of buprenorphine/naloxone sublingual tablets for the treatment of opioid dependence (according to DSM-IV-TR criteria) was conducted in China and enrolled adult Chinese nationals who were opioid-dependent [19]. In that study, 130 participants were randomized to receive buprenorphine/naloxone dosages ranging from 2 mg/0.5 mg to 24 mg/6 mg daily, based on individual participants’ withdrawal symptoms and abstinence from illicit opioids, and 130 participants were randomized to treatment with placebo. Buprenorphine/naloxone treatment was superior to placebo on the primary outcome of treatment failures. Buprenorphine/naloxone treatment also significantly reduced withdrawal and craving symptoms, increased consecutive days of abstinence, and decreased illicit opioid usage, compared with placebo [19]. Due to the wide range of therapeutic dosages, both in the phase III study and in real-world usage, a PK evaluation was undertaken to better understand the PK profile after administration of buprenorphine/naloxone sublingual tablets in the Chinese population. Two phase I studies were conducted to assess the PK profiles of buprenorphine, norbuprenorphine (major metabolite of buprenorphine), and naloxone following administration of buprenorphine/naloxone sublingual tablets in (i) healthy Chinese volunteers under a naltrexone block and (ii) Chinese patients in withdrawal treatment for opioid dependence. The results of these two phase I studies are reported here and discussed in comparison with data previously obtained in Western populations.
2 Methods
2.1 Study Designs
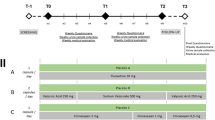
Study 1 (RB-CN-10-0012) was a phase I, open-label, parallel-group, single ascending-dose (SAD) study evaluating the PK profiles and safety of buprenorphine, norbuprenorphine, and naloxone following administration of buprenorphine/naloxone sublingual tablets under a naltrexone block in healthy Chinese volunteers. The study consisted of eight inpatient days, with administration on day 1 of one of six possible buprenorphine/naloxone doses ranging from 2 mg/0.5 mg to 24 mg/6 mg.
Study 2 (RB-CN-10-0015) was a phase I, open-label, parallel-group, multiple ascending-dose (MAD) study evaluating the steady-state PK profiles and safety of buprenorphine, norbuprenorphine, and naloxone following repeated daily administration of buprenorphine/naloxone sublingual tablets in Chinese patients who were in withdrawal treatment for opioid dependence. The study comprised three stages: (i) dose titration with buprenorphine/naloxone sublingual tablets for days 1–5 or days 1–7, (ii) stable doses of buprenorphine/naloxone at either 16 mg/4 mg or 24 mg/6 mg for 9 days, and (iii) dose reduction for 10–15 days.
For both studies, a follow-up visit to assess safety and tolerability was conducted 7 days after the end of treatment, either in the clinic (study 1) or via telephone (study 2).
2.2 Treatments and Dosing
Study 1 participants were allocated to one of six dose cohorts: 2 mg/0.5 mg (n = 8); 4 mg/1 mg (n = 8); 8 mg/2 mg (n = 16); 12 mg/3 mg (n = 16); 16 mg/4 mg (n = 18); and 24 mg/6 mg (n = 16). Buprenorphine/naloxone sublingual tablets were provided as buprenorphine 2 mg and naloxone 0.5 mg, or buprenorphine 8 mg and naloxone 2 mg [Reckitt Benckiser Healthcare (UK) Ltd., now Indivior Inc.].
Oral naltrexone 100 mg (Beijing Huasa Pharmaceutical Company, China) was administered 12 h and 1 h prior to buprenorphine/naloxone dosing; oral naltrexone 50 mg was administered 12 and 24 h following buprenorphine/naloxone dosing. Naltrexone is a well-tolerated µ-opioid receptor antagonist and was administered to prevent or attenuate buprenorphine-related effects in opioid-naïve participants (e.g., nausea, vomiting, euphoria, and at higher doses, respiratory depression).
Study 2 participants received buprenorphine/naloxone sublingual tablets with dosages titrated based on Clinical Opiate Withdrawal Scale (COWS) and Opioid Craving Visual Analog Scale (VAS) scores for either 5 days or 7 days. Patients were stabilized at randomized daily dosages of buprenorphine/naloxone 16 mg/4 mg (n = 15) or 24 mg/6 mg (n = 12) for 9 consecutive days. Following dose stabilization, buprenorphine/naloxone dose was gradually reduced to zero over 10–15 days based on clinical symptoms (e.g., withdrawal, craving); if a patient’s dose could not be reduced to zero, methadone maintenance treatment was started or the patient was transferred to a detoxification center.
2.3 Participants
Both studies were conducted in China. In study 1, eligible participants were healthy men or women aged 18–65 years (inclusive), with a body mass index (BMI) of 18.5–30 kg/m2 (inclusive), and weighing at least 50 kg. Study 1 participants were required to have urine drug screens negative for opioids and other drugs of abuse.
In study 2, participants were men or women, aged 18–55 years (inclusive), with a BMI of 18.5–30 kg/m2 (inclusive), and weighing at least 45 kg. Study 2 participants were in withdrawal treatment for opioid dependence and may have had positive urine drug screens for morphine or methadone before taking study treatment.
Participants in both studies were excluded if they had a history or current presence of disease, co-existing major psychiatric illness, or any significant condition known to interfere with the absorption, distribution, metabolism, or excretion of buprenorphine or naloxone. Other exclusion criteria included clinically significant abnormal findings on the physical exam, electrocardiogram (ECG), or vital signs, and treatment with cytochrome P450 3A4 or cytochrome P450 2C8 inhibitors or inducers (e.g., barbiturates, carbamazepine, erythromycin, phenytoin, thiazolidinediones, rifampicin) within 30 days.
Both studies were conducted in compliance with the guidelines of the Declaration of Helsinki and the principles of Good Clinical Practice. Study protocols, amendments, informed consent forms, and all other study materials were reviewed and approved by the Ethics Committee of the People’s Liberation Army 307 Hospital. All participants provided written informed consent.
2.4 Blood Sampling Procedures
In study 1, blood samples were drawn prior to buprenorphine/naloxone dosing, and at 5 min, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 2, 3, 4, 6, 8, 10, 12, 24, 36, 48, 72, 96, 120, and 144 h post-dose. In study 2, blood samples were drawn on day 9 prior to buprenorphine/naloxone dosing, and at 5 min, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 2, 3, 4, 6, 8, 10, 12, and 24 h post-dose. In study 2, blood samples were also collected prior to buprenorphine/naloxone dosing on days 7 and 8 to evaluate achievement of steady-state concentrations.
All PK blood samples were immediately placed into an ice bath after collection. Blood samples were then centrifuged at approximately 3000 rpm for 10 min at below 4 °C. Plasma concentrations of buprenorphine, norbuprenorphine, and naloxone were determined using liquid chromatography electrospray tandem mass spectrometry (LC-ESI-MS/MS) assays. The assays were fully validated for linearity, selectivity, recovery, matrix effect, accuracy, precision, and stability before their application to sample analysis. The lower limit of quantitation was 0.0250 ng/mL for both buprenorphine and norbuprenorphine, and 0.002 ng/mL for naloxone. During the method validation, the accuracy (overall bias) ranged from − 6.8 to 3.5% for buprenorphine, from − 9.6 to 1.8% for norbuprenorphine, and from 2.0 to 4.7% for naloxone. The precision ranged from 4.6 to 5.6% for buprenorphine, from 3.4 to 13.3% for norbuprenorphine, and from 3.7 to 6.2% for naloxone. The overall accuracy and precision for quality control samples during the sample analyses were all within 15%. All the plasma samples were analyzed within the established stability window.
2.5 Pharmacokinetic Outcomes
PK parameters analyzed included the area under the plasma concentration–time curve (AUC) from time zero to the time of the last quantifiable concentration (AUC0–last), AUC from time zero extrapolated to infinity (AUC0–inf), AUC over the daily dosing interval at steady-state (AUC0–24,ss), the maximum observed plasma concentration (Cmax), the time to reach Cmax (Tmax), plasma terminal half-life (t½), apparent clearance (CL/F) and apparent volume of distribution (Vz/F). In study 2, pre-dose concentrations (Ctrough) were assessed from day 7 to day 10 to evaluate achievement of steady state. Individual plasma concentration–time data for buprenorphine, norbuprenorphine, and naloxone were analyzed using non-compartmental analysis by WinNonlin (Version 6.3, Pharsight Corporation, Mountain View, CA, USA).
2.6 Safety and Tolerability Assessments
Safety assessments included monitoring of adverse events (AEs), vital signs, 12-lead ECGs, physical examinations, clinical laboratory tests, and use of concomitant medications.
2.7 Statistical Analysis
The safety population was defined as all participants who were enrolled in the study and received at least one dose of study medication. The PK population was defined as all participants who received at least one dose of buprenorphine/naloxone and had an adequate number of blood draws to perform PK evaluation.
In study 1, a power model was used to evaluate dose proportionality of AUC0–last, AUC0–inf, and Cmax within the 2- to 24-mg dose range for buprenorphine and its metabolite, and the 0.5- to 6-mg dose range for naloxone: \(\ln \left( {{\text{PK}}\,{\text{parameter}}} \right) = \beta_{0} + \beta_{1} \ln \left( {\text{dose}} \right)\), where β0 and β1 were estimated. Dose proportionality was established when the 90% confidence interval of the β1 estimate fell within \(\left[ {1 + \frac{{\ln \left( {0.8} \right)}}{\ln \left( r \right)}, 1 + \frac{{\ln \left( {1.25} \right)}}{\ln \left( r \right)}} \right]\); here r = high dose/low dose [20].
In study 2, steady state was assessed for buprenorphine, norbuprenorphine, and naloxone using the Helmert transformation approach [21] within an analysis of variance (ANOVA), with natural log-transformed Ctrough concentrations as the dependent variable and day as a fixed effect. A series of contrasts were used to compare the mean Ctrough value of the first tested day (day 7) to the pooled mean over all remaining time points (days 8, 9, and 10) for each dose level. Testing continued until the contrast was not statistically significant at the 0.1 level; the first time point at which the contrast was not significant indicated that steady state was attained.
No formal sample size calculation was performed for either study. In study 1, at the request of the China Food and Drug Administration (CFDA), a sample size of eight participants for the 2-mg/0.5-mg and 4-mg/1-mg dose groups with six completers in each group, and a sample size of 16 participants for the 8-mg/2-mg, 12-mg/3-mg, 16-mg/4-mg, and 24-mg/6-mg dose groups with 12 completers in each group was considered appropriate for this study. Therefore, a total of 80 participants were targeted for enrollment. In study 2, a sample size of 48 participants (24 randomly assigned to each group) was targeted, assuming a 50% drop out rate, to reach a minimum of 12 participants per group.
Statistical analyses were performed using SAS® System Version 9.2 (SAS Institute, Inc., Cary, NC, USA).
3 Results
3.1 Participant Disposition and Demographics
Study 1 enrolled 82 participants, all of whom were included in the safety population (Fig. S1A, see electronic supplementary material [ESM]). Six participants discontinued after receiving a single dose of naltrexone and prior to receiving buprenorphine/naloxone sublingual tablet(s) due to withdrawal of consent. A total of 76 (92.6%) participants completed the study, but one participant vomited before complete melting of the buprenorphine/naloxone sublingual tablet and was therefore not included in the PK analysis (n = 75, PK population). The majority were Han ethnicity (96.3%), 51.2% were women, and the mean (SD) age was 27.7 (5.2) years (Table 1).
Study 2 enrolled 32 participants, all of whom were included in the safety population, and 25 (78.1%) were included in the PK population (Fig. S1B, see ESM). Five participants (15.6%) discontinued during dose titration; four withdrew consent and one discontinued due to AEs (nausea, vomiting, and abdominal pain). Two additional participants withdrew due to AEs (one with somnolence, dizziness on stable dosage day 1; one with headache on stable dosage day 2). Most participants were men (87.5%), of Han ethnicity (100%), and older than study 1 participants with a mean (SD) age of 38.6 (7.0) years (Table 1).
3.2 Pharmacokinetic Analysis
3.2.1 Study 1 (SAD)
The mean plasma concentration-time profiles following a single administration of buprenorphine/naloxone sublingual tablets are shown for buprenorphine (Fig. 1a, b), norbuprenorphine (Fig. 1c, d), and naloxone (Fig. 1e, f) over the dose range investigated. PK parameters for all three analytes are summarized in Table 2. Buprenorphine was rapidly absorbed with a median Tmax of 1 h. Norbuprenorphine plasma concentration peaked at a median Tmax of 1–1.75 h after buprenorphine/naloxone dosing. Plasma terminal half-life (t½) ranged on average from 22 to 39 h for buprenorphine and from 32 to 44 h for norbuprenorphine. Naloxone was rapidly absorbed with a median Tmax of 0.5 h; the average naloxone plasma t½ ranged between 1.4 and 10 h.

Mean (+ SD) plasma concentration–time curves of buprenorphine, norbuprenorphine, and naloxone following a single administration of buprenorphine/naloxone sublingual tablets at various doses in healthy Chinese participants (study 1, PK population). SD standard deviation
Statistical evaluation of dose proportionality in study 1 indicated that the plasma exposure to buprenorphine as assessed by AUC and Cmax increased less than dose proportionally over the investigated dose range of 2–24 mg (Table S1). Similar results were observed for naloxone over the dose range of 0.5–6 mg. For both buprenorphine 2–24 mg and naloxone 0.5–6 mg, the point estimates of β1 were all below 1 and the 90% confidence interval (CI) did not fall within the prespecified adjusted ranges. Regarding norbuprenorphine, there was no clear dose proportional relationship for plasma exposure as assessed by AUC and Cmax. The point estimates of β1 were close to 1 or slightly higher than 1; however, 90% CIs were not fully included in the prespecified ranges.
3.2.2 Study 2 (MAD)
Based on statistical analysis, steady-state for buprenorphine, norbuprenorphine, and naloxone was achieved by day 7 in both 16-mg/4-mg and 24-mg/6-mg dose groups. Mean steady-state plasma concentration-time profiles on day 9 are shown for buprenorphine (Fig. 2a), norbuprenorphine (Fig. 2b), and naloxone (Fig. 2c) for each dose group. Pharmacokinetic parameters for all three analytes are summarized in Table 3. Buprenorphine was rapidly absorbed with a median Tmax of 0.75–1 h. Norbuprenorphine concentration peaked at a median Tmax of 0.75–1 h. A 50% increase in buprenorphine dose from 16 to 24 mg resulted in a 26% increase in mean buprenorphine AUC0–24,ss, a 75% increase in mean buprenorphine Cmax,ss, a 37% increase in mean norbuprenorphine AUC0–24,ss, and a 75% increase in mean norbuprenorphine Cmax,ss. Naloxone Tmax occurred at a median 0.5–0.75 h. A 50% increase in naloxone dose from 4 to 6 mg resulted in a 20% increase in mean Cmax,ss of naloxone; naloxone AUC0–24,ss was similar between the two doses.

Mean (+ SD) plasma concentrations on day 9 following repeated daily administration of buprenorphine/naloxone sublingual tablets in Chinese participants with OUD (study 2) on a linear scale. OUD opioid use disorder, SD standard deviation
3.3 Safety
In study 1, 161 AEs were reported by 54 participants; all were mild and resolved (Table 4). In study 2, 17 AEs were reported by 10 participants; 16 were mild and 1 moderate (elevated hepatic enzymes, not clinically significant), all resolved. The most common AEs were vomiting, nausea, and dizziness in study 1 and dizziness and somnolence in study 2. In study 2, one participant discontinued due to AEs (nausea, vomiting, and abdominal pain) during dose titration. During stable dosage, two participants discontinued due to AEs (somnolence, dizziness, in one participant, stable dosage day 1; headache in one participant, stable dosage day 2). No serious AEs or deaths occurred in either study. No clinically significant changes in vital signs, ECGs, laboratory values, and physical examinations occurred.
4 Discussion
This PK evaluation was undertaken to better understand the effect of buprenorphine/naloxone sublingual tablets in the Chinese population. Two phase I studies were conducted in healthy Chinese volunteers under a naltrexone block (single doses; study 1) and in Chinese patients in withdrawal treatment for opioid dependence (multiple doses; study 2). In both phase I studies, buprenorphine reached peak plasma concentrations rapidly at 0.75–1 h post-dose. As expected, buprenorphine AUC at steady state (AUC0–24,ss) was similar to buprenorphine AUC0–inf after a single dose (Table 5). Buprenorphine Cmax at steady state (Cmax,ss) was somewhat higher compared with the single dose, reflecting the slight accumulation of buprenorphine over repeated daily administration.
In study 1, buprenorphine/naloxone sublingual tablets were administered to healthy Chinese volunteers under a naltrexone block. Naltrexone is commonly co-administrated with buprenorphine in healthy volunteers to prevent or attenuate buprenorphine-related effects [22]. Any PK interaction between the two compounds is unlikely because, unlike buprenorphine, naltrexone is not metabolized through either the cytochrome P450 or uracil diphosphate-glucuronosyl transferase pathways [23]. Additionally, the results that buprenorphine AUC0–inf in study 1 closely matches the buprenorphine AUC0–24,ss in study 2 (patients without co-administration of naltrexone) support the lack of PK interaction between naltrexone and buprenorphine.
After single buprenorphine/naloxone doses within the range of 2–24 mg for buprenorphine, buprenorphine AUC values in Chinese participants were very close to those reported for Western populations based on the literature [22] and historical studies conducted by the sponsor [24] (Fig. S2A, see ESM). Larger differences were observed for buprenorphine Cmax; however, Cmax values in Chinese participants were similar to those reported in some Western studies (Fig. S2A, see ESM) [22]. Data from the multiple-dose study confirmed those findings, with similar Cmax,ss and AUC0–24,ss between Chinese participants and Western subjects in a published study [25] (Table 5). Moderate between-subject variability was observed for Cmax (25–46%) and AUC0–inf (24–37%) in the present studies, which was also in agreement with the variability reported for studies in Western populations (Cmax: 25–54%; AUC0–inf: 23–43%). In both Chinese and Western populations, buprenorphine plasma exposure (AUC, Cmax) increased with the dose, albeit slightly less than dose proportionally. Similar values for buprenorphine Tmax and plasma t½ were observed (Tmax of 0.75–2 h and t½ of 24–45 h in Western populations [22, 24]).
Plasma concentrations of norbuprenorphine, the major metabolite of buprenorphine, peaked in the Chinese participants at 1–1.75 h, consistent with previous data for Western populations (median Tmax of 1–1.75 h [24]). Norbuprenorphine plasma t½ ranged between 32 and 44 h on average in Chinese participants, similar to the 35–48 h reported in Western populations [24]. Steady-state norbuprenorphine AUC was approximately 35–40% lower than AUC0–inf after a single dose, which was unexpected. These differences may be explained, at least in part, by the between-subject variability involving genetic differences in UDP-glucuronosyltransferase (UGT) isoforms.
Buprenorphine and its metabolite, norbuprenorphine, are glucuronidated through different UGT isoforms [26]. Norbuprenorphine is glucuronidated primarily by UGT1A3 (63%) and UGT1A1 (34%), while only 10% of buprenorphine is glucuronidated by UGT1A1. Approximately 13–27% of the Asian population has allelic nucleotide changes that produce decreased activity or reduced expression of UGT1A1. Furthermore, evaluation of liver protein content of individual UGT isoforms has shown 2.5- to 25.2-fold individual variability, similar to the mRNA expression level of UGT [27]. This individual variation in UGT1A1 activity and expression likely contributed to the variability in the PK profile of norbuprenorphine across subjects and across the two studies, while having only a mild effect on buprenorphine metabolism. Importantly, there was a large overlap between individual AUCs from the multiple-dose study (AUC0–24,ss) and the single-dose study (AUC0–inf). Also, no differences were found in the PK of buprenorphine, which is the main driver of clinical efficacy. Indeed, although norbuprenorphine has shown some pharmacological activity in vitro, it is expected to have negligible contribution to brain µ-opioid receptor occupancy given its limited ability to cross the blood–brain barrier [28].
As anticipated, the PK profile of naloxone was similar following single and repeated doses, reflecting negligible accumulation due to the short plasma t½. Naloxone plasma t½ was 1.4–2.1 h on average over the 0.5–4 mg dose range; a longer t½ of 10 h was identified at the highest dose of 6 mg as a result of the sensitive bioanalytical method but had a minor contribution to the overall elimination and accumulation of naloxone. Despite some variability in Cmax, naloxone PK in Chinese participants was consistent with previous observations in Western populations (Fig. S2B, see ESM). Importantly, buprenorphine-to-naloxone ratios observed in Chinese participants for Cmax (18.1–23.3) and AUC0–inf (68.9–89.9) were constant over the dose range investigated and were consistent with previous data in Western studies (Cmax: 13.3–24.0; AUC0–inf: 60.9–96.6) (Fig. S2C, see ESM). The high buprenorphine-to-naloxone ratios for Cmax and AUC indicate that naloxone plasma concentrations are low compared with buprenorphine when buprenorphine/naloxone is administered sublingually.
Overall, the PK of buprenorphine, its metabolite, and naloxone observed in the two Chinese studies was consistent with the known PK profiles reported in Western populations. One limitation of this comparison is the use of historical or published results in Western subjects.
5 Conclusions
Despite the limitation of using historical/published results for comparison with Western populations, the PK profiles of buprenorphine and naloxone after single and repeated doses in Chinese participants were consistent, overall, with the known PK profiles of buprenorphine and naloxone in Western populations. The variability observed between single-dose and steady-state PK profiles of norbuprenorphine, the metabolite of buprenorphine, may reflect individual differences in the activity and expression of UGT1A1; however, these differences have little to no effect on buprenorphine metabolism or its clinical efficacy. The PK results presented here suggest that no differences in buprenorphine dosing are needed for patients with OUD in China.
References
Doosti F, Dashti S, Tabatabai SM, Hosseinzadeh H. Traditional Chinese and Indian medicine in the treatment of opioid-dependence: a review. Avicenna J Phytomed. 2013;3(3):205–15.
Li J, Li X. Current status of drug use and HIV/AIDS prevention in drug users in China. J Food Drug Anal. 2013;21(4):S37–41.
Ling W, Casadonte P, Bigelow G, et al. Buprenorphine implants for treatment of opioid dependence: a randomized controlled trial. JAMA. 2010;304(14):1576–83.
Marienfeld C, Liu P, Wang X, Schottenfeld R, Zhou W, Chawarski MC. Evaluation of an implementation of methadone maintenance treatment in China. Drug Alcohol Depend. 2015;157:60–7.
Rosenthal RN, Lofwall MR, Kim S, Chen M, Beebe KL, Vocci FJ. Effect of buprenorphine implants on illicit opioid use among abstinent adults with opioid dependence treated with sublingual buprenorphine: a randomized clinical trial. JAMA. 2016;316(3):282–90.
Zhang SX, Chin K. A people’s war: China’s struggle to contain its illicit drug problem. 2018. https://www.brookings.edu/wp-content/uploads/2016/07/A-Peoples-War-final.pdf. Accessed 11 Oct 2018.
Zhang B, Chen Y, Cheng K, Shen X, Liu S. Efficacy of acupuncture for psychological symptoms associated with opioid addiction: a systematic review and meta-analysis. Evid Based Complement Altern Med eCAM. 2014;2014:313549.
Shi J, Liu YL, Fang YX, Xu GZ, Zhai HF, Lu L. Traditional Chinese medicine in treatment of opiate addiction. Acta Pharmacol Sin. 2006;27(10):1303–8.
Mattick RP, Breen C, Kimber J, Davoli M. Buprenorphine maintenance versus placebo or methadone maintenance for opioid dependence. Cochrane Database Syst Rev. 2014;2:Cd002207.
Tang YL, Hao W. Improving drug addiction treatment in China. Addiction (Abingdon, England). 2007;102(7):1057–63.
Sullivan SG, Wu Z. Rapid scale up of harm reduction in China. Int J Drug Policy. 2007;18(2):118–28.
Sullivan SG, Wu Z, Rou K, et al. Who uses methadone services in China? Monitoring the world’s largest methadone programme. Addiction (Abingdon, England). 2015;110(Suppl 1):29–39.
Hoffman K, Peyton ML, Sumner M. Safety of a rapidly dissolving buprenorphine/naloxone sublingual tablet (BNX-RDT) for treatment of opioid dependence: a multicenter, open-label extension study. J Addict Med. 2017;11(3):217–23.
Schottenfeld RS, Chawarski MC, Mazlan M. Maintenance treatment with buprenorphine and naltrexone for heroin dependence in Malaysia: a randomised, double-blind, placebo-controlled trial. Lancet (London, England). 2008;371(9631):2192–200.
Woody GE. Advances in the treatment of opioid use disorders. F1000Research. 2017;6:87.
Lofwall MR, Walsh SL. A review of buprenorphine diversion and misuse: the current evidence base and experiences from around the world. J Addict Med. 2014;8(5):315–26.
Fudala PJ, Bridge TP, Herbert S, et al. Office-based treatment of opiate addiction with a sublingual-tablet formulation of buprenorphine and naloxone. N Engl J Med. 2003;349(10):949–58.
Harris DS, Jones RT, Welm S, et al. Buprenorphine and naloxone co-administration in opiate-dependent patients stabilized on sublingual buprenorphine. Drug Alcohol Depend. 2000;61:85–94.
Wang XJH, Zhao M, Li J, Gray F, Sheng L, Li Y, Li X, Ling W, Li W, Hao W. Treatment of opioid dependence with buprenorphine/naloxone sublingual tablets: a Phase 3 randomized, double-blind, placebo-controlled trial. Asia Pac Psychiatry. 2019;11(1):e12344.
Smith BP, Vandenhende FR, DeSante KA, et al. Confidence interval criteria for assessment of dose proportionality. Pharm Res. 2000;17(10):1278–83.
Maganti L, Panebianco D, Maes A. Evaluation of methods for estimating time to steady state with examples from phase 1 studies. AAPS J. 2008;10(1):141–7.
McAleer SD, Mills RJ, Polack T, et al. Pharmacokinetics of high-dose buprenorphine following single administration of sublingual tablet formulations in opioid naive healthy male volunteers under a naltrexone block. Drug Alcohol Depend. 2003;72(1):75–83.
Bai SA, Xiang Q, Finn AF. Evaluation of the pharmacokinetics of single-and multiple-dose buprenorphine buccal film in healthy volunteers. Clin Ther. 2016;38(2):358–69.
Investigator’s Brochure, edition date October 14th, 2013. Reckitt Benckiser Pharmaceuticals Inc (unpublished).
Compton P, Ling W, Chiang CN, et al. Pharmacokinetics of buprenorphine: a comparison of sublingual tablet versus liquid after chronic dosing. J Addict Med. 2007;1(2):88–95.
Rouguieg K, Picard N, Sauvage FL, Gaulier JM, Marquet P. Contribution of the different UDP-glucuronosyltransferase (UGT) isoforms to buprenorphine and norbuprenorphine metabolism and relationship with the main UGT polymorphisms in a bank of human liver microsomes. Drug Metab Dispos Biol Fate Chem. 2010;38(1):40–5.
Oda S, Fukami T, Yokoi T, Nakajima M. A comprehensive review of UDP-glucuronosyltransferase and esterases for drug development. Drug Metab Pharmacokinet. 2015;30(1):30–51.
Ohtani M. Basic pharmacology of buprenorphine. Eur J Pain Suppl. 2007;1(1):69–73.
Acknowledgements
Manuscript preparation, including editing and formatting the manuscript, incorporating author comments, preparing tables and figures, and coordinating submission requirements, was provided by Mary Clare Kane, PhD, of Prescott Medical Communications Group (Chicago, IL, USA). This editorial support was funded by Indivior, Inc.
Author information
Authors and Affiliations
Contributions
RD, ZL, JJ and designed the study; RD, DL, and ZL implemented the study. LL and JJ analyzed all PK blood samples and calculated the PK parameters. RD, HW, JJ, and ZL analyzed the data. FG contributed to data review and interpretation and was a contributing author to the manuscript. YL closely monitored all bioanalytical work including method validation and sample analysis, contributed to the analysis of pharmacokinetic data, the interpretation of the study data, and the drafting, critical review and revision of the manuscript for important intellectual content. CL contributed to the analysis of pharmacokinetic data, the interpretation of the study data, and the drafting, critical review and revision of the manuscript for important intellectual content. MY contributed to interpretation of the data and manuscript. SML was accountable for all scientific and medical governance of the clinical trial, including its conduct, analysis, interpretation and reporting.
Corresponding author
Ethics declarations
Funding
Funded by Reckitt Benckiser, Inc., now Indivior, Inc., North Chesterfield, VA, USA.
Conflict of Interest
RD, HW, DL, LL, JJ, and ZL declare no competing interests. FG, YL, CL, MY, and SML are employees of Indivior, Inc and declare no competing interests.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dong, R., Wang, H., Li, D. et al. Pharmacokinetics of Sublingual Buprenorphine Tablets Following Single and Multiple Doses in Chinese Participants With and Without Opioid Use Disorder. Drugs R D 19, 255–265 (2019). https://doi.org/10.1007/s40268-019-0277-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-019-0277-9